
Differentiating between Natural and Modified Cellulosic Fibres Using ATR-FTIR Spectroscopy

 , and
, and
Abstract
1. Introduction
- −
- no sample preparation;
- −
- no band saturation phenomena;
- −
- time and cost saving;
- −
- few micrograms or less are generally necessary;
- −
- non-destructive, as the sample can be re-used for further investigations (although the pressure applied during the analysis can induce morphological modifications);
- −
- extensive database available, as literature dealing with transmission FTIR can be generally extended to ATR spectra.
1.1. Aim of the Study
1.2. Theoretical Background
2. Materials and Methods
2.1. Reference Materials
2.2. The Morigi Collection of Traditional Japanese Armours
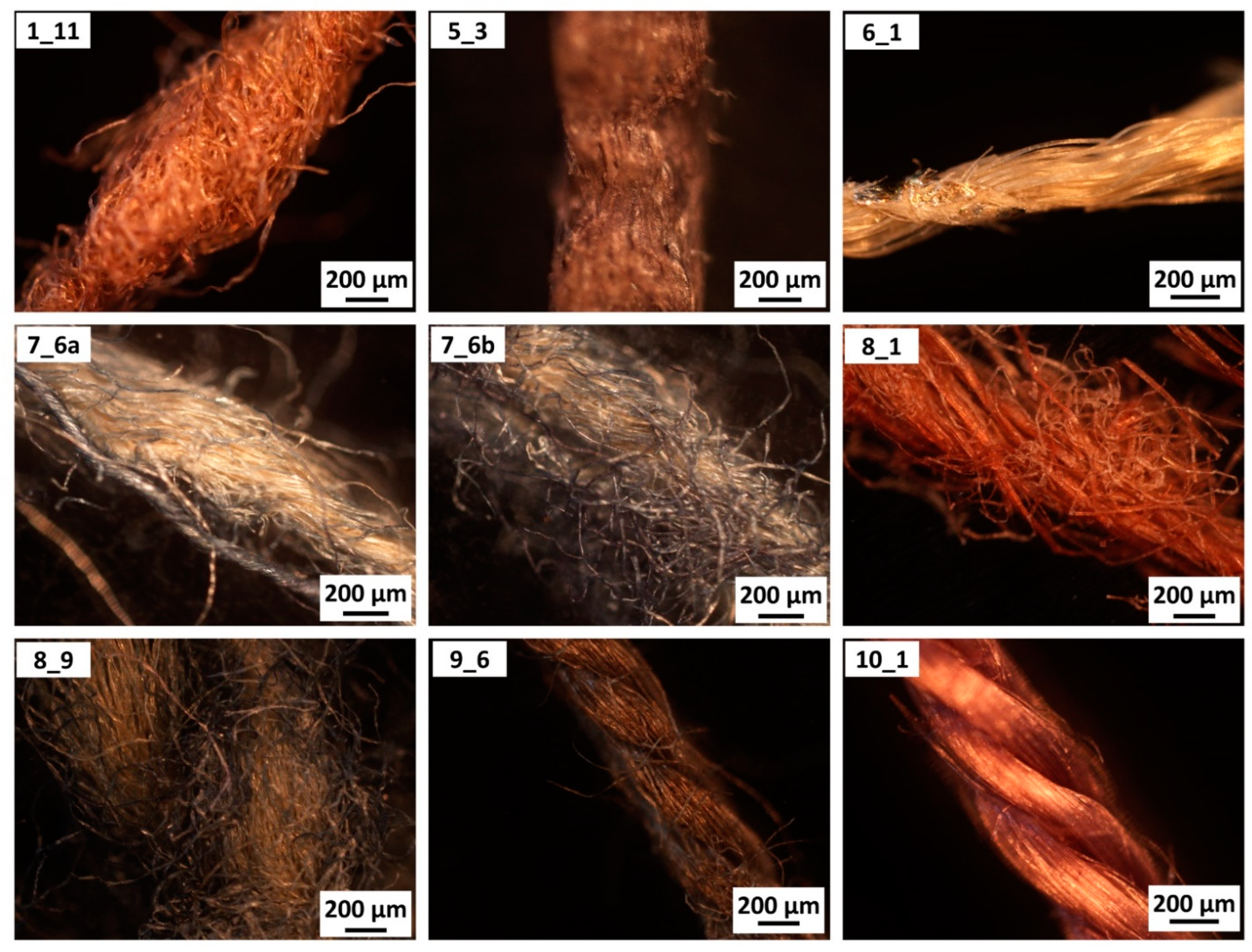
2.3. Sampling
2.4. Optical Microscopy
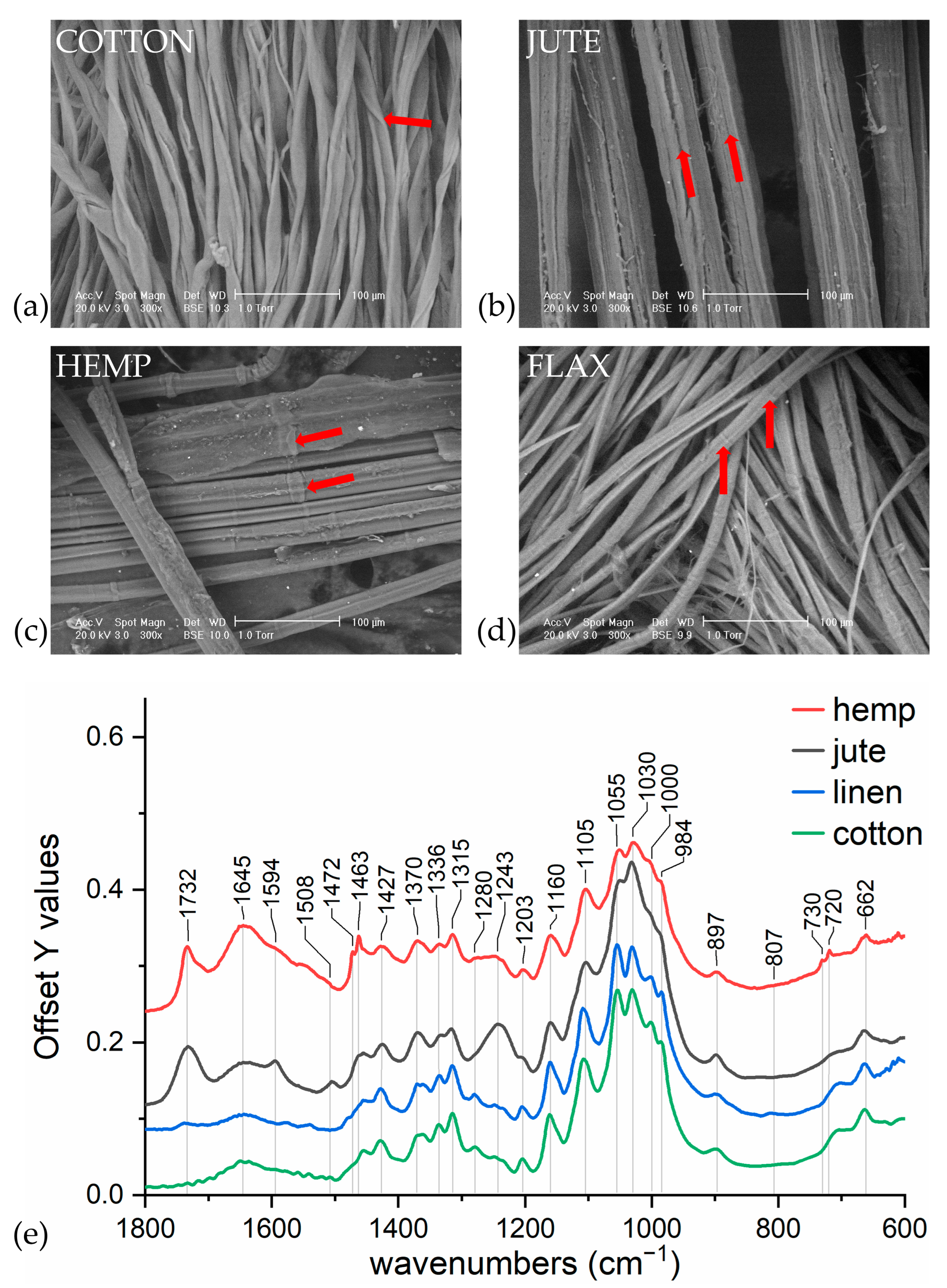
2.5. Scanning Electron Microscopy (SEM)
2.6. Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy
2.7. Data Treatment and Elaboration
2.8. Spectral Analysis and Curve-Fitting
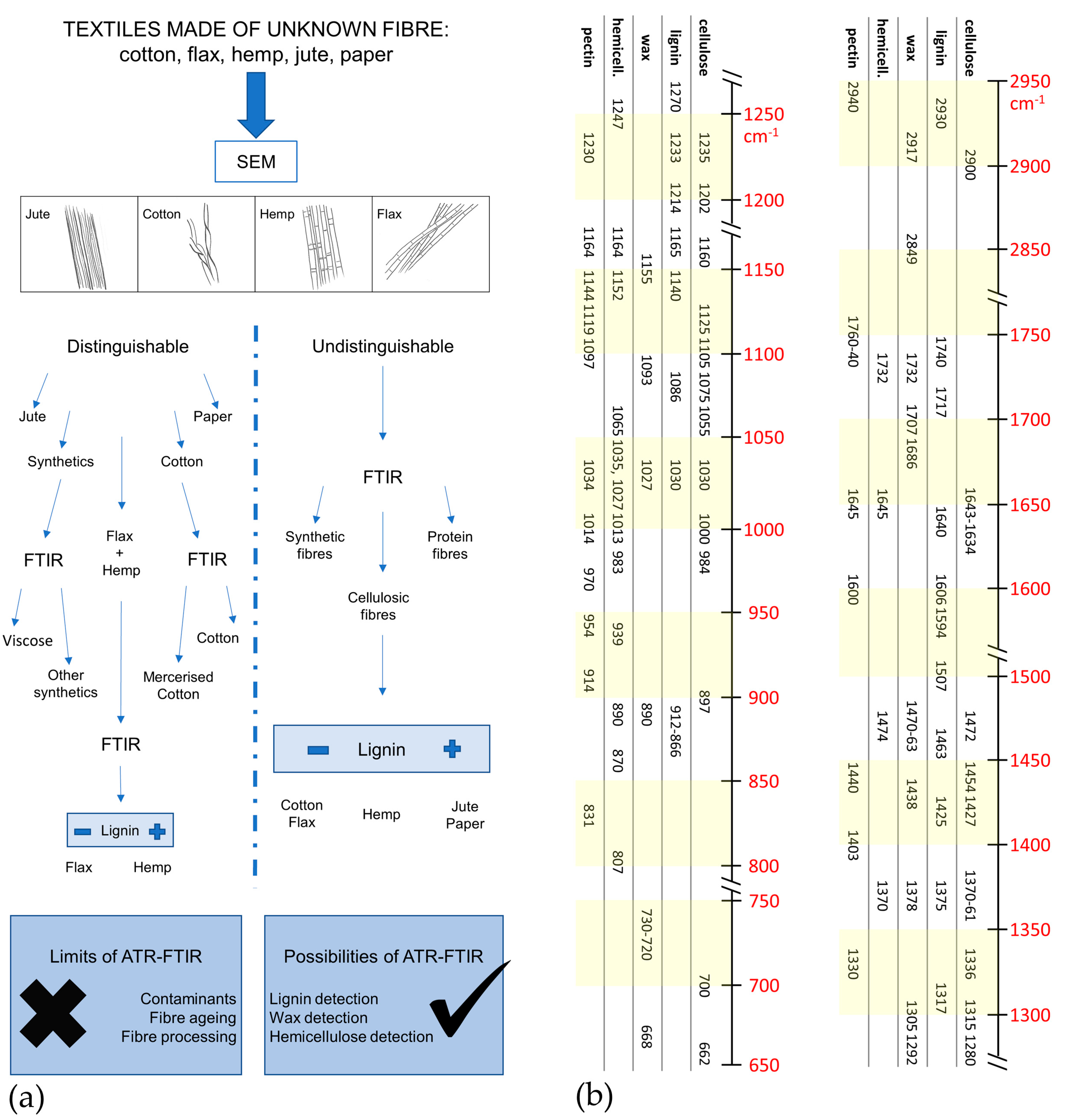
3. Results and Discussion
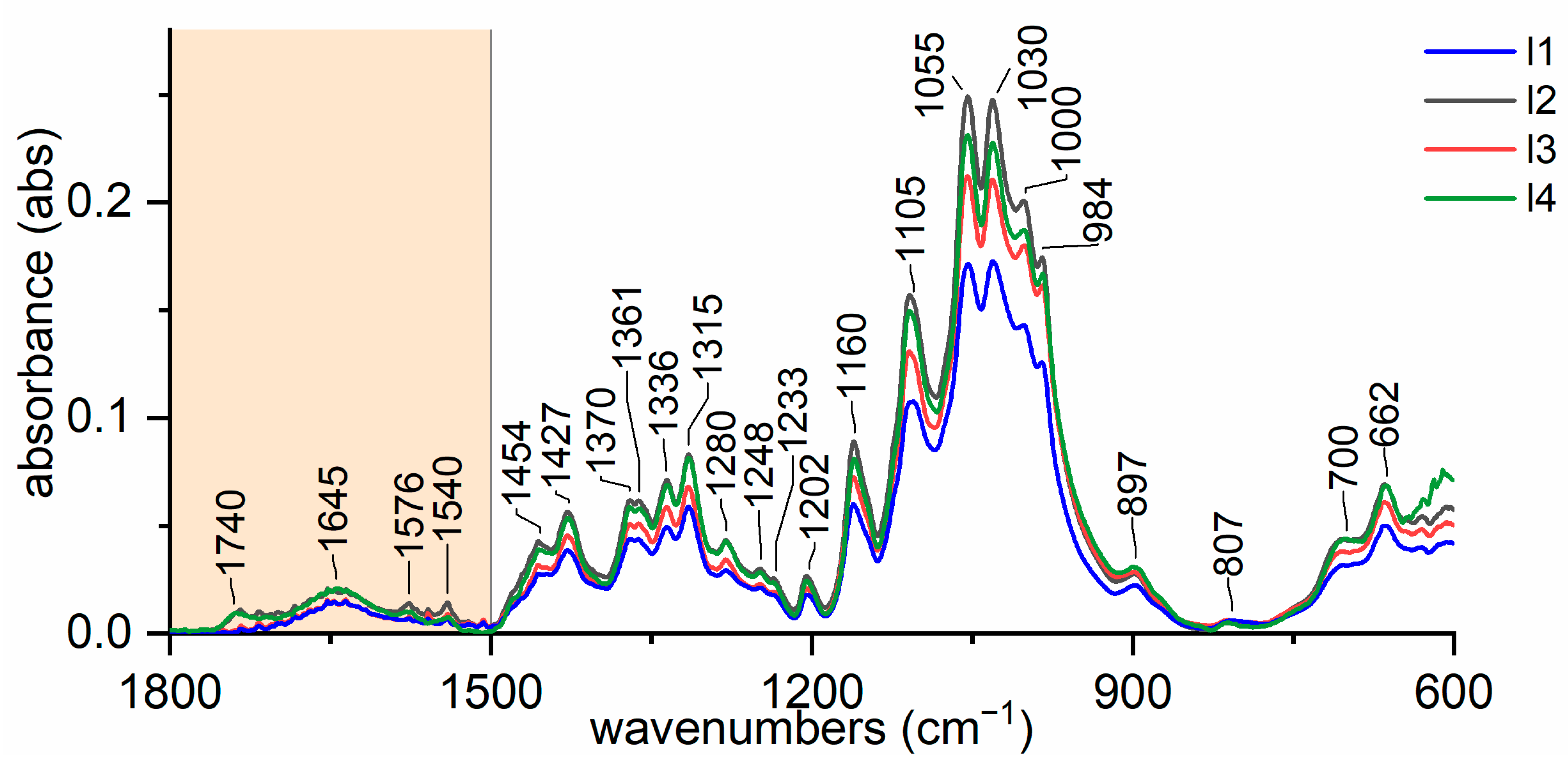
3.1. Variability of Natural Cellulose Fibres
- −
- for lignin: 2920, 1740, 1717, 1606, 1594, 1507 cm−1;
- −
- for wax: 2926, 2854, 1732, 1707, 1686, 730–20 cm−1;
- −
- for hemicellulose: 1247, 1013, 939, and 807 cm−1;
- −
- for pectin: 1740, 1600, 1141, 1097, 1014, 954, 914, and 831 cm−1.
