1. Introduction
Many food, pharmaceutical, and cosmetic products are emulsion-based, where fine oil droplets are dispersed in an aqueous phase. The droplet sizes and their distribution play an important role in stability, texture, color, and rheology. The production of emulsions with very fine droplets requires a high energy input, which is usually introduced by high-pressure systems [
1]. To prevent immediate re-coalescence within seconds after the breakup of oil droplets, a high concentration of emulsifier molecules with fast adsorption kinetics and good emulsion-stabilizing properties are needed.
Natural polymers, such as hydrocolloids, are often used as emulsifiers in these products due to their emulsion-stabilizing functionality and high consumer acceptance. They adsorb onto the surface of the dispersed oil phase and form a thick protective layer that prevents oil droplets from coalescing [
2,
3]. However, there are some limitations and difficulties in using hydrocolloids due to their molecular structure. Most hydrocolloids possess high molecular weight, a broad molecular weight distribution, and a complex structure. Due to their size and complex structure, most hydrocolloids have a slow adsorption mechanism, leading to increased coalescence during high-pressure processing and large droplet sizes, especially in products of increased oil content. The viscosity-enhancing properties of hydrocolloids—helpful in preventing creaming—unfortunately often prevent the use of high-pressure systems above a certain hydrocolloid concentration. As a result, hydrocolloids have limited efficacy and usability in high-pressure homogenization processes. To overcome these difficulties, hydrocolloids can be modified by adjusting their molecular size and structure. Enzymatic degradation allows for a specific modification. Various enzymes from nature provide a toolbox that can be used for a targeted cleavage of linkages.
Sugar beet pectins (SBPs) can be used as a model hydrocolloid for investigations in this regard. SBPs are extracted from sugar beet pulp, a by-product of the sugar industry, and possess good emulsifying properties. Many authors have studied the relationship between particular structural features and their influence on the emulsifying properties of SBPs. It has often been described that the hydrophobic groups of the molecules, such as proteins and ferulic acids, serve as anchors to the oil phase and facilitate adsorption onto the interface [
4,
5,
6,
7,
8]. In addition, it was shown that smaller droplets were obtained with SBPs that possessed a higher proportion of neutral sugar side chains [
9]. In contrast to the side chains, the hydrophilic homogalacturonans (the backbone of (1,4)-linked
α-
D-galacturonic acid units) protrude into the aqueous phase and lead to steric and electrostatic stabilization [
6]. However, the homogalacturonan seems to have less influence on the droplet size, compared to the neutral sugar side chains and hydrophobic groups [
9].
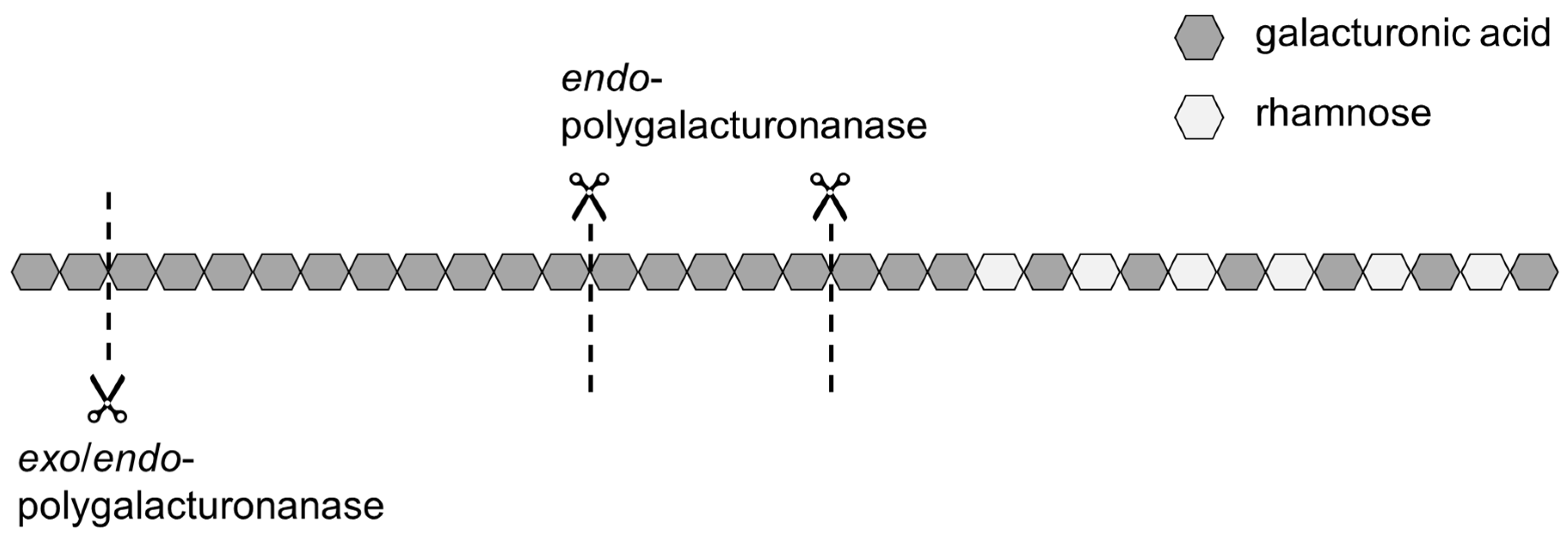
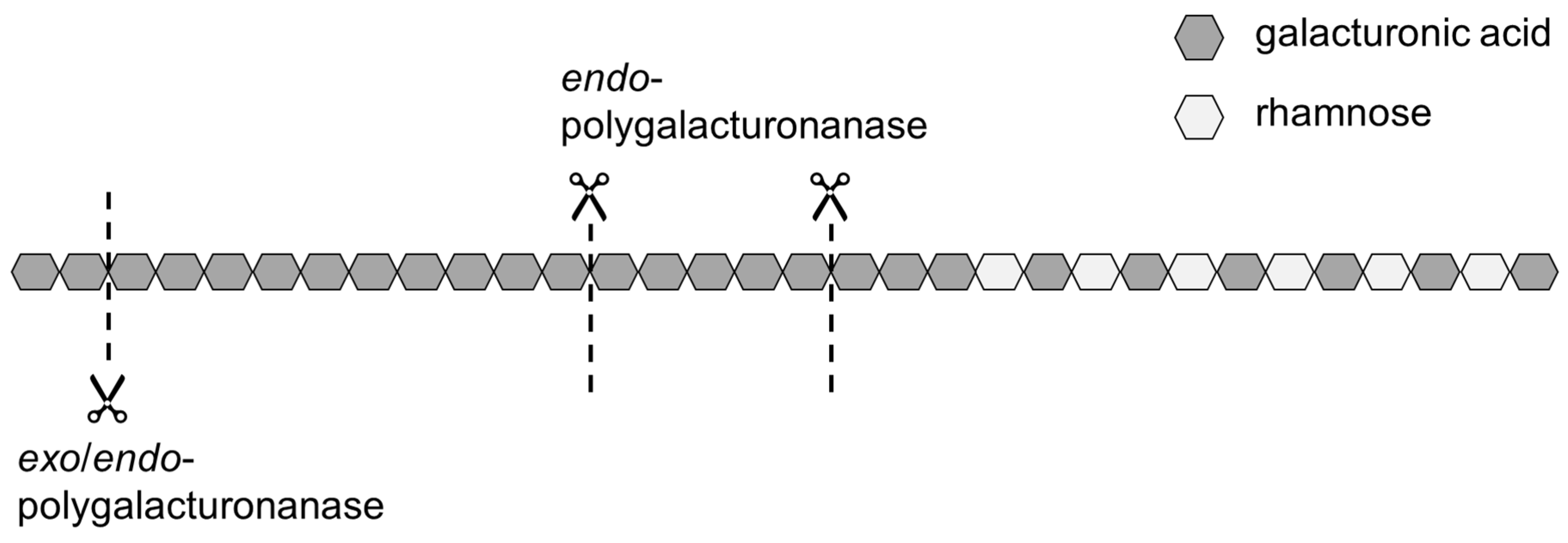
Therefore, modifying the homogalacturonan without degrading the side chains and functional groups might be useful to improve the adsorption kinetics of SBPs. A way to modify the homogalacturonan is the use of polygalacturonanases, such as
exo- and
endo-polygalacturonanase (
exo-PG and
endo-PG, respectively). Both are hydrolytic enzymes, which catalyze the hydrolysis of the O-glycosyl bond of α-
D-(1→ 4)polygalacturonan but in a different pattern. Whereas
endo-PG catalyzes the random cleavage of the O-glycosyl bond between unesterified α-1,4-
D-linked galacturonic acid residues,
exo-PG exclusively catalyzes the terminal cleavage of dimers from the non-reducing end [
10,
11]. As a result of the cleavage, the proportion of GalA in the molecule decreases and the proportion of neutral sugars increases [
6,
8,
12].
Figure 1 shows schematically where the two enzymes operate on the pectic backbone.