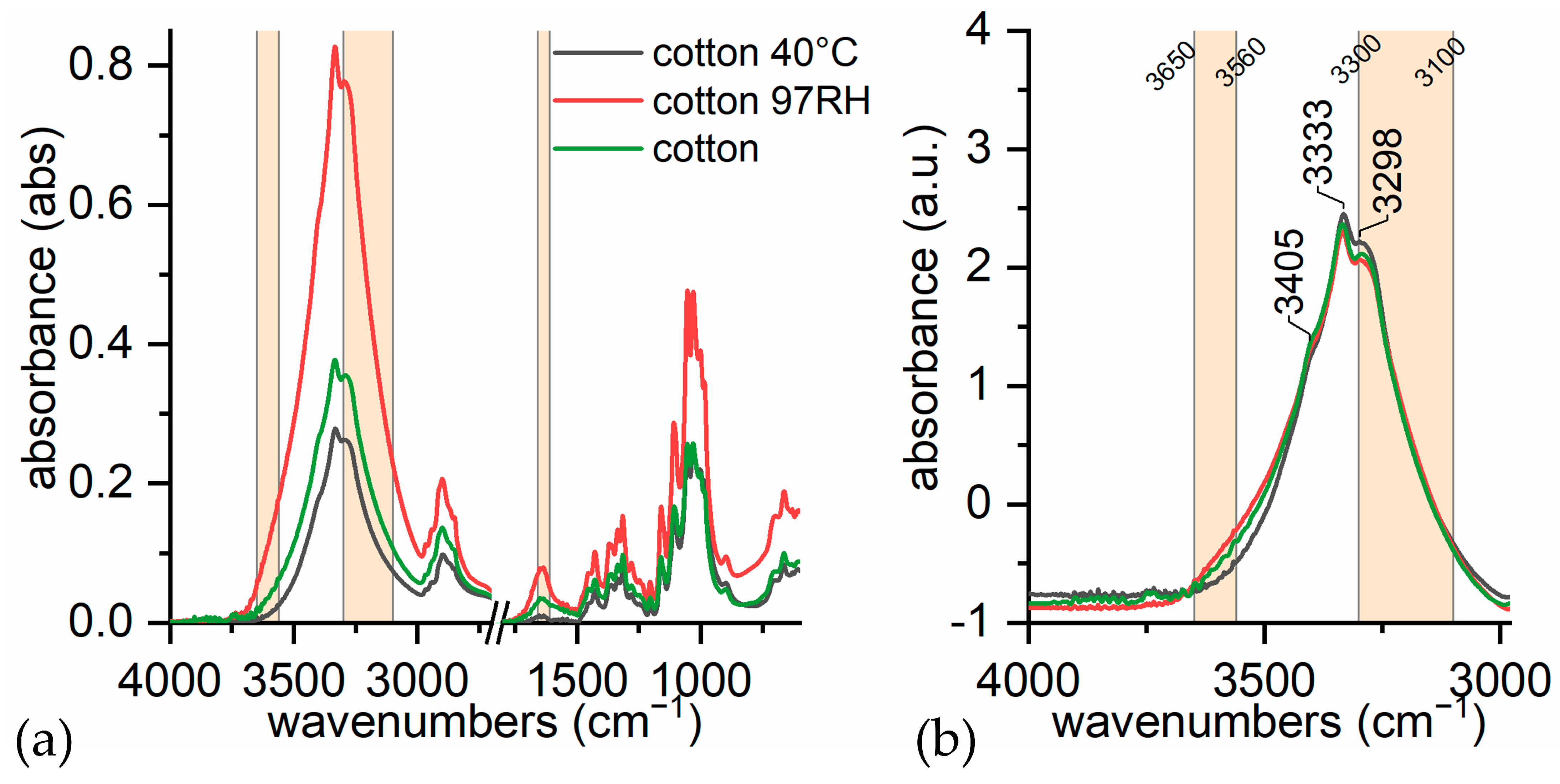
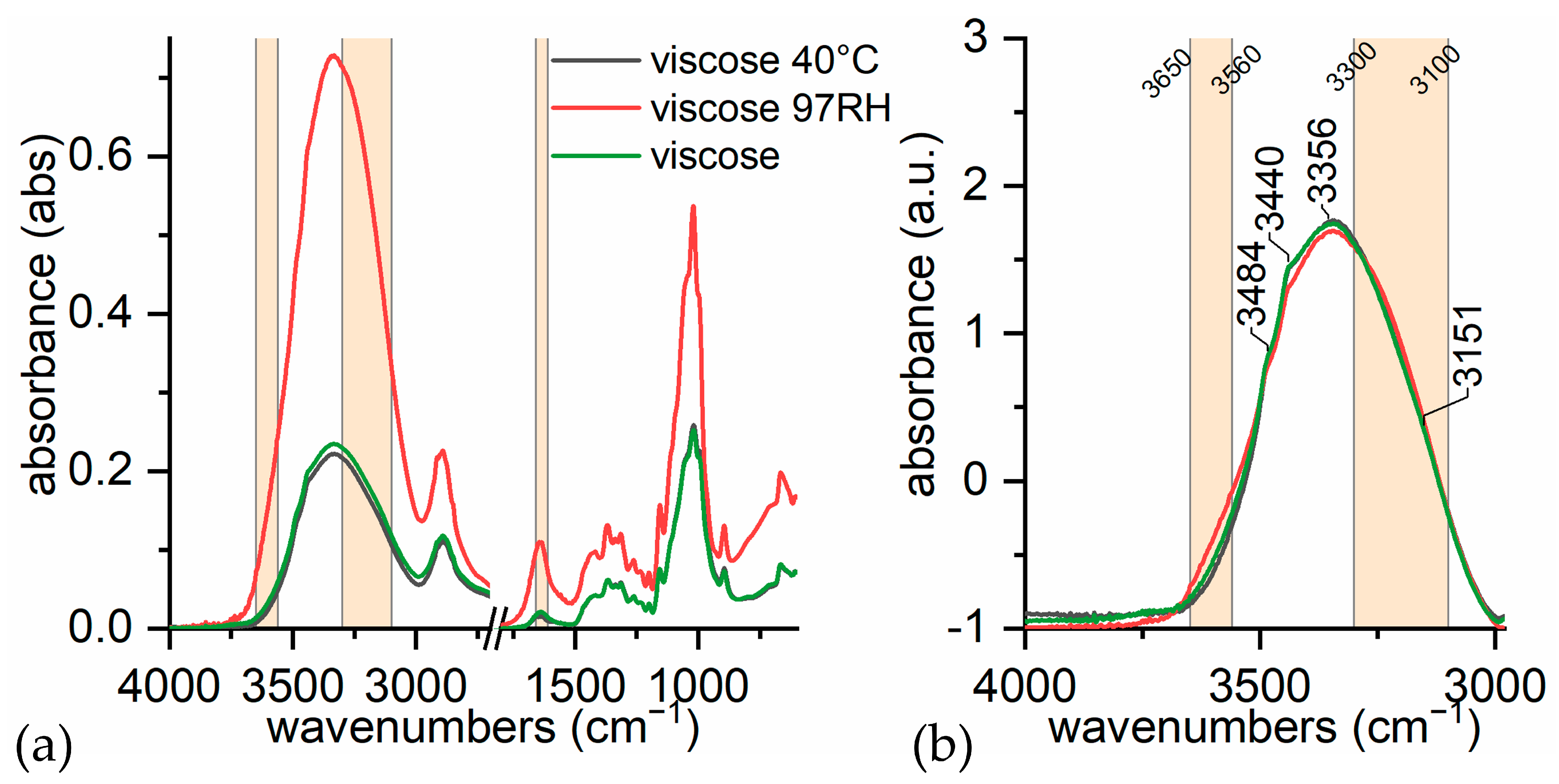
- (i)
- in dry conditions, the contributions from loosely bound water (3580 and 3555 cm−1) are much lower;
- (ii)
- at room humidity, contributions from water absorption are higher, both at 1640 cm−1 and at 3300 cm−1;
- (iii)
- under high humidity conditions (RH 97% for 65 h), the peak at 1640 cm−1 is higher and more resolute (1636 cm−1). The band at 3300 cm−1 is not very different from the sample at room humidity, while the contribution at 3200 cm−1 is slightly higher. This is in accordance with Berthold’s work [94], which refers that uncharged cellulose does not contain freezing water even at RH 97%.
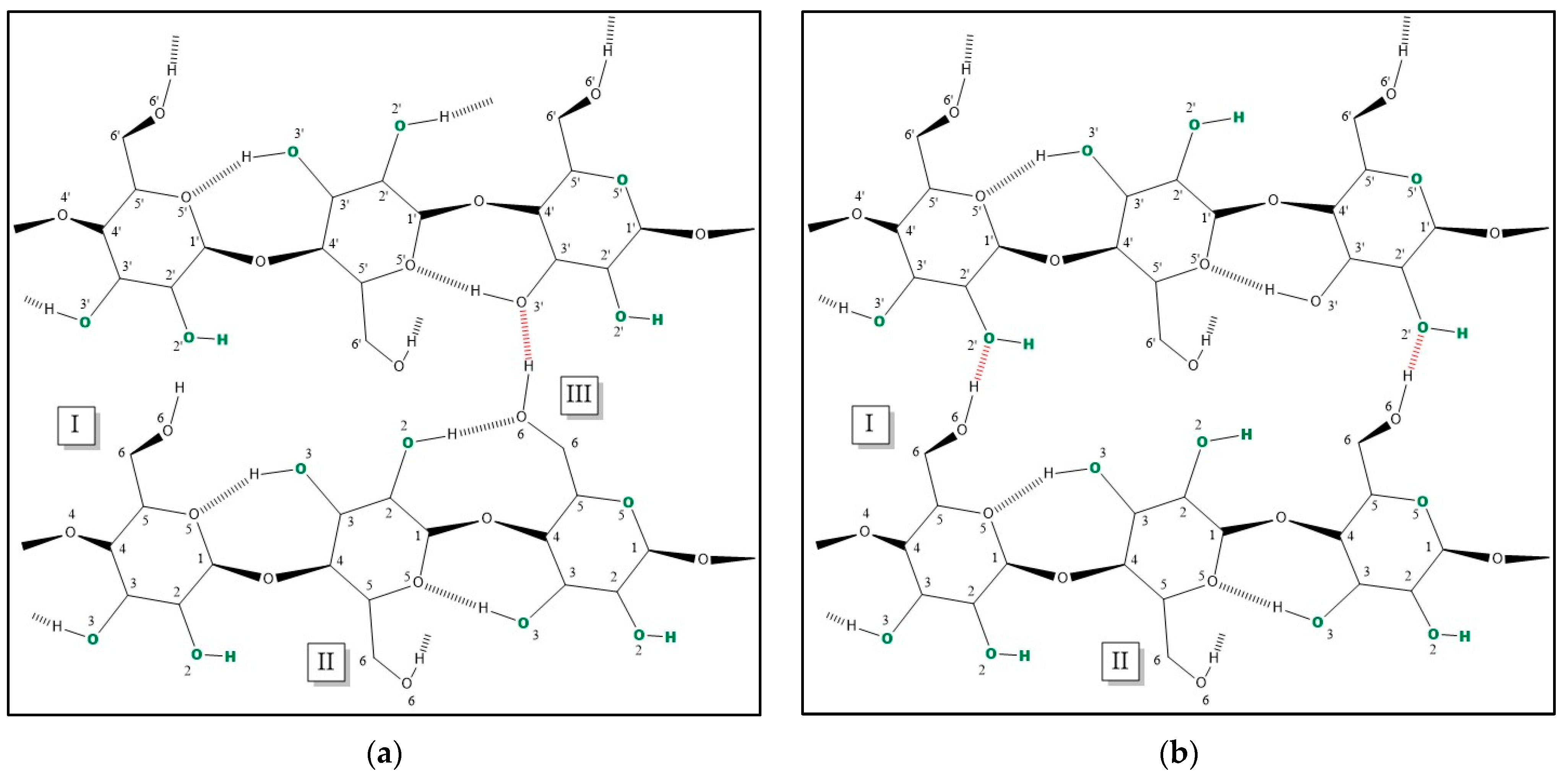
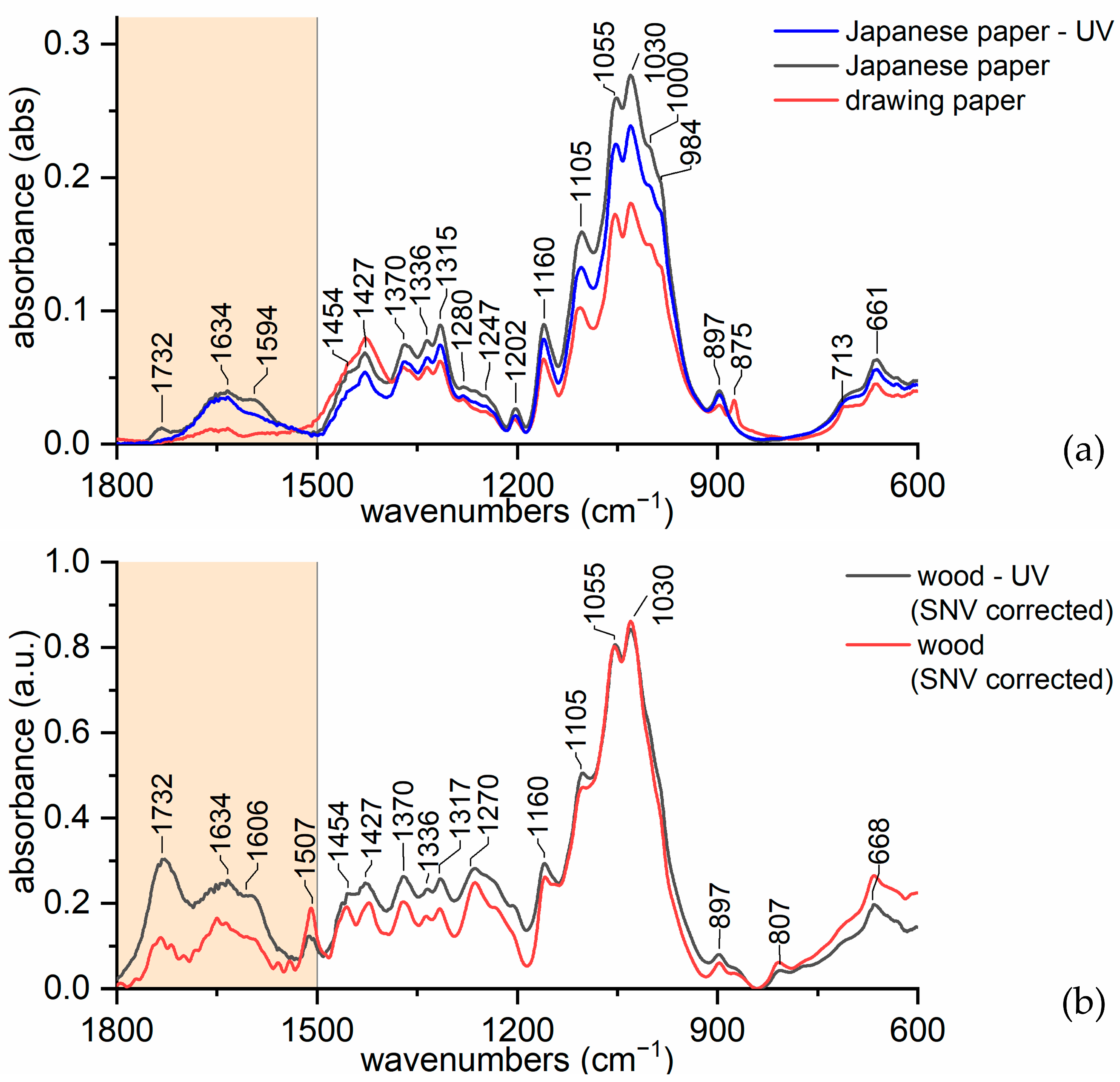
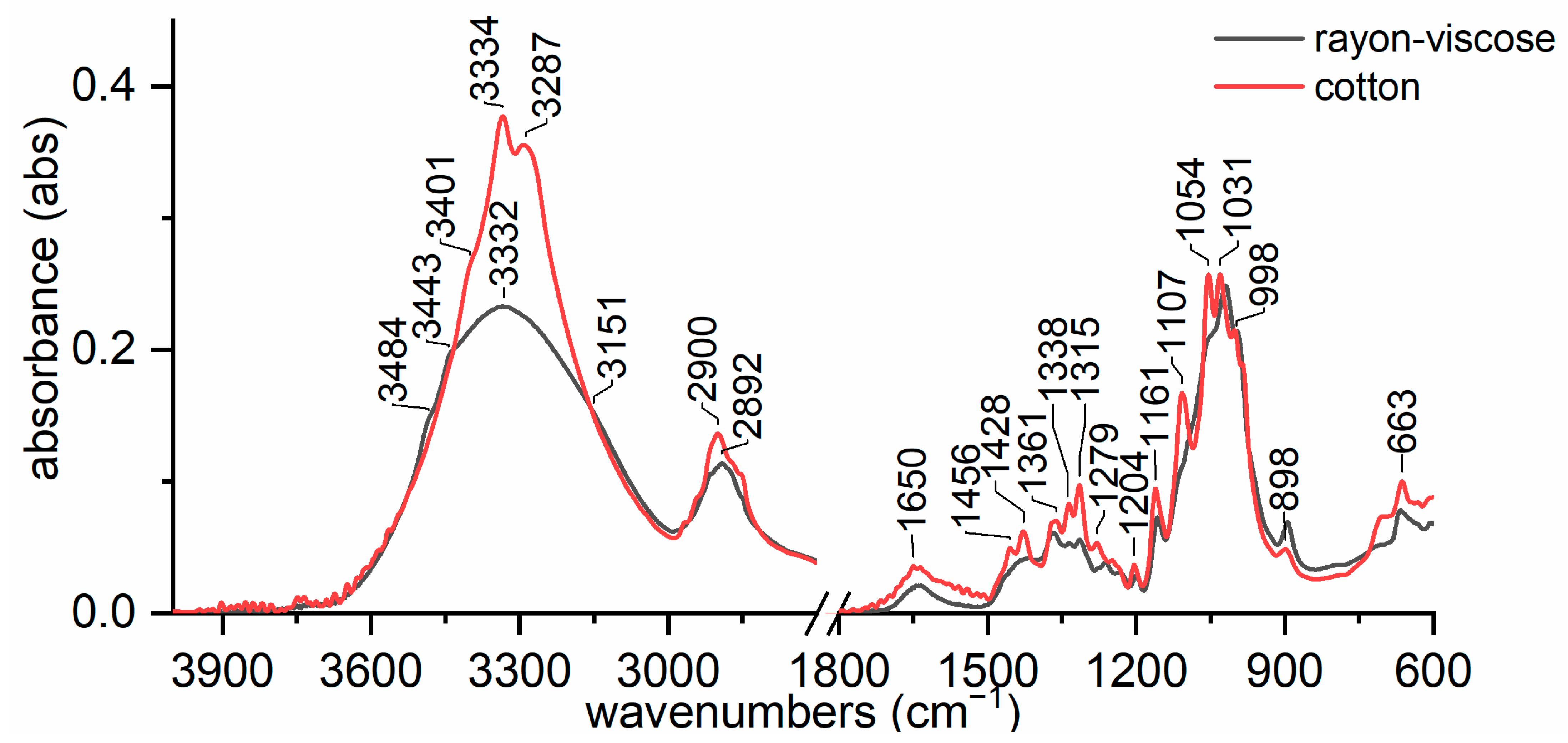
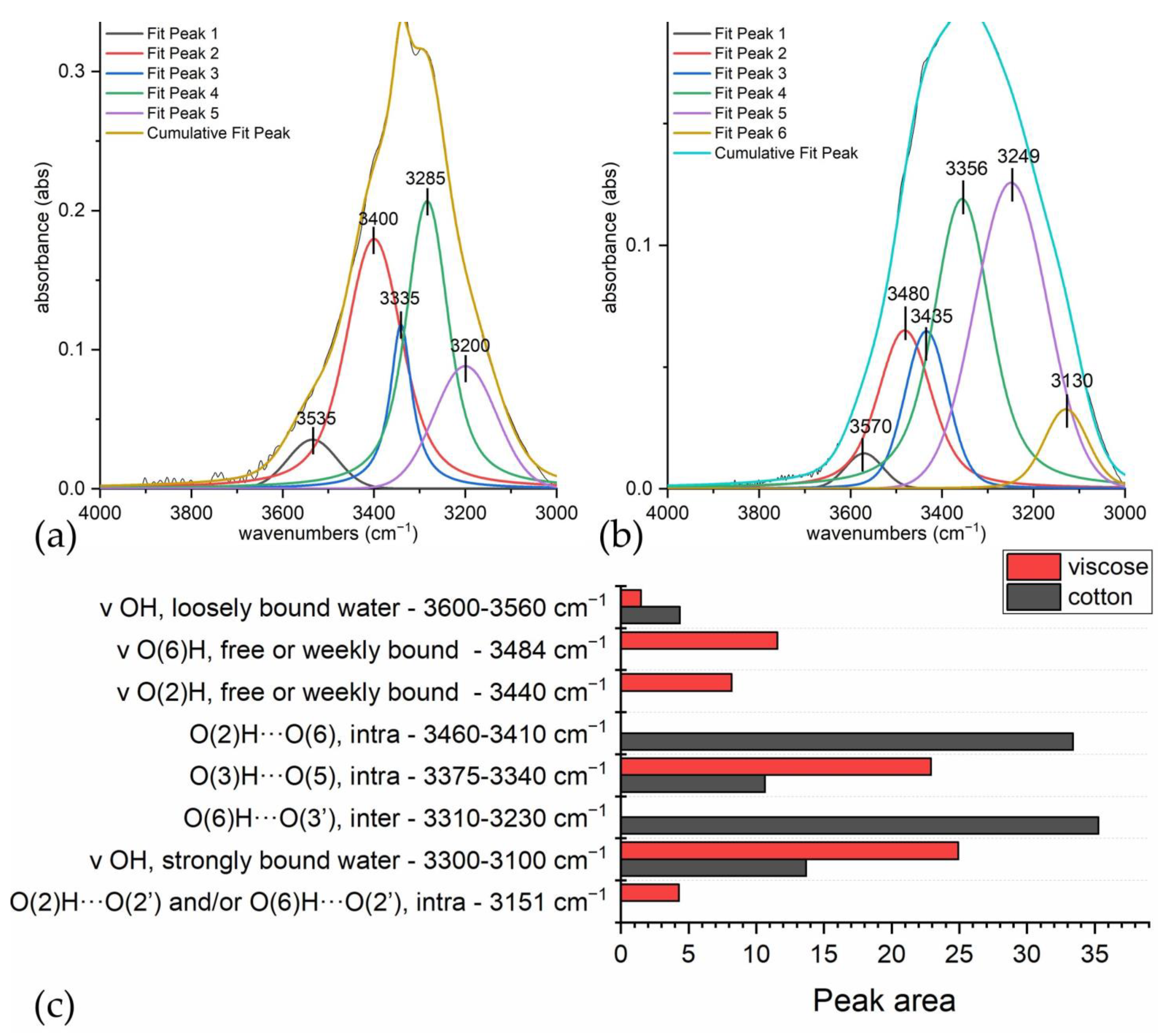
3.2. Discriminating Natural and Modified Cellulose Fibres
- (i)
- 3280 cm−1 (O(6)H⋯⋯O(3′), intermolecular) disappears as it is replaced by 3151 cm−1 (O(6)H⋯⋯O(2′), intramolecular) and 3480 cm−1.