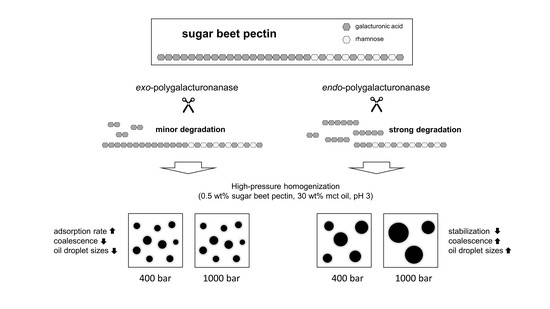
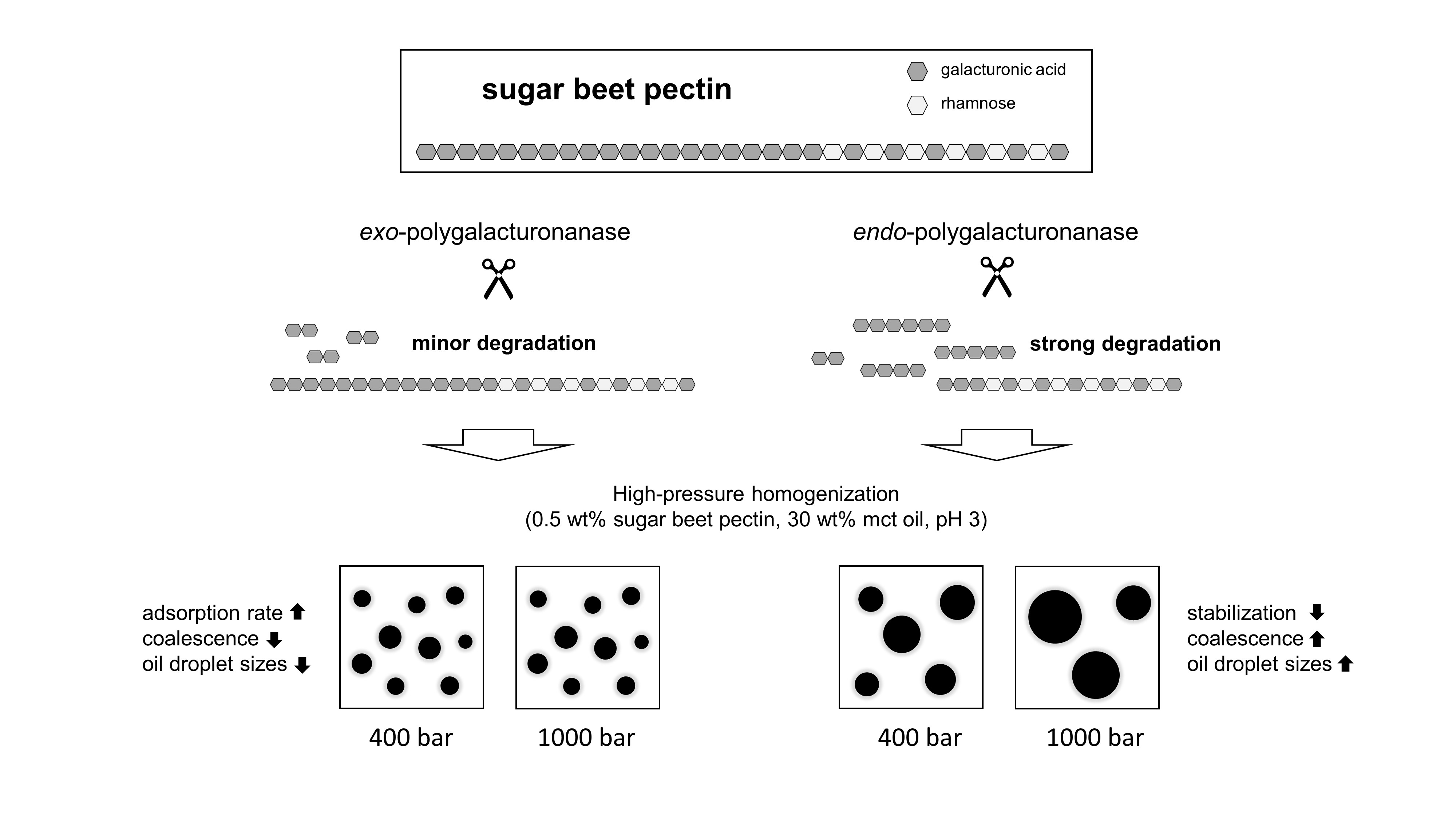
The use of endo-PG leads to stronger fragmentation than the use of exo-PG, and thus to smaller SBPs with hydrophobic groups distributed over several fragments. On the one hand, a smaller size could have a positive effect on the adsorption rate, but, on the other hand, it could have a negative effect on the emulsion stabilizing properties.
Various enzymes to modify the structure of SBPs were used by Funami et al. [
6]. They showed that an enzymatic treatment with 10 U/g SBP
endo-PG led to larger emulsion droplets, compared to the unmodified control sample. Chen et al. [
8] showed that the emulsifying properties of SBPs declined after enzymatic digestion with 10 U/g SBP
endo-PG. This was determined by comparing droplet sizes of SBP-stabilized emulsions, creaming indices, and interfacial activities. SBPs with increased molecular weight due to laccase-mediated conjugation, on the other hand, showed better functionality and emulsion stabilization [
13]. This was thought to be due to the formation of thick interfacial layers at the oil–water interface. Other authors demonstrated that pectin fractions of very low molecular weight resulted in lower interfacial activity and emulsions with larger droplet sizes due to insufficient steric stabilization [
14,
15,
16].
Therefore, the enzymatic modification should not be excessive, as otherwise cleavage might lead to a loss of the emulsifying and emulsion-stabilizing properties.
The objective of this study was to determine if SBPs could be modified to improve their ability to reduce coalescence during high pressure homogenization. For this purpose, the size of SBPs was varied by the enzymatic cleavage of the homogalacturonan using exo- and endo-PG. The effect of the altered size on the adsorption of SBPs onto the oil–water interface was investigated using drop profile analysis tensiometry. Then, a high-pressure homogenizer was used to produce pectin-stabilized emulsions. The homogenization pressure was varied to achieve different degrees of droplet break-up and collision-induced coalescence rates, respectively. By analyzing the droplet size distributions, conclusions could be drawn about the coalescence that occurred immediately after droplet breakup.
2. Materials and Methods
2.1. Materials
Commercial sugar beet pectin was provided by Herbstreith & Fox KG (Neuenbürg, Germany). According to Bindereif et al. [
9], pectins molecular characteristics were as follows: molecular weight, 104 ± 5 kDa; galacturonic acid, 66.1 ± 1.8%; protein content, 4.1 ± 0.2%; degree of methylation, 52.1 ± 1.7%; degree of acetylation, 23.1 ± 0.5%; trans-ferulic acid, 706 ± 21 mg/100 g.
The enzymes endo-polygalacturonanase (EC 3.2.1.15 from Aspergillus aculeatus) and exo-polygalacturonanase (EC 3.2.1.82 from Yersinia enterocolitica) were purchased from Megazyme (Bray, Ireland).
Medium chain triglyceride oil (mct) was purchased from Schumann & Sohn GmbH (Karlsruhe, Germany). According to supplier information, the mct oil was composed of 60% C8 and 40% C10 chains and had a density of 0.95 kg/L at room temperature. Unless stated otherwise, all chemicals used were of analytical grade and ultrapure water was used throughout the experiments.
2.2. Enzymatic Modification of Pectins