- (ii)
- 3340 cm−1 (O(3)H⋯⋯O(5), intramolecular) does not change significantly (which also applies to 1055 cm−1, which is the C(3)O stretching, and 1315 cm−1, which is the O(3)H bending); the shift could be related to a higher water uptake [61].
- (ii)
- 3410 cm−1 (O(2)H⋯⋯O(6), intramolecular) disappears as it is replaced by 3151 cm−1 (O(2)H⋯⋯O(2′), intramolecular), 3450 and 3480 cm−1.
- (iv)
- 3450 cm−1 (O(2)H, secondary alcohol) is more evident because in viscose, the amount of water H bonded O(2)H is higher than in cotton.
- (v)
- 3480 cm−1 (O(6)H, primary alcohol) is more evident because in viscose the number of water H bonded O(6)H is higher than in cotton.
3.3. Case Study: Traditional Samurai Armours
4. Conclusions
- (i)
- As FTIR is highly sensitive to lignin, it could be considered a non-destructive alternative for Herzberg stain test, for the evaluation of the lignin content (paper).
- (ii)
- The lignin content can distinguish jute and paper, which are richer in it, from hemp, showing a lower content and from cotton and flax, which contains negligible amount of it.
- (iii)
- Wax content can be useful to distinguish hemp from other fibres, unless fibre processing eliminated wax content.
- (iv)
- Hemicellulose can distinguish flax from other fibres, unless fibre ageing eliminated hemicellulose content.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Britannica, The Editors of Encyclopedia. “Natural Fibres”. Encyclopedia Britannica, 2020. Available online: https://www.britannica.com/topic/natural-fiber (accessed on 20 September 2022).
- Mwaikambo, L.Y. Review of the History, Properties and Application of Plant Fibres. Afr. J. Sci. Technol. 2006, 7, 120–133. [Google Scholar]
- Fike, J. The History of Hemp. In Industrial Hemp as a Modern Commodity Crop; Williams, D.W., Ed.; American Society of Agronomy: Madison, WI, USA, 2019; pp. 1–25. [Google Scholar]
- Broudy, E. The Book of Looms A History of the Handloom from Ancient Times to the Present; University Press of New England: Hanover, NH, USA, 1993. [Google Scholar]
- Melelli, A.; Jamme, F.; Beaugrand, J.; Bourmaud, A. Evolution of the Ultrastructure and Polysaccharide Composition of Flax Fibres over Time: When History Meets Science. Carbohydr. Polym. 2022, 291, 119584. [Google Scholar] [CrossRef] [PubMed]
- Biagiotti, J.; Puglia, D.; Kenny, J.M. A Review on Natural Fibre-Based Composites-Part I. J. Nat. Fibers 2004, 1, 37–68. [Google Scholar] [CrossRef]
- Liu, X.; Renard, C.M.G.C.; Bureau, S.; le Bourvellec, C. Revisiting the Contribution of ATR-FTIR Spectroscopy to Characterize Plant Cell Wall Polysaccharides. Carbohydr. Polym. 2021, 262, 117935. [Google Scholar] [CrossRef]
- España, L.; Heredia-Guerrero, J.A.; Segado, P.; Benítez, J.J.; Heredia, A.; Domínguez, E. Biomechanical Properties of the Tomato (Solanum lycopersicum) Fruit Cuticle during Development Are Modulated by Changes in the Relative Amounts of Its Components. New Phytol. 2014, 202, 790–802. [Google Scholar] [CrossRef]
- Tsien, T.-H. Raw Materials for Old Papermaking in China. J. Am. Orient. Soc. 1973, 93, 510–519. [Google Scholar] [CrossRef]
- Tang, Y.; Smith, G.J.; Weston, R.J.; Kong, X. Chinese Handmade Mulberry Paper: Generation of Reactive Oxygen Species and Sensitivity to Photodegradation. J. Cult. Herit. 2017, 28, 82–89. [Google Scholar] [CrossRef]
- Feng, J.; Ma, J.; Pang, X.; Tang, B. Non-Destructive Quantitative Analysis of Nano-Mechanics of Aged Xuan Paper. J. Cult. Herit. 2020, 46, 155–158. [Google Scholar] [CrossRef]
- Quattrini, M.V.; Ioele, M.; Sodo, A.; Priori, G.F.; Radeglia, D. A Seventeenth Century Japanese Painting: Scientific Identification of Materials and Techniques. Stud. Conserv. 2014, 59, 328–340. [Google Scholar] [CrossRef]
- Roberts, J.C. The Chemistry of Paper; The Royal Society of Chemistry: Letchworth, UK, 1996. [Google Scholar]
- Britannica, T.E. Rayon. Available online: https://www.britannica.com/technology/rayon-textile-fibre (accessed on 20 September 2022).
- Timar-Balazsy, A.; Eastop, D. Materials. In Chemical Principles in Textile Conservation; Routledge: New York, NY, USA, 1998; pp. 3–66. [Google Scholar]
- Łojewski, T.; Miśkowiec, P.; Missori, M.; Lubańska, A.; Proniewicz, L.M.; Łojewska, J. FTIR and UV/Vis as Methods for Evaluation of Oxidative Degradation of Model Paper: DFT Approach for Carbonyl Vibrations. Carbohydr. Polym. 2010, 82, 370–375. [Google Scholar] [CrossRef]
- Małachowska, E.; Dubowik, M.; Boruszewski, P.; Przybysz, P. Accelerated Ageing of Paper: Effect of Lignin Content and Humidity on Tensile Properties. Herit. Sci. 2021, 9, 132. [Google Scholar] [CrossRef]
- Potthast, A.; Schiehser, S.; Rosenau, T.; Sixta, H.; Kosma, P. Effect of UV Radiation on the Carbonyl Distribution in Different Pulps. Holzforschung 2004, 58, 597–602. [Google Scholar] [CrossRef]
- Małachowska, E.; Dubowik, M.; Boruszewski, P.; Łojewska, J.; Przybysz, P. Influence of Lignin Content in Cellulose Pulp on Paper Durability. Sci. Rep. 2020, 10, 19998. [Google Scholar] [CrossRef]
- Barclay, L.R.C.; Xi, F.; Norris, J.Q. Antioxidant Properties of Phenolic Lignin Model Compounds. J. Wood Chem. Technol. 1997, 17, 73–90. [Google Scholar] [CrossRef]
- Hermans, P.H.; Weidinger, A. Quantitative X-Ray Investigations on the Crystallinity of Cellulose Fibers. A Background Analysis. J. Appl. Phys. 1948, 19, 491–506. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Hatakeyama, T. Interaction between Water and Hydrophilic Polymers. Thermochim. Acta 1998, 308, 3–22. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Tanaka, M.; Kishi, A.; Hatakeyama, H. Comparison of Measurement Techniques for the Identification of Bound Water Restrained by Polymers. Thermochim. Acta 2012, 532, 159–163. [Google Scholar] [CrossRef]
- Célino, A.; Gonçalves, O.; Jacquemin, F.; Fréour, S. Qualitative and Quantitative Assessment of Water Sorption in Natural Fibres Using ATR-FTIR Spectroscopy. Carbohydr. Polym. 2014, 101, 163–170. [Google Scholar] [CrossRef]
- Olsson, A.M.; Salmén, L. The Association of Water to Cellulose and Hemicellulose in Paper Examined by FTIR Spectroscopy. Carbohydr. Res. 2004, 339, 813–818. [Google Scholar] [CrossRef]
- Cotugno, S.; Larobina, D.; Mensitieri, G.; Musto, P.; Ragosta, G. A Novel Spectroscopic Approach to Investigate Transport Processes in Polymers: The Case of Water-Epoxy System. Polymers 2001, 42, 6431–6438. [Google Scholar] [CrossRef]
- Célino, A.; Fréour, S.; Jacquemin, F.; Casari, P. The Hygroscopic Behavior of Plant Fibers: A Review. Front. Chem. 2014, 1, 43–55. [Google Scholar] [CrossRef]
- Célino, A.; Fréour, S.; Jacquemin, F.; Casari, P. Characterization and Modeling of the Moisture Diffusion Behavior of Natural Fibers. J. Appl. Polym. Sci. 2013, 130, 297–306. [Google Scholar] [CrossRef]
- Nakamura, K.; Hatakeyama, T.; Hatakeyama, H. Effect of Bound Water on Tensile Properties of Native Cellulose. Text. Res. J. 1983, 53, 682–688. [Google Scholar] [CrossRef]
- Yan, Y.; Wen, C.; Jin, M.; Duan, L.; Zhang, R.; Luo, C.; Xiao, J.; Ye, Z.; Gao, B.; Liu, P.; et al. FTIR Spectroscopy in Cultural Heritage Studies: Non-Destructive Analysis of Chinese Handmade Papers. Chem. Res. Chin. Univ. 2019, 35, 586–591. [Google Scholar] [CrossRef]
- Carr, D.; Cruthers, N.; Smith, C.; Myers, T. Identification of Selected Vegetable Textile Fibres. Stud. Conserv. 2008, 53, 75–87. [Google Scholar] [CrossRef]
- Garside, P. The Role of Fibre Identification in Textile Conservation. In Identification of Textile Fibres; Cambridge Woodhead Publishing: Boca Raton, FL, USA, 2009; pp. 335–365. [Google Scholar] [CrossRef]
- Coletti, F.; Romani, M.; Ceres, G.; Zammit, U.; Guidi, M.C. Evaluation of Microscopy Techniques and ATR-FTIR Spectroscopy on Textile Fibers from the Vesuvian Area: A Pilot Study on Degradation Processes That Prevent the Characterization of Bast Fibers. J. Archaeol. Sci. Rep. 2021, 36, 102794. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, Y.; Zhang, X. A Combination of Techniques to Study Chinese Traditional Lajian Paper. J. Cult. Herit. 2019, 38, 75–81. [Google Scholar] [CrossRef]
- Rowe, W.F. Identification of Natural Fibers. In Handbook for the Analysis of Micro-Particles in Archaeological Samples. Interdisciplinary Contributions to Archaeology; Henry, A., Ed.; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Bergfjord, C.; Holst, B. A Procedure for Identifying Textile Bast Fibres Using Microscopy: Flax, Nettle/Ramie, Hemp and Jute. Ultramicroscopy 2010, 110, 1192–1197. [Google Scholar] [CrossRef]
- Corti, C.; Motella De Carlo, S.; Rampazzi, L. The Intimate Soul of the Pyres: New Archaeological Data from the Terre Di Rogo (Pyre Debris) of Pre-Roman Necropolis in Padua (Northern Italy). PLoS ONE 2022. [Google Scholar] [CrossRef]
- Sisko, M.; Pfaffli, I. Fiber Atlas—Identification of Papermaking Fibers; Springer: Espoo, Finland, 1995. [Google Scholar]
- Edwards, H.G.M.; Nikhassan, N.F.; Farwell, D.W.; Garside, P.; Wyeth, P. Raman Spectroscopic Analysis of a Unique Linen Artefact: The HMS Victory Trafalgar Sail. J. Raman Spectrosc. 2006, 37, 1193–1200. [Google Scholar] [CrossRef]
- Akyuz, S.; Akyuz, T.; Cakan, B.; Basaran, S. Investigations of the Historic Textiles Excavated from Ancient Ainos (Enez-Turkey) by Multiple Analytical Techniques Dedicated to Professor Simion Simon. J. Mol. Struct. 2014, 1073, 37–43. [Google Scholar] [CrossRef]
- Margariti, C. The Application of FTIR Microspectroscopy in a Non-Invasive and Non-Destructive Way to the Study and Conservation of Mineralised Excavated Textiles. Herit. Sci. 2019, 7, 63. [Google Scholar] [CrossRef]
- Liu, J.; Guo, D.; Zhou, Y.; Wu, Z.; Li, W.; Zhao, F.; Zheng, X. Identification of Ancient Textiles from Yingpan, Xinjiang, by Multiple Analytical Techniques. J. Archaeol. Sci 2011, 38, 1763–1770. [Google Scholar] [CrossRef]
- Cybulska, M.; Jedraszek-Bomba, A.; Kuberski, S.; Wrzosek, H. Methods of Chemical and Physicochemical Analysis in the Identification of Archaeological and Historical Textiles. Fibres Text. East. Eur. 2008, 16, 67–73. [Google Scholar]
- Margariti, C. The Effects of Micro-Organisms in Simulated Soil Burial on Cellulosic and Proteinaceous Textiles and the Morphology of the Fibres. Stud. Conserv. 2021, 66, 282–297. [Google Scholar] [CrossRef]
- Kavkler, K.; Gunde-Cimermann, N. FTIR Spectroscopy of Biodegraded Historical Textiles. Polym. Degrad. Stab. 2011, 96, 574–580. [Google Scholar] [CrossRef]
- Kavkler, K.; Demšar, A. Application of FTIR and Raman Spectroscopy to Qualitative Analysis of Structural Changes in Cellulosic Fibres. Tekstilec 2012, 55, 19–31. [Google Scholar]
- Uring, P.; Chabas, A.; Alfaro, S.C. Textile Ageing Due to Atmospheric Gases and Particles in Indoor Cultural Heritage. Environ. Sci. Pollut. Res. 2021, 28, 66340–66354. [Google Scholar] [CrossRef]
- Uring, P.; Chabas, A.; Alfaro, S. Dust Deposition on Textile and Its Evolution in Indoor Cultural Heritage. Eur. Phys. J. Plus. 2019, 134, 255. [Google Scholar] [CrossRef]
- Bicchieri, M.; Biocca, P.; Colaizzi, P.; Pinzari, F. Microscopic Observations of Paper and Parchment: The Archaeology of Small Objects. Herit. Sci. 2019, 7, 47. [Google Scholar] [CrossRef]
- Garside, P.; Wyeth, P. Identification of Cellulosic Fibres by FTIR Spectroscopy: Thread and Single Fibre Analysis by Attenuated Total Reflectance. Stud. Conserv. 2004, 48, 269–275. [Google Scholar] [CrossRef]
- Garside, P.; Wyeth, P. Identification of Cellulosic Fibres by FTIR Spectroscopy: Differentiation of Flax and Hemp by Polarized ATR-FTIR. Stud. Conserv. 2006, 51, 205–211. [Google Scholar] [CrossRef]
- Garside, P.; Wyeth, P. Use of Polarized Spectroscopy as a Tool for Examining the Microstructure of Cellulosic Textile Fibers. Appl. Spectrosc. 2007, 61, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.; Kokot, S. Discrimination of Cellulosic Fabrics by Diffuse Reflectance Infrared Fourier Transform Spectroscopy and Chemometrics. Vib. Spectrosc. 1995, 9, 161–167. [Google Scholar] [CrossRef]
- Saito, K.; Yamagata, T.; Kanno, M.; Yoshimura, N.; Takayanagi, M. Discrimination of Cellulose Fabrics Using Infrared Spectroscopy and Newly Developed Discriminant Analysis. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 257, 119772. [Google Scholar] [CrossRef]
- Titok, V.; Leontiev, V.; Yurenkova, S.; Nikitinskaya, T.; Barannikova, T.; Khotyleva, L. Infrared Spectroscopy of Fiber Flax. J. Nat. Fibers 2010, 7, 61–69. [Google Scholar] [CrossRef]
- Manian, A.P.; Braun, D.E.; Široká, B.; Bechtold, T. Distinguishing Liquid Ammonia from Sodium Hydroxide Mercerization in Cotton Textiles. Cellulose 2022, 29, 4183–4202. [Google Scholar] [CrossRef]
- Oh, S.Y.; Dong, I.Y.; Shin, Y.; Hwan, C.K.; Hak, Y.K.; Yong, S.C.; Won, H.P.; Ji, H.Y. Crystalline Structure Analysis of Cellulose Treated with Sodium Hydroxide and Carbon Dioxide by Means of X-Ray Diffraction and FTIR Spectroscopy. Carbohydr. Res. 2005, 340, 2376–2391. [Google Scholar] [CrossRef]
- Grdadolnik, J. Atr-ftir spectroscopy: Its advantages and limitations. Acta Chim. Slov. 2002, 49, 631–642. [Google Scholar]
- Maréchal, Y.; Chanzy, H. The Hydrogen Bond Network in I(β) Cellulose as Observed by Infrared Spectrometry. J. Mol. Struct. 2000, 523, 183–196. [Google Scholar] [CrossRef]
- Lindman, B.; Karlström, G.; Stigsson, L. On the Mechanism of Dissolution of Cellulose. J. Mol. Liq. 2010, 156, 76–81. [Google Scholar] [CrossRef]
- Cichosz, S.; Masek, A. IR Study on Cellulose with the Varied Moisture Contents: Insight into the Supramolecular Structure. Materials 2020, 13, 4573. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.F.; Preston, J.M. 32—Some Effects of Temperature on Wet Viscose Rayon: Part III-Discussion. J. Text. Inst. Trans. 1956, 47, T419–T421. [Google Scholar] [CrossRef]
- Fengel, D. Influence of Water on the OH Valency Range in Deconvoluted FTIR Spectra of Cellulose. Holzforschung 1993, 47, 103–108. [Google Scholar] [CrossRef]
- Hofstetter, K.; Hinterstoisser, B.; Salmén, L. Moisture Uptake in Native Cellulose—The Roles of Different Hydrogen Bonds: A Dynamic FT-IR Study Using Deuterium Exchange. Cellulose 2006, 13, 131–145. [Google Scholar] [CrossRef]
- Nakamura, K.; Hatakeyama, T.; Hatakeyama, H. Studies on Bound Water of Cellulose by Differential Scanning Calorimetry. Text. Res. J. 1981, 51, 607–613. [Google Scholar] [CrossRef]
- Skoglund, G. Traditional Manufacture of Hemp and Hop Textiles: Why Botany and Agronomy Matter. J. Anthropol. Archaeol. 2021, 9, 1–16. [Google Scholar] [CrossRef]
- Feller, R.L. Accelerated Ageing—Photochemical and Thermal Aspects; Berland, D., Ed.; The Getty Conservation Institute: Ann Arbor, MI, USA, 1994. [Google Scholar]
- Menges, F. Spectragryph—Optical Spectroscopy Software, Version 1.2.16.1, 2022. Available online: http://www.effemm2.de/spectragryph/ (accessed on 30 October 2022).