Pectins were enzymatically modified by endo-PG and exo-PG. As a first step of the treatment, 2.0 wt% SBP was dissolved in water at 60 °C using an Ultraturrax T-25 digital (IKA® Werke GmbH & Co. KG, Staufen, Germany) at a rotational speed of 10,000 rpm (tip speed 6.54 m/s) for 30 sec. After cooling to room temperature, the pH was adjusted to pH 5.5 for endo-PG experiments and pH 6.0 for exo-PG experiments using 0.5 M NaOH. Then, the solutions were left to equilibrate overnight for at least 15 h under gentle stirring. Then, the temperature was set to 50 °C (for endo-PG) or 60 °C (for exo-PG), respectively. To these solutions, 3 U/g pectin of endo-PG or exo-PG were added. After specific incubation times (10 min, 1 h, or 24 h, respectively), the enzymes were inactivated by heating the solution to 95 °C and holding the temperature for 5 min to stop the enzymatic reaction. The same procedure was performed for 24 h without the addition of enzymes in order to obtain reference samples. The solutions were then deep-frozen (−18 °C), freeze-dried, and ground to particles <0.5 mm. Pectins without enzymatic modification are referred to as exo-reference (reference pectin for treatments with exo-PG) and endo-reference (reference pectin for treatments with endo-PG). Furthermore, the modified pectins are referred to as exo-24h (treatment with exo-PG for 24 h), endo-10min, endo-1h, and endo-24h (treatment with endo-PG for 10 min, 1 h, and 24 h, respectively).
2.3. Molecular Size Distribution
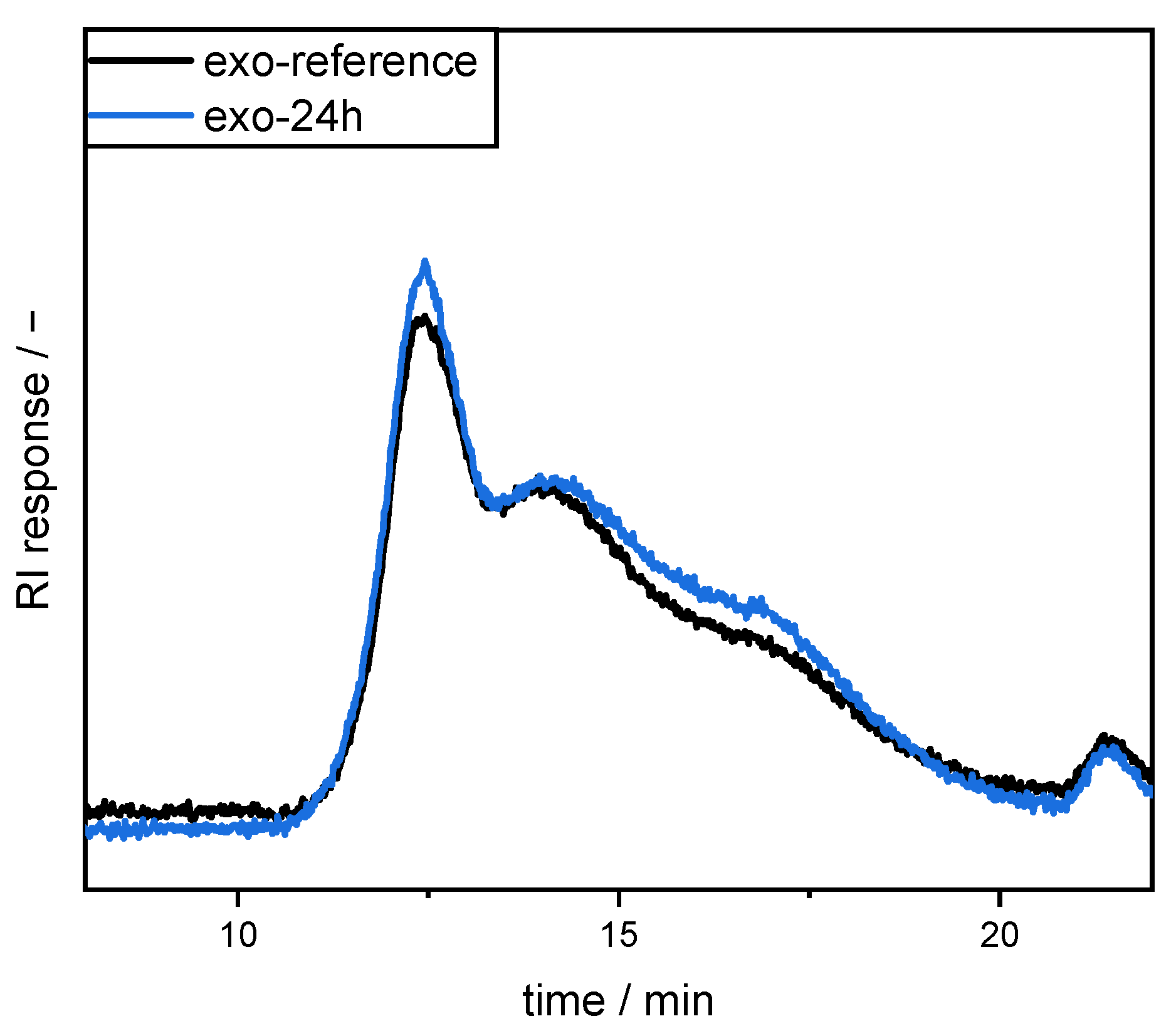
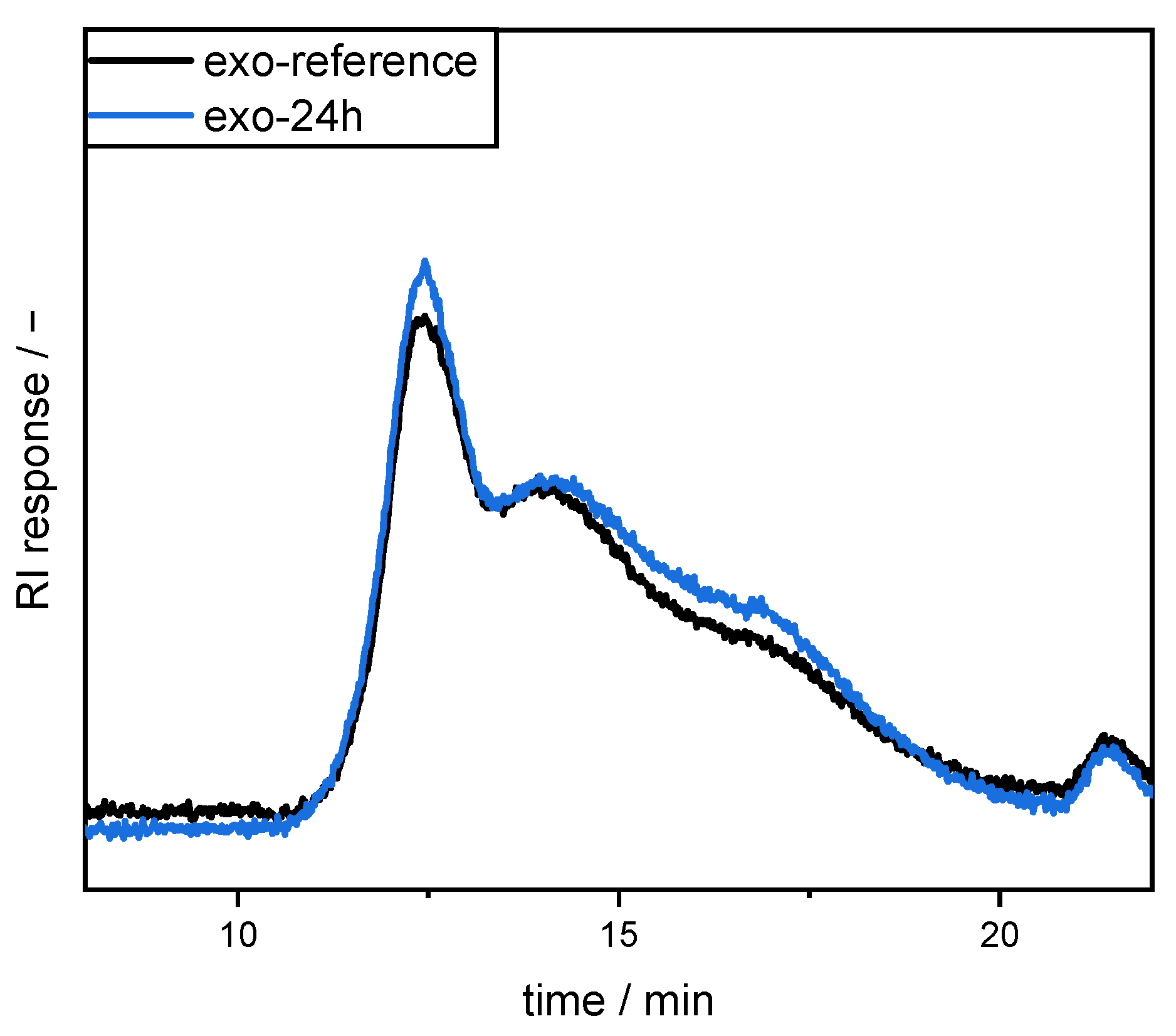
High performance size exclusion chromatography (HPSEC) coupled with a refractive index (RI) detector (L-7490 LaChrom RI, Merck, Germany) was used to compare the molecular size distribution of the pectins. For this purpose, SBP samples were dissolved in 50 mM sodium nitrate (2 g/L). After centrifugation, the supernatant was used for SEC analysis on a Merck Hitachi L-7000 system equipped with a PWXL guard column (12 µm, 40 × 6 mm) connected in series with a TSKgel G6000PWXL (300 × 7.8 mm, 13 µm particle size) and a G4000PWXL (300 × 7.8 mm, 10 µm particle size, Tosoh Bioscience GmbH, Griesheim, Germany) column. Sodium nitrate (50 mM) was used as eluent at 40 °C (0.5 mL/min).
2.4. Preparation of Pectin Solutions
Sugar beet pectins (0.1 wt% or 0.5 wt%) were dissolved in water at 60 °C using an Ultraturrax T-25 digital (IKA® Werke GmbH & Co. KG, Staufen, Germany) at a rotational speed of 10,000 rpm for 30 s. The solution was cooled down, and the pH was adjusted to 3 using 1 M HCl at room temperature. Then, the pectin solutions were left to equilibrate for at least 15 h under constant magnetic stirring. The pH was checked again and readjusted to pH 3 if necessary.
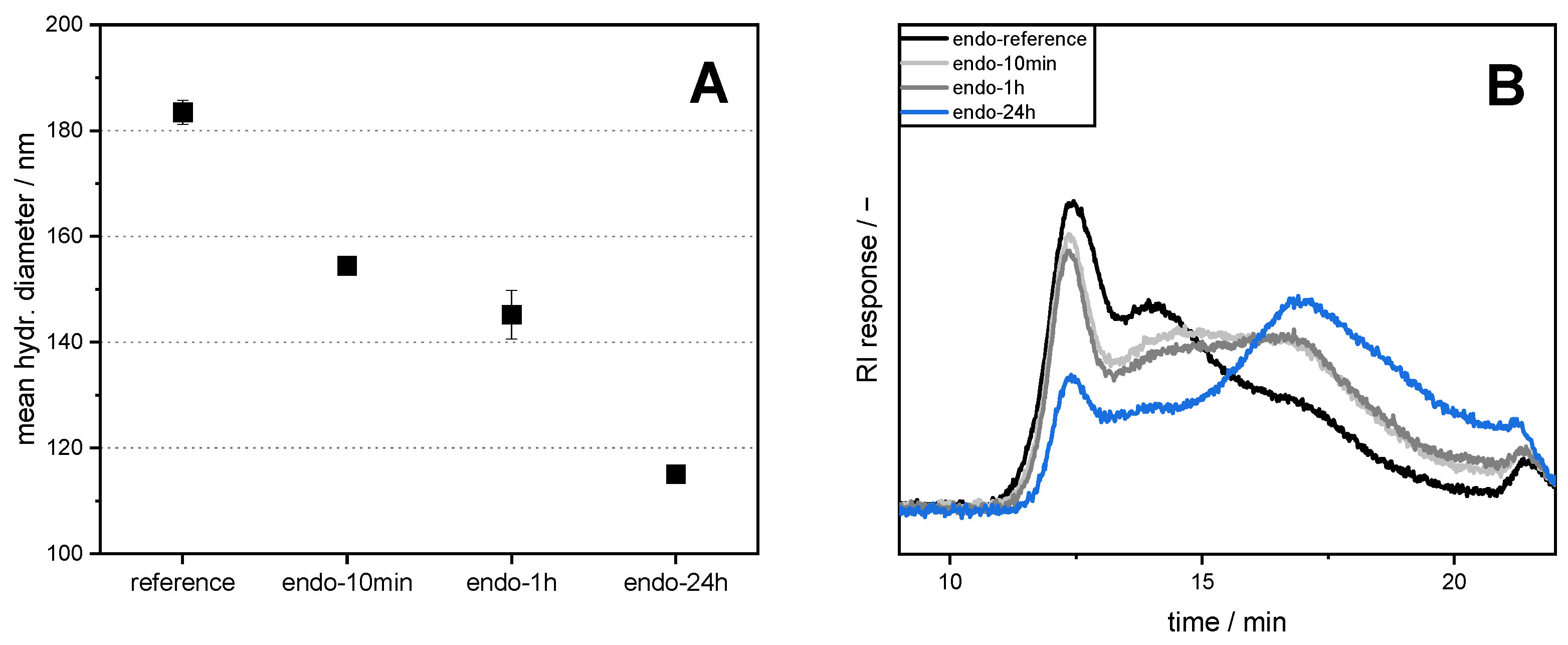
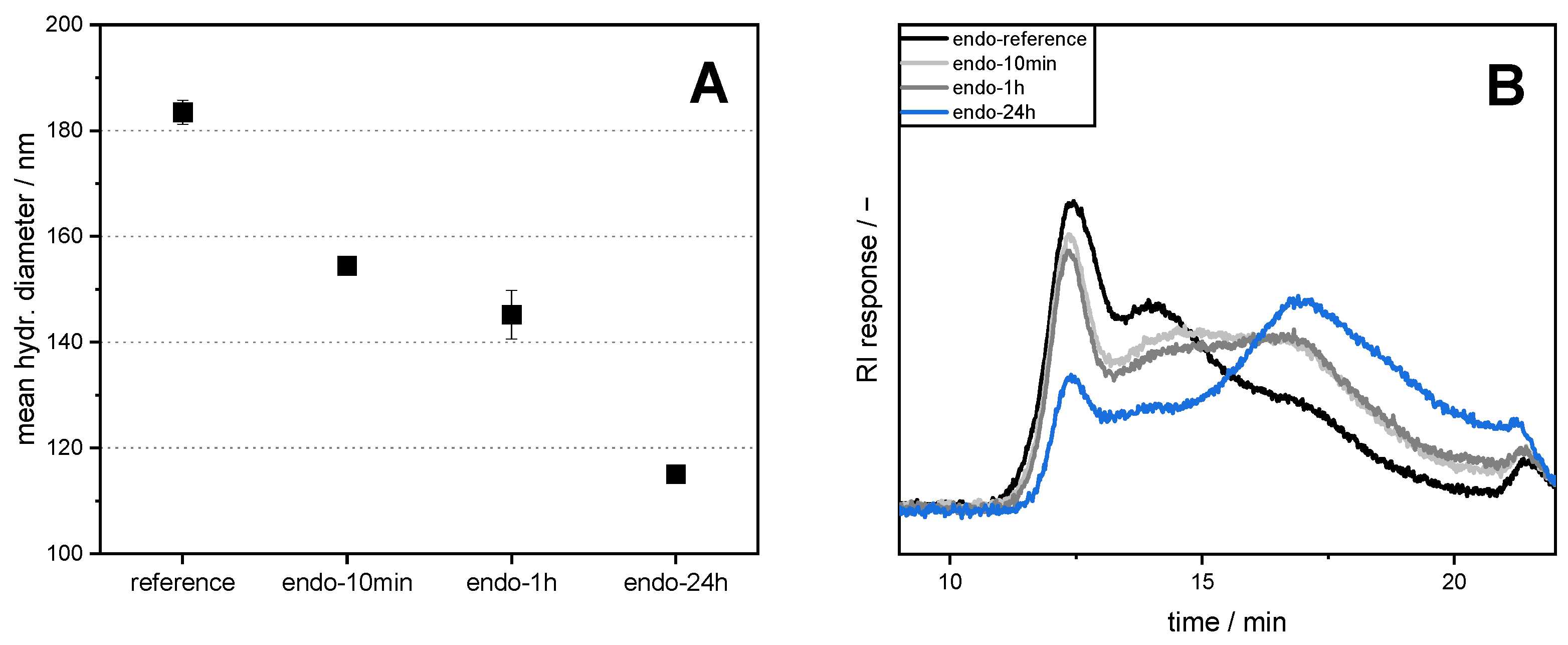
2.5. Measurement of Hydrodynamic Diameter
Aqueous pectin solutions with a concentration of 0.1 wt% SBP were prepared, as described in
Section 2.4. Then, the hydrodynamic diameters were measured, as described in Bindereif et al. [
17]. In brief, SBP solutions at pH 3 were diluted further to yield 8 diluted samples (0.005–0.1 wt%). After an additional equilibration time of at least 15 h, three measurements of 10 runs each with 90 s run time per sample were performed. Refractive indices were set at
n = 1.5470 for pectin and n = 1.333 for water. The hydrodynamic diameter was determined by plotting the measured z-average diameter versus SBP concentration and extrapolation to an SBP concentration of zero.