- Barnes, R.J.; Dhanoa, M.S.; Lister, S.J. Standard Normal Variate Transformation and De-Trending of near-Infrared Diffuse Reflectance Spectra. Appl. Spectrosc. 1989, 43, 772–777. [Google Scholar] [CrossRef]
- Reilly, J.T.; Walsh, J.M.; Greenfield, M.L.; Donohue, M.D. Analysis of FT-IR Spectroscopic Data: The Voigt Profile. Spectrochim. Acta A 1992, 48, 1459–1479. [Google Scholar] [CrossRef]
- Bradley, M.S. Lineshapes in IR and Raman Spectroscopy: A Primer. Spectroscopy 2015, 30, 42–46. [Google Scholar]
- Schwanninger, M.; Rodrigues, J.C.; Pereira, H.; Hinterstoisser, B. Effects of Short-Time Vibratory Ball Milling on the Shape of FT-IR Spectra of Wood and Cellulose. Vib. Spectrosc. 2004, 36, 23–40. [Google Scholar] [CrossRef]
- Quaglierini, C. CHIMICA DELLE FIBRE TESSILI; ZANICHELLI: Bologna, Italy, 1989. [Google Scholar]
- Casoli, A.; Cremonesi, P.; Isca, C.; Groppetti, R.; Pini, S.; Senin, N. Evaluation of the Effect of Cleaning on the Morphological Properties of Ancient Paper Surface. Cellulose 2013, 20, 2027–2043. [Google Scholar] [CrossRef]
- Synytsya, A.; Čopíková, J.; Matějka, P.; Machovič, V. Fourier Transform Raman and Infrared Spectroscopy of Pectins. Carbohydr. Polym. 2003, 54, 97–106. [Google Scholar] [CrossRef]
- Liang, C.Y.; Marchessault, R.H. Infrared Spectra of Crystalline Polysaccharides. II. Native Celluloses in the Region from 640 to 1700 cm−1. J. Polym. Sci. 1959, 39, 269–278. [Google Scholar] [CrossRef]
- Pandey, K.K. A Study of Chemical Structure of Soft and Hardwood and Wood Polymers by FTIR Spectroscopy. J. Appl. Polym. Sci. 1999, 71, 1969–1975. [Google Scholar] [CrossRef]
- Chen, H.; Ferrari, C.; Angiuli, M.; Yao, J.; Raspi, C.; Bramanti, E. Qualitative and Quantitative Analysis of Wood Samples by Fourier Transform Infrared Spectroscopy and Multivariate Analysis. Carbohydr. Polym. 2010, 82, 772–778. [Google Scholar] [CrossRef]
- Colom, X.; Carrillo, F.; Nogués, F.; Garriga, P. Structural Analysis of Photodegraded Wood by Means of FTIR Spectroscopy. Polym. Degrad. Stab. 2003, 80, 543–549. [Google Scholar] [CrossRef]
- Blackwell, J.; Vasko, P.D.; Koenig, J.L. Infrared and Raman Spectra of the Cellulose from the Cell Wall of Valonia Ventricosa. J. Appl. Phys. 1970, 41, 4375–4379. [Google Scholar] [CrossRef]
- Farber, C.; Li, J.; Hager, E.; Chemelewski, R.; Mullet, J.; Rogachev, A.Y.; Kurouski, D. Complementarity of Raman and Infrared Spectroscopy for Structural Characterization of Plant Epicuticular Waxes. ACS Omega 2019, 4, 3700–3707. [Google Scholar] [CrossRef]
- Kimura, F.; Umemura, J.; Takenaka, T. FTIR–ATR Studies on Langmuir–Blodgett Films of Stearic Acid with 1–9 Monolayers. Langmuir 1986, 2, 96–101. [Google Scholar] [CrossRef]
- Merk, S.; Blume, A.; Riederer, M. Phase Behaviour and Crystallinity of Plant Cuticular Waxes Studied by Fourier Transform Infrared Spectroscopy. Planta 1997, 204, 44–53. [Google Scholar] [CrossRef]
- Nikonenko, N.A.; Buslov, D.K.; Sushko, N.I.; Zhbankov, R.G. Spectroscopic Manifestation of Stretching Vibrations of Glycosidic Linkage in Polysaccharides. J. Mol. Struct. 2005, 752, 20–24. [Google Scholar] [CrossRef]
- Pereira, S.C.; Maehara, L.; Machado, C.M.M.; Farinas, C.S. Physical-Chemical-Morphological Characterization of the Whole Sugarcane Lignocellulosic Biomass Used for 2G Ethanol Production by Spectroscopy and Microscopy Techniques. Renew Energy 2016, 87, 607–617. [Google Scholar] [CrossRef]
- Attard, T.M.; Bainier, C.; Reinaud, M.; Lanot, A.; McQueen-Mason, S.J.; Hunt, A.J. Utilisation of Supercritical Fluids for the Effective Extraction of Waxes and Cannabidiol (CBD) from Hemp Wastes. Ind. Crops Prod. 2018, 112, 38–46. [Google Scholar] [CrossRef]
- le Troedec, M.; Sedan, D.; Peyratout, C.; Bonnet, J.P.; Smith, A.; Guinebretiere, R.; Gloaguen, V.; Krausz, P. Influence of Various Chemical Treatments on the Composition and Structure of Hemp Fibres. Compos. Part A Appl. Sci. Manuf. 2008, 39, 514–522. [Google Scholar] [CrossRef]
- Mwaikambo, L.Y.; Ansell, M.P. Chemical Modification of Hemp, Sisal, Jute, and Kapok Fibers by Alkalization. J. Appl. Polym. Sci. 2002, 84, 2222–2234. [Google Scholar] [CrossRef]
- Sawpan, M.A.; Pickering, K.L.; Fernyhough, A. Effect of Various Chemical Treatments on the Fibre Structure and Tensile Properties of Industrial Hemp Fibres. Compos. Part A Appl. Sci. Manuf. 2011, 42, 888–895. [Google Scholar] [CrossRef]
- Li, Y.; Pickering, K.L. Hemp Fibre Reinforced Composites Using Chelator and Enzyme Treatments. Compos. Sci. Technol. 2008, 68, 3293–3298. [Google Scholar] [CrossRef]
- Guerriero, G.; Mangeot-Peter, L.; Legay, S.; Behr, M.; Lutts, S.; Siddiqui, K.S.; Hausman, J.F. Identification of Fasciclin-like Arabinogalactan Proteins in Textile Hemp (Cannabis sativa L.): In Silico Analyses and Gene Expression Patterns in Different Tissues. BMC Genom. 2017, 18, 741. [Google Scholar] [CrossRef]
- Brunello, V.; Corti, C.; Sansonetti, A.; Tedeschi, C.; Rampazzi, L. Non-Invasive FTIR Study of Mortar Model Samples: Comparison among Innovative and Traditional Techniques. Eur. Phys. J. Plus. 2019, 134, 270. [Google Scholar] [CrossRef]