2.6. Measurement of Dynamic Interfacial Tension
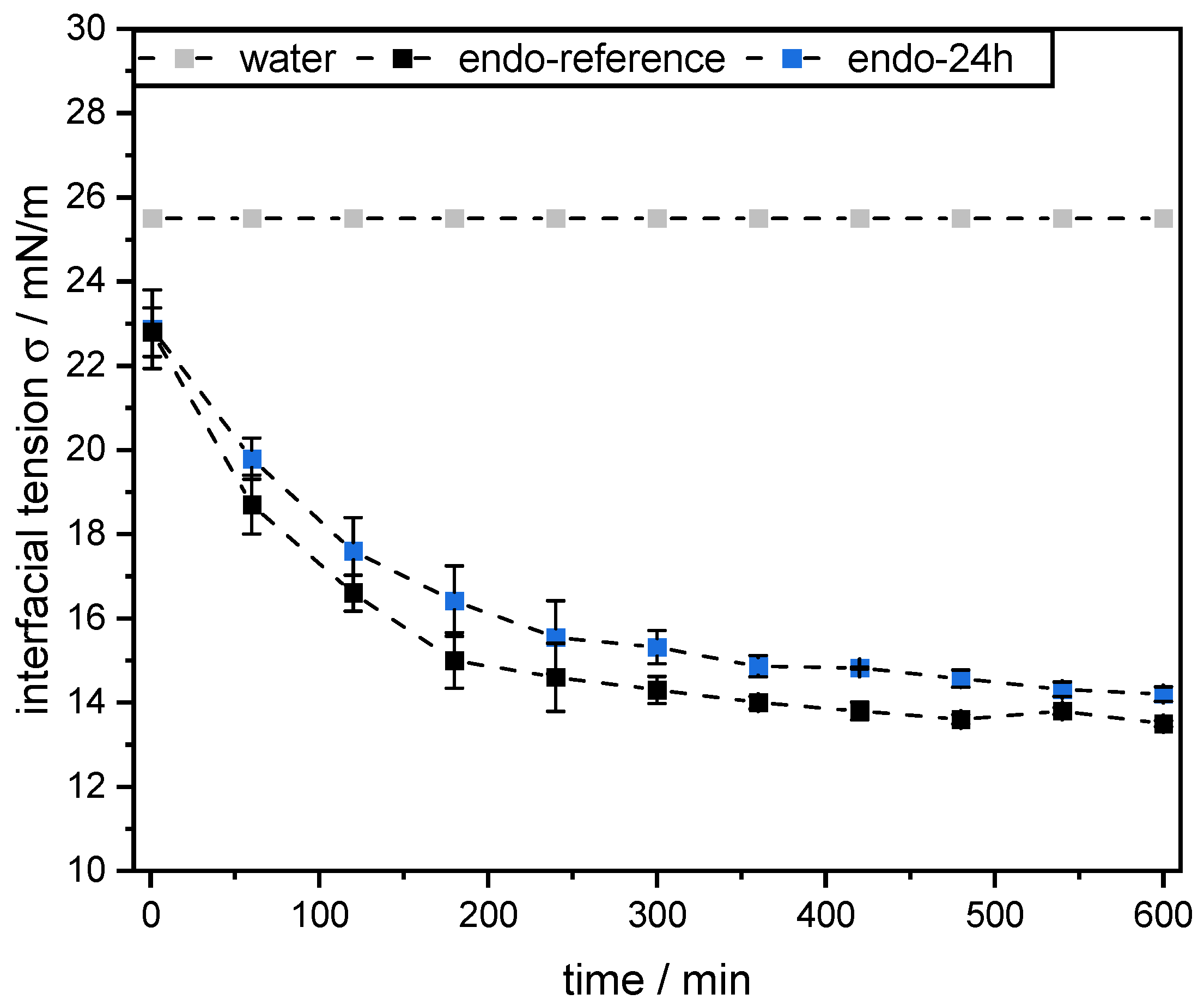
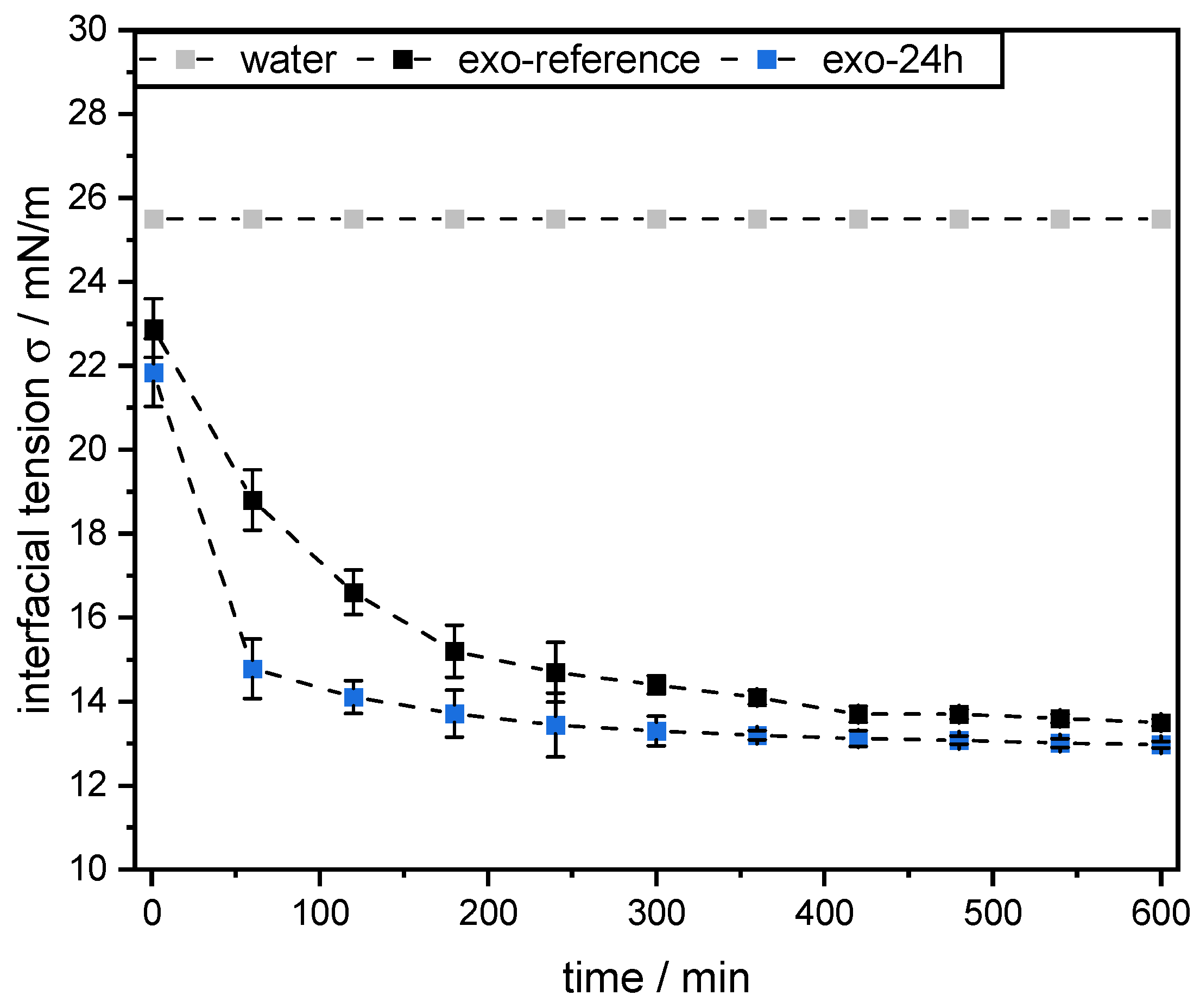
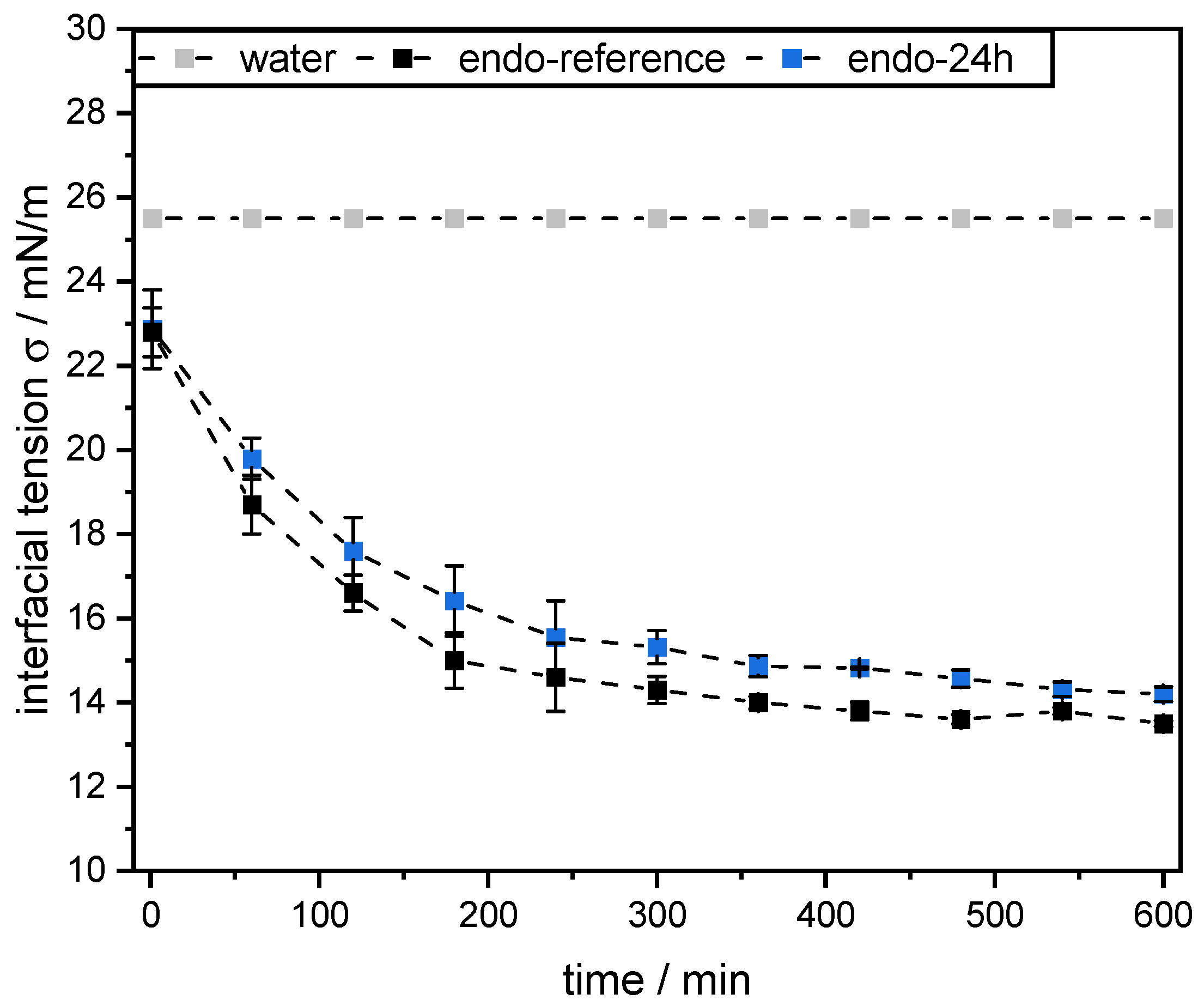
Dynamic interfacial tension measurements at the mct oil–water interface were performed using the pendant drop technique (OCA 15 LJ, DataPhysics Instruments GmbH, Germany). Therefore, a droplet of mct oil with a volume of 12.5 µL was formed at the tip of the tensiometer capillary in a 0.1 wt% SBP solution (pH 3, see
Section 2.4). All measurements were conducted in triplicate at 25 °C for 10 h. As a reference, the interfacial tension of the mct oil–water interface without the addition of pectins was also determined.