- Jeong, M.J.; Kang, K.Y.; Bacher, M.; Kim, H.J.; Jo, B.M.; Potthast, A. Deterioration of Ancient Cellulose Paper, Hanji: Evaluation of Paper Permanence. Cellulose 2014, 21, 4621–4632. [Google Scholar] [CrossRef]
- Berthold, J. Types of Adsorbed Water in Relation to the Ionic Groups and Their Counter-Ions for Some Cellulose Derivatives. Polymers 1994, 35, 5729–5736. [Google Scholar] [CrossRef]
- Fengel, D. Characterization of Cellulose by Deconvoluting the OH Valency Range In FTIR Spectra. Holzforschung 1992, 46, 283–288. [Google Scholar] [CrossRef]
- Geminiani, L.; Campione, F.P.; Corti, C.; Motella, S.; Rampazzi, L.; Recchia, S.; Luraschi, M. Unveiling the Complexity of Japanese Metallic Threads. Heritage 2021, 4, 4017–4039. [Google Scholar] [CrossRef]
- EL Oudiani, A.; Chaabouni, Y.; Msahli, S.; Sakli, F. Crystal Transition from Cellulose i to Cellulose II in NaOH Treated Agave Americana L. Fibre. Carbohydr. Polym. 2011, 86, 1221–1229. [Google Scholar] [CrossRef]
- Wakelyn, P.J. Cotton Fiber Chemistry and Technology; Lewin, M., Ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2006. [Google Scholar]
- Arnold, L.N.; Rippon, J.A. Continuous Mercerisation of Cotton Sliver under Tension Offers Potential for New Products. Int. Dye 2010, 195, 12–16. [Google Scholar]
- Kafle, K.; Greeson, K.; Lee, C.; Kim, S.H. Cellulose Polymorphs and Physical Properties of Cotton Fabrics Processed with Commercial Textile Mills for Mercerization and Liquid Ammonia Treatments. Text. Res. J. 2014, 84, 1692–1699. [Google Scholar] [CrossRef]
- Šajn Gorjanc, D.; Zupin, Ž. Responses of Fabric from Lyocell/Natural Bamboo Yarn to Loading. J. Text. Inst. 2017, 108, 1707–1714. [Google Scholar] [CrossRef]
- Comnea-Stancu, I.R.; Wieland, K.; Ramer, G.; Schwaighofer, A.; Lendl, B. On the Identification of Rayon/Viscose as a Major Fraction of Microplastics in the Marine Environment: Discrimination between Natural and Manmade Cellulosic Fibers Using Fourier Transform Infrared Spectroscopy. Appl. Spectrosc. 2017, 71, 939–950. [Google Scholar] [CrossRef]
- Yan, C.F.; Yu, H.Y.; Yao, J.M. One-Step Extraction and Functionalization of Cellulose Nanospheres from Lyocell Fibers with Cellulose II Crystal Structure. Cellulose 2015, 22, 3773–3788. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Armour | Microsamples | Armour Type | Presumed Dating |
|---|---|---|---|
| 2017.Mor.1 | 1_11 | kinsei gusoku | late 16th |
| 2017.Mor.5 | 5_3 | kinsei gusoku | 20th |
| 2017.Mor.6 | 6_1 | kinsei gusoku | early 20th |
| 2017.Mor.7 | 7_6a, 7_6b | kinsei gusoku | 17th |
| 2017.Mor.8 | 8_1 | kinsei gusoku | 17th |
| 2017.Mor.8 | 8_9 | kinsei gusoku | early 19th |
| 2017.Mor.9 | 9_6 | mukashi gusoku | late 19th |
| 2017.Mor.10 | 10_1 | kinsei gusoku | early 20th |
| Water (cm−1) | Cotton (cm−1) | Viscose (cm−1) | Assignment |
|---|---|---|---|
| 3600–3560 | ν OH, loosely bound water [20] | ||
| 3484, sh | ν O(6)H, free or weekly bound (primary alcohol) | ||
| 3440, sh | ν O(2)H, free or weekly bound (secondary alcohol) [45] | ||
| 3460–3410 | O(2)H⋯⋯O(6), intramolecular [44,45,47,49] | ||
| 3375–3340 | 3356 | O(3)H⋯⋯O(5), intramolecular [44,45,47,49] | |
| 3310–3230 | O(6)H⋯⋯O(3′), intermolecular [44,45,47,49] | ||
| 3300–3100 | ν OH, strongly bound water [20] | ||
| 3151 | O(2)H⋯⋯O(2′) and/or O(6)H⋯⋯O(2′), intramolecular [44,45,47,49] | ||
| 1643–1634 | δ OH of adsorbed water [37,56,57,63] |
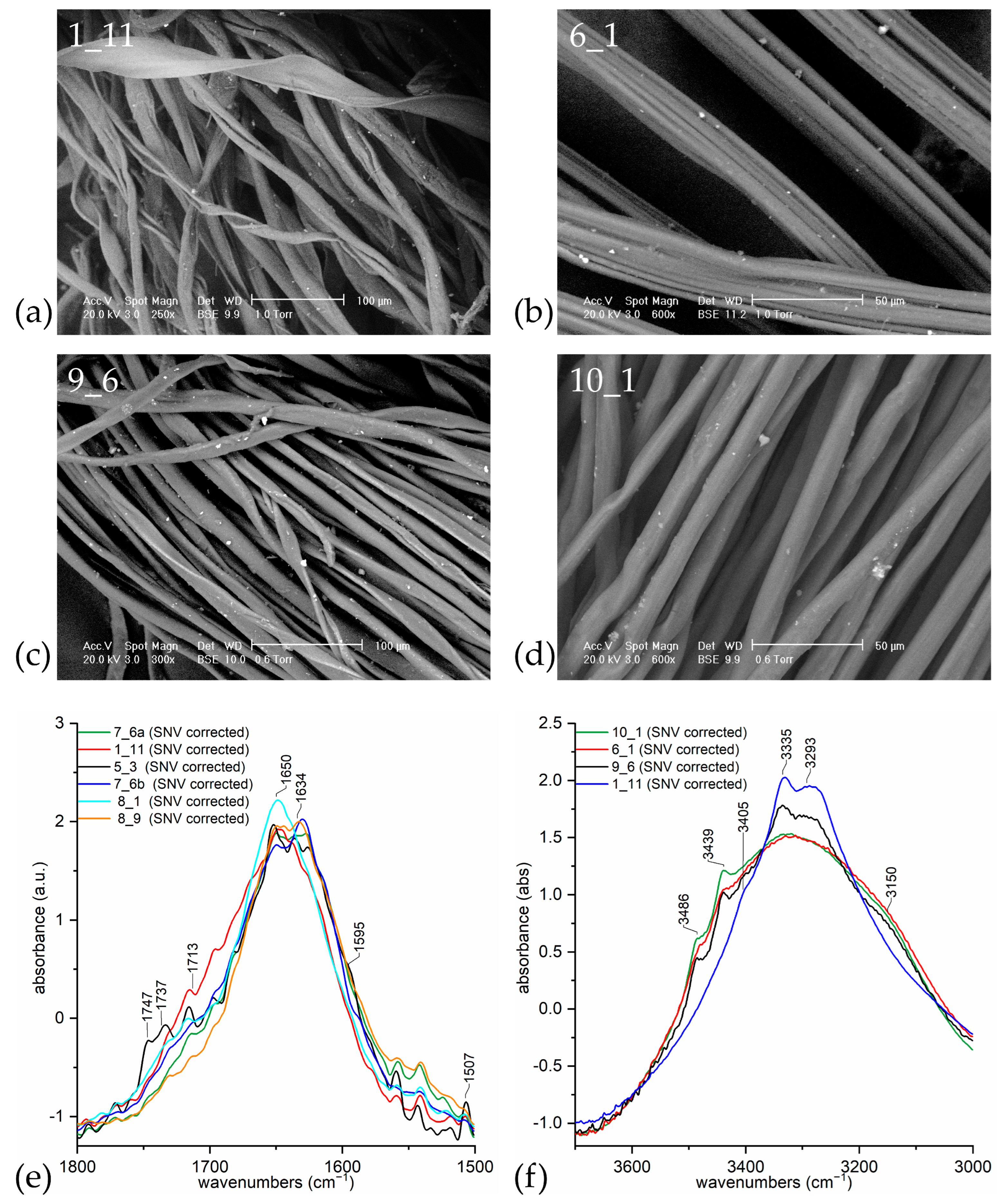
| Sample | Recognised Material (by Microscopy) | Recognised Material (by ATR-FTIR) |
|---|---|---|
| 1_11 | cotton | cellulosic (no lignin) |
| 5_3 | paper | cellulosic (with lignin) |
| 6_1 | synthetic | rayon-viscose |
| 7_6a | cotton | cellulosic (no lignin) |
| 7_6b | cotton | cellulosic (no lignin) |
| 8_1 | bast fibre | cellulosic (no lignin) |
| 8_9 | cotton | cellulosic (no lignin) |
| 9_6 | swelled cotton | mercerised cotton (NaOH) |
| 10_1 | swelled cotton | mercerised cotton (NH3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geminiani, L.; Campione, F.P.; Corti, C.; Luraschi, M.; Motella, S.; Recchia, S.; Rampazzi, L. Differentiating between Natural and Modified Cellulosic Fibres Using ATR-FTIR Spectroscopy. Heritage 2022, 5, 4114-4139. https://doi.org/10.3390/heritage5040213
Geminiani L, Campione FP, Corti C, Luraschi M, Motella S, Recchia S, Rampazzi L. Differentiating between Natural and Modified Cellulosic Fibres Using ATR-FTIR Spectroscopy. Heritage. 2022; 5(4):4114-4139. https://doi.org/10.3390/heritage5040213
Chicago/Turabian StyleGeminiani, Ludovico, Francesco Paolo Campione, Cristina Corti, Moira Luraschi, Sila Motella, Sandro Recchia, and Laura Rampazzi. 2022. "Differentiating between Natural and Modified Cellulosic Fibres Using ATR-FTIR Spectroscopy" Heritage 5, no. 4: 4114-4139. https://doi.org/10.3390/heritage5040213
APA StyleGeminiani, L., Campione, F. P., Corti, C., Luraschi, M., Motella, S., Recchia, S., & Rampazzi, L. (2022). Differentiating between Natural and Modified Cellulosic Fibres Using ATR-FTIR Spectroscopy. Heritage, 5(4), 4114-4139. https://doi.org/10.3390/heritage5040213