2.7. Viscosity Measurement
The viscosities of pectin solutions, mct oil, and emulsions were determined with a Physica MCR 301 rheometer equipped with a double gap geometry DG26.7 (Anton Paar, Graz, Austria). Rotational measurements were conducted at a temperature of 25 °C by applying a logarithmic shear rate profile recording 7 measurement points for each decade starting at 0.1 s−1 and rising up to 1000 s−1. Since SBP solutions and emulsions showed shear thinning behavior, the viscosity at 100 s−1 was used for comparison.
2.8. Preparation of Pectin-Stabilized Emulsions
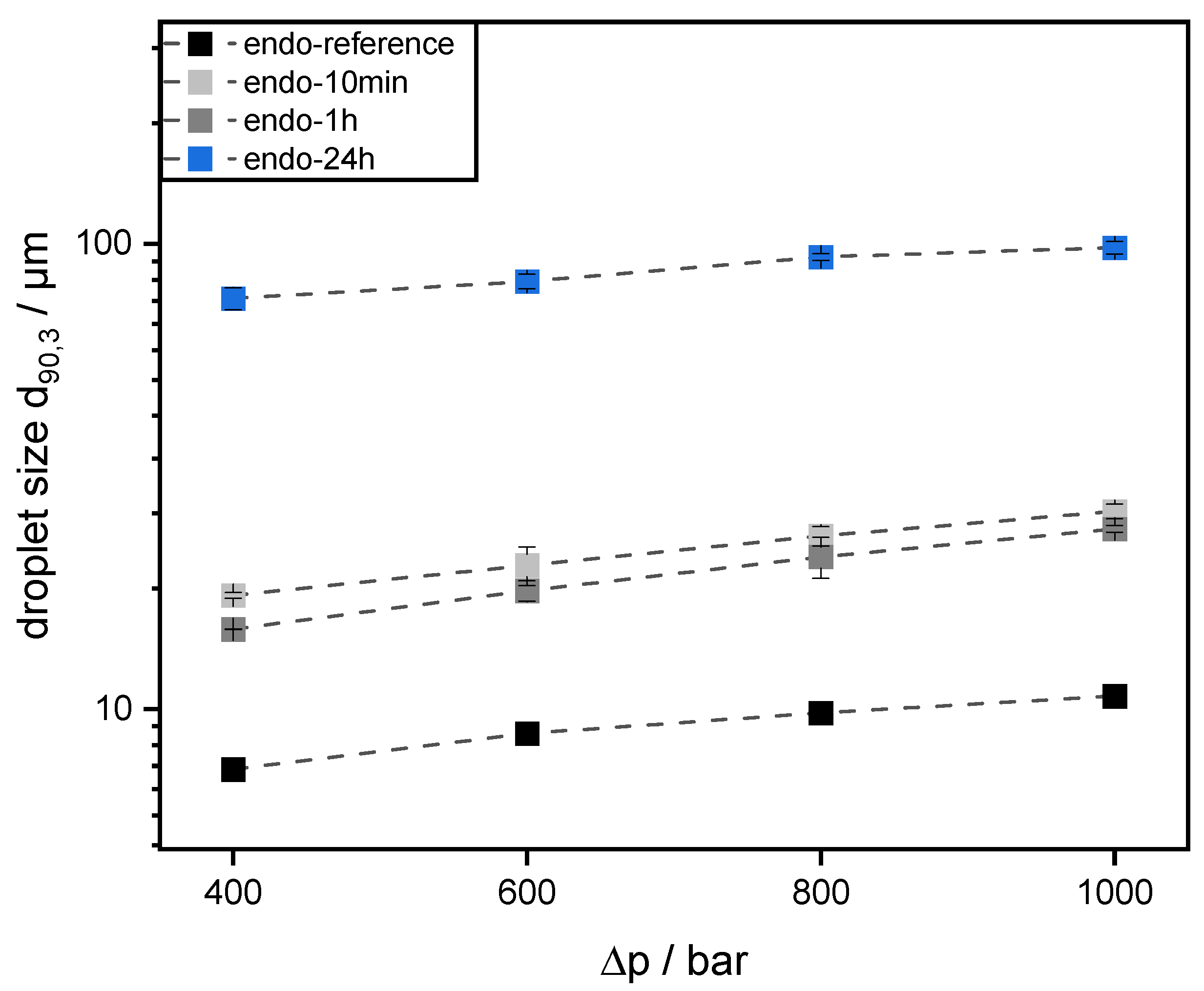
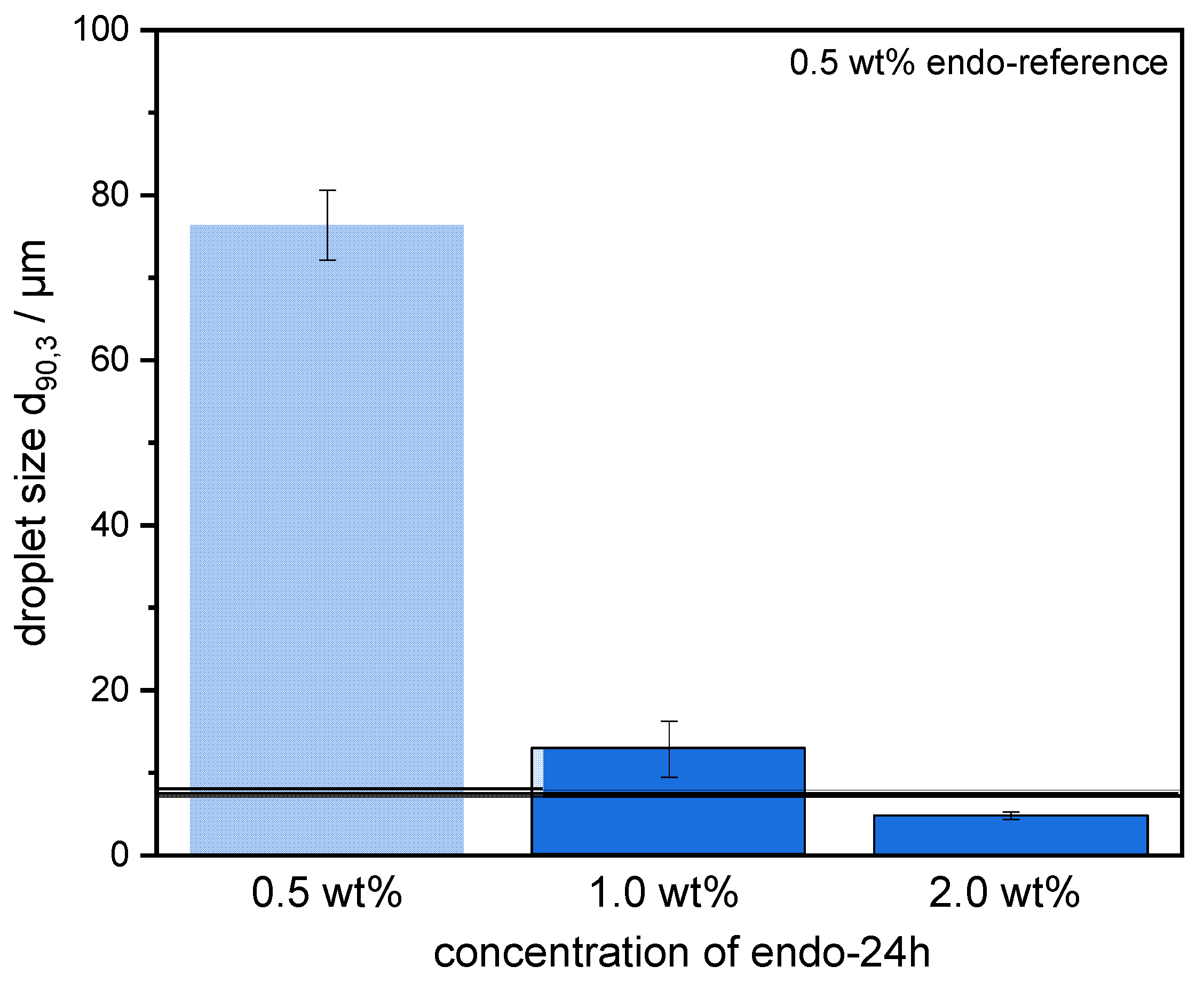
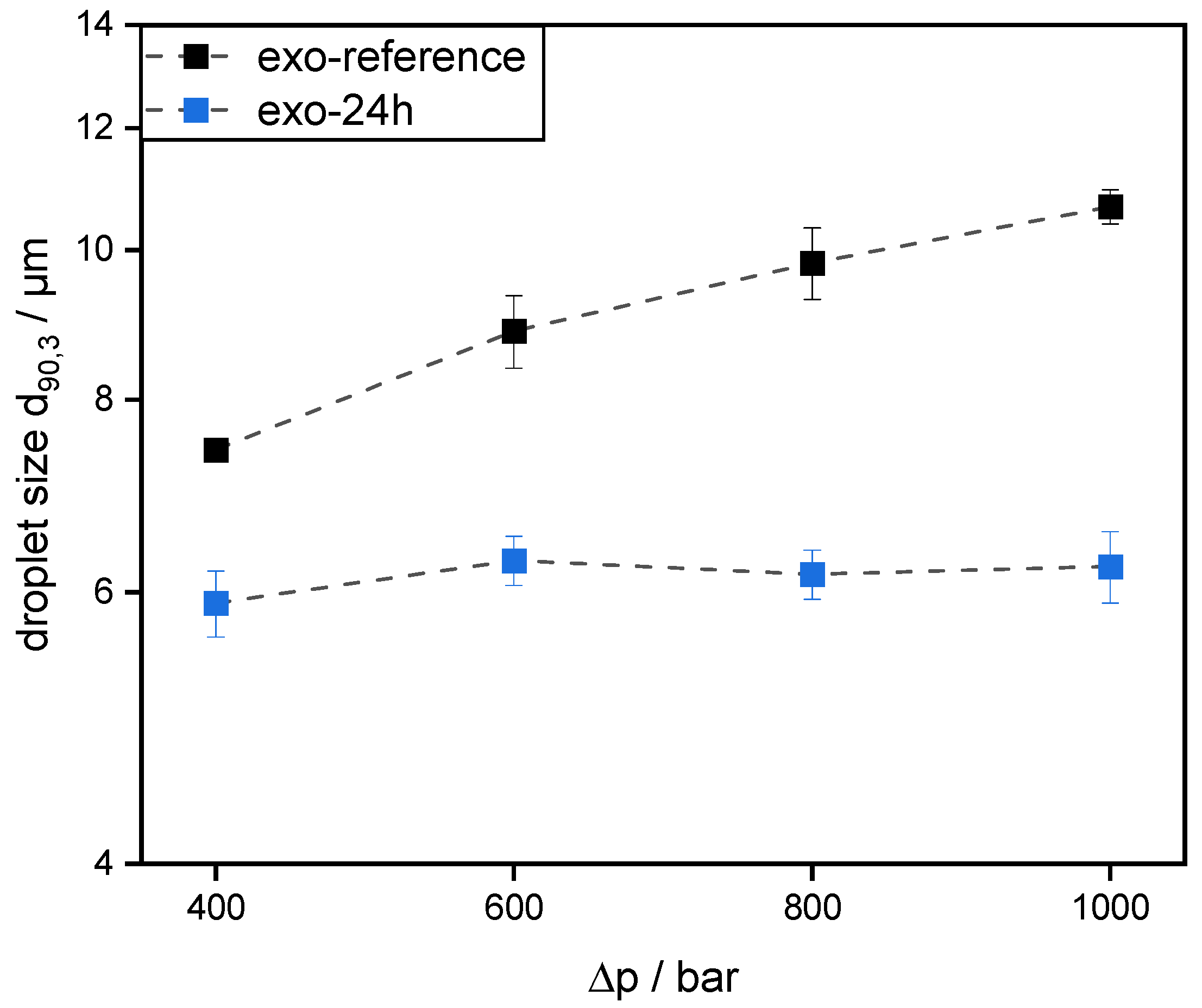
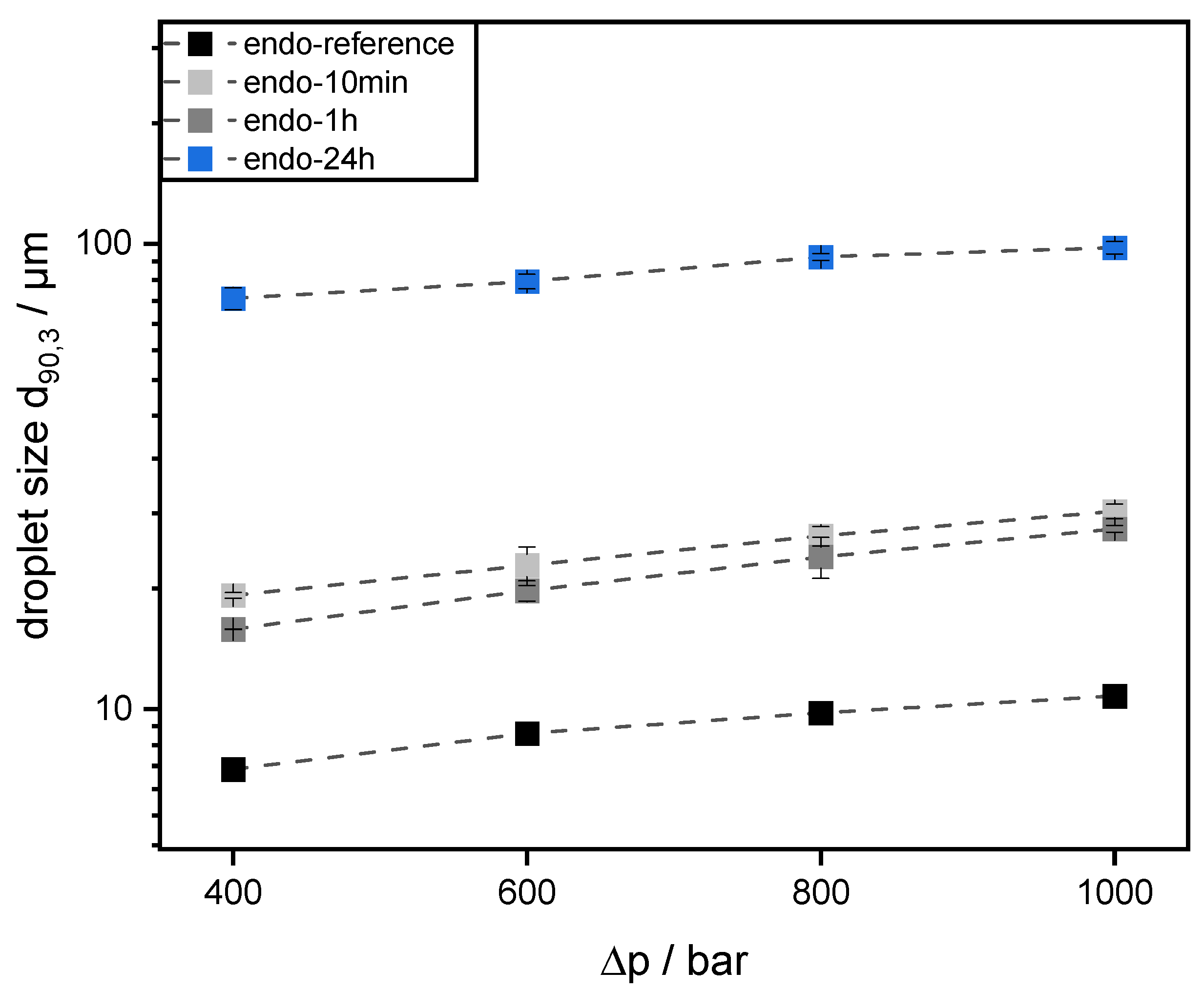
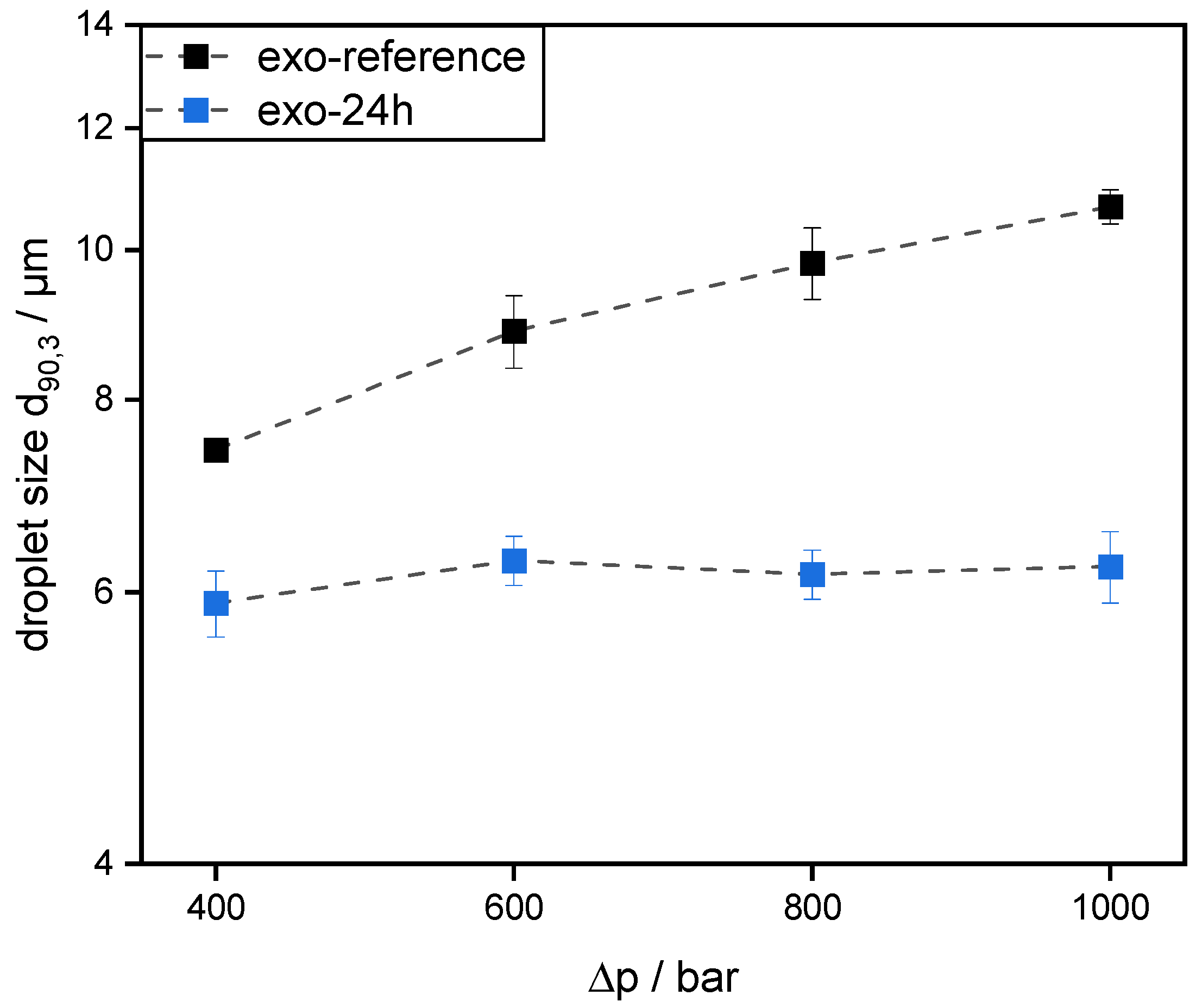
Emulsions were prepared using 30 wt% mct oil as dispersed phase and 70 wt% pectin solution (0.5 wt% SBP) as continuous aqueous phase. The pectin solutions were prepared, as described in 2.4. Then, mct oil was added to prepare an emulsion premix by using an Ultraturrax T-25 digital at 15,000 rpm (tip speed 9.81 m/s) for one minute. For further homogenization, the premix was transferred to a high-pressure homogenizer, Microfluidizer® MF 110 EH, equipped with a Y-type interaction chamber and with a microchannel diameter of 75 µm (Microfluidics Corporation, Newton, MA, USA). The interaction chamber is followed by an auxiliary processing module (APM) with a microchannel of 200 µm. The emulsions were homogenized at 400 bar, 600 bar, 800 bar, and 1000 bar. With increasing homogenizing pressure Δp, droplet break-up is increased. Droplet size decreases and the number of fine droplets increases. Since the number of droplets increases significantly more than their contact area, the collision rate increases strongly with increasing homogenization pressure.
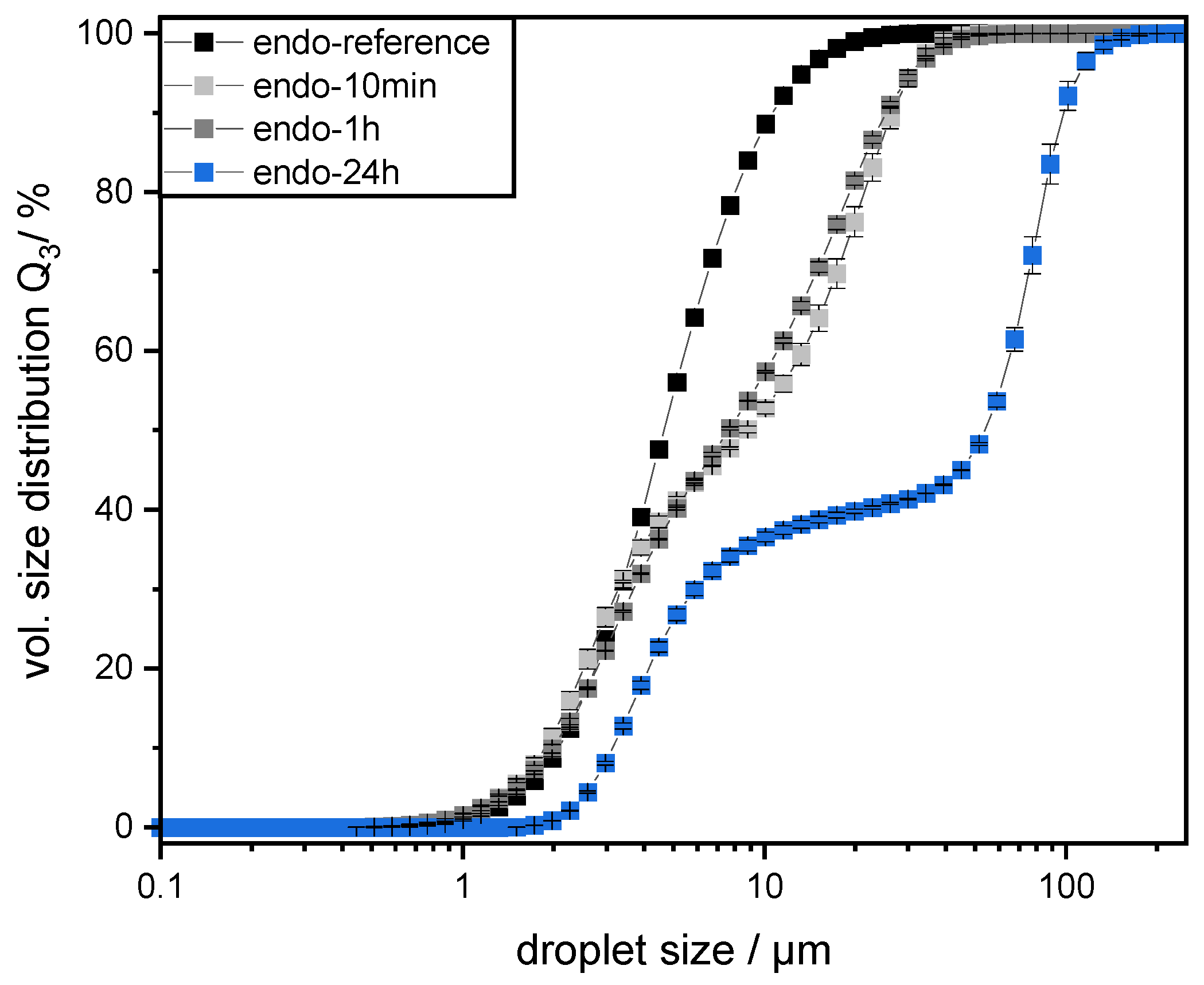
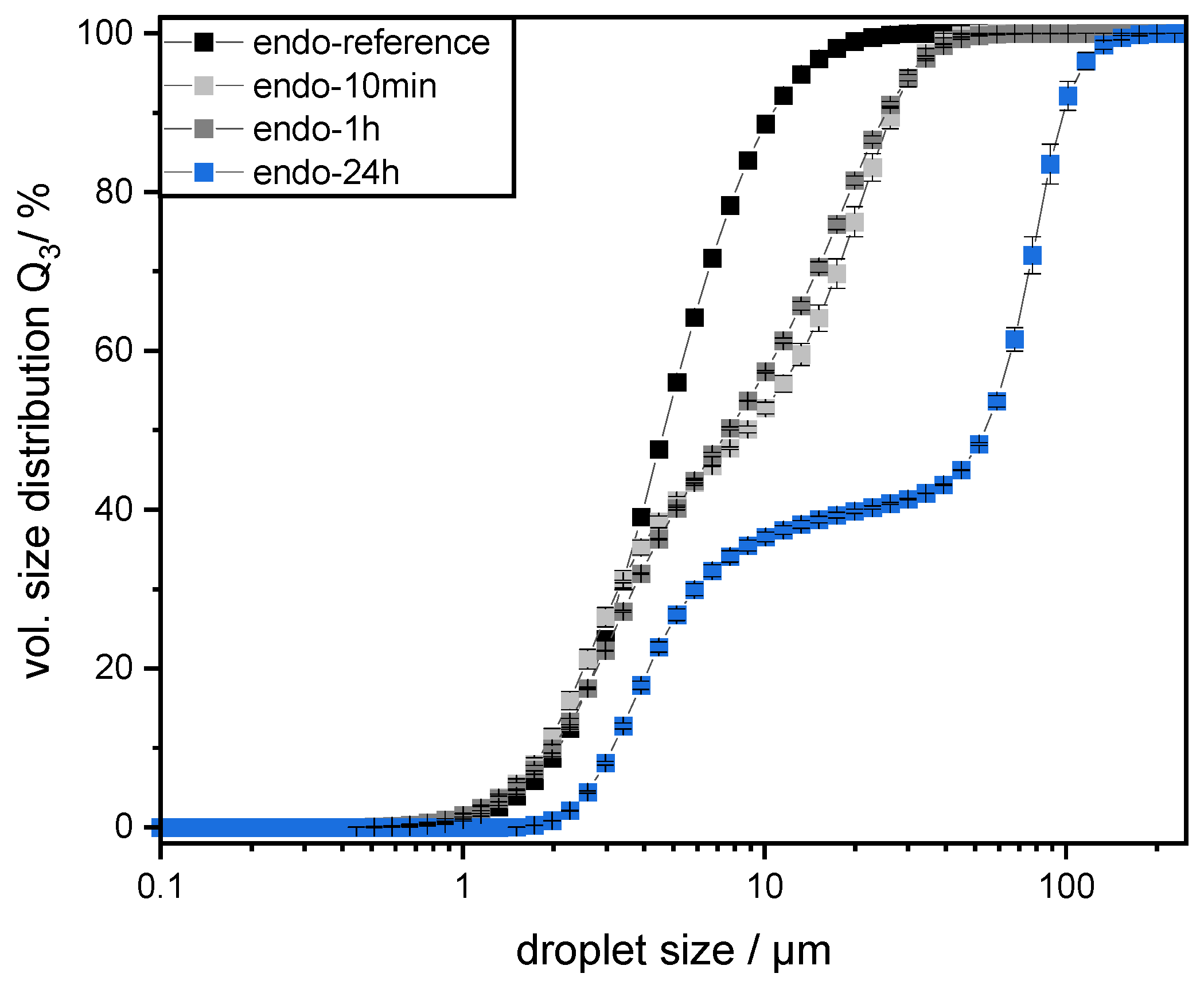
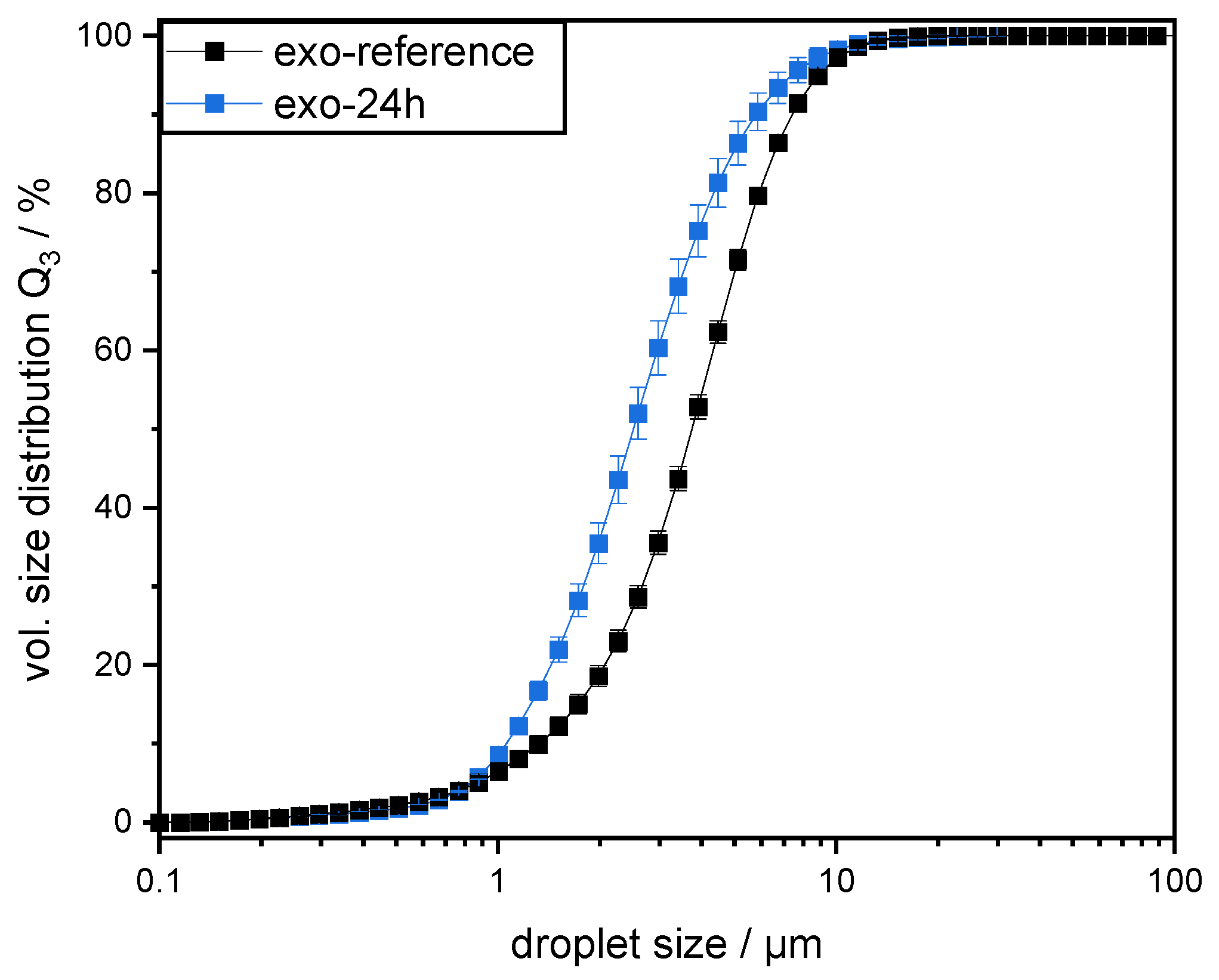
2.9. Measurement of Droplet Size Distribution
Prepared emulsions were characterized by measuring their droplet size distributions (DSD) by static laser light scattering using a particle analyzer (HORIBA LA-950, Retsch Technology, Haan, Germany). The refractive indices were set at
n = 1.333 for water and
n = 1.4494 for mct oil for all measurements [
18]. The measurements were conducted in triplicate at room temperature immediately after sample preparation. The results are depicted as the cumulative volume distribution Q
3 and the 90th percentile of the volumetric cumulative size distribution (d
90,3).
2.10. Statistical Analysis
All measurements and emulsions were prepared at least three times if not stated otherwise, per independent experiment. Statistical analysis, calculation of averages, and standard deviations were performed by using the software OriginPro 2020 (OriginLab Corp., Northampton, MA, USA).
4. Conclusions
In the present study, we attempted to improve the emulsifying properties of SBPs in order to reduce coalescence during high-pressure homogenization. For this purpose, SBPs were subjected to enzymatic treatment to increase their adsorption rate due to smaller hydrodynamic sizes. As shown in the literature, neutral sugar side chains and functional groups play an important role for the emulsion-stabilizing properties. Therefore, two enzymes (endo-PG and exo-PG) were used to cleave glycosidic linkages in the homogalacturonan without degrading other structural features. HPSEC and hydrodynamic diameter measurements showed that fragmentation increased with increasing incubation time. Furthermore, the use of endo-PG resulted in significantly smaller SBPs than the use of exo-PG. This could be attributed to the different nature of the enzymatic cleavage of endo-PG, compared to exo-PG.
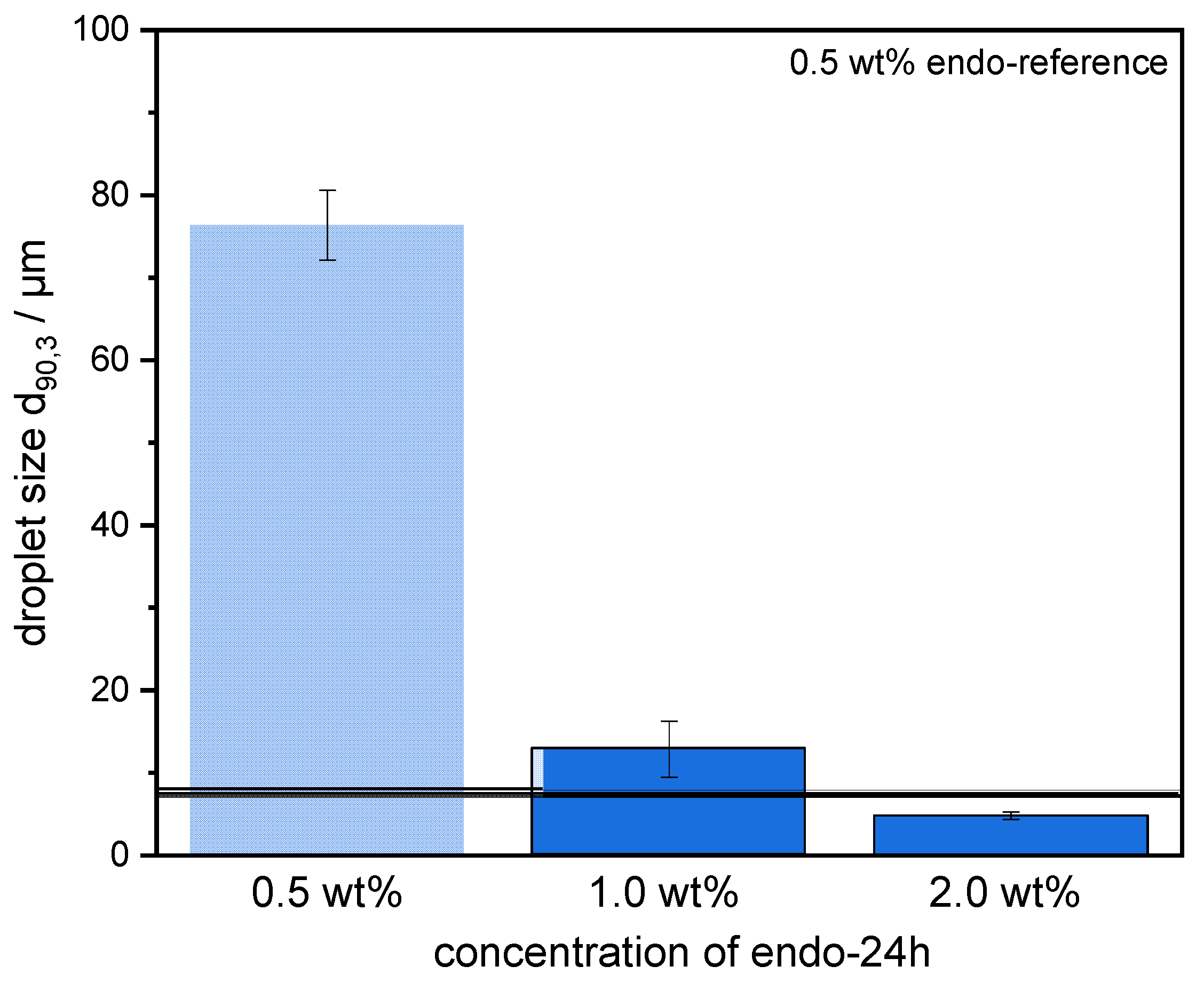
The preparation of emulsions (0.5 wt% SBP, 30 wt% mct oil, pH 3) and measurement of droplet size distributions provided information on the coalescence that occurred during emulsification. It was observed that emulsions prepared with SBPs modified with endo-PG showed larger droplet sizes than nonmodified SBPs due to enhanced coalescence. The coalescence-dominated emulsification process was also evident when the homogenization pressure was increased. With increasing Δp, larger droplets were obtained. This could be attributed to the reduced emulsion stabilizing properties of SBPs due to their strong fragmentation after enzymatic modification.
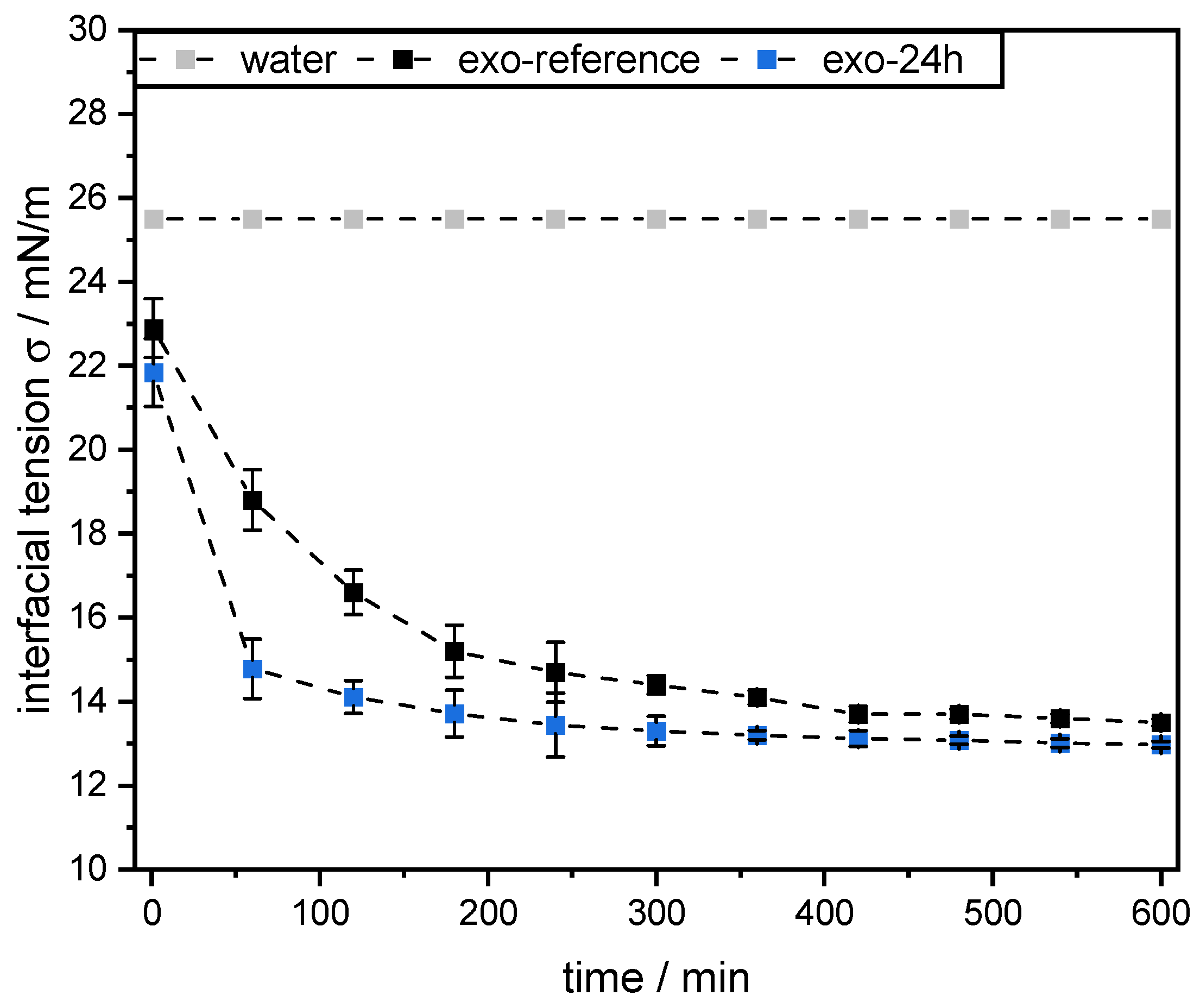
However, this was not the case for SBPs incubated with exo-PG. An enzymatic treatment of 24 h significantly improved their emulsifying properties. Faster adsorption, confirmed by droplet profile analysis tensiometer, resulted in less coalescence during emulsification. As a result, smaller droplets were obtained than with the reference pectins. In addition, increasing the homogenization pressure did not result in larger droplets, despite higher collision rates.
Overall, the present study showed that SBPs can be modified to alter their emulsifying properties. This allows coalescence to be reduced or increased during high-pressure homogenization, depending on modification. Based on this knowledge, other pectins and hydrocolloids could also be enzymatically modified to modify their emulsifying properties. However, this is only possible in a targeted manner if the structures that are decisive for the emulsifying properties are known. Structures that are insignificant for emulsion stabilization can be cleaved off to decrease the viscosity, increase the adsorption rate, and optimize the conformation at the interface. For this purpose, however, it is important to first establish the structure–property functions of the respective hydrocolloids in order to specifically modify the emulsifying properties.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}