
A Study on the Early Degradation of the Non-Additive Polypropylene–Polyethylene Composite Sampled between the Polymerization Reactor and the Deactivation-Degassing Tank

 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Instrumental Analysis
2.3. Thermogravimetric Analyzer
2.4. Gas Chromatography (GC-MS)
2.5. Pyrolysis Setup
2.6. Thermodynamical Analysis
2.7. Kinetic Modeling
2.7.1. Method 1 and 2: Coats–Redfern and Horowitz–Metzger
2.7.2. Method 3: Flynn–Wall–Ozawa (FWO)
2.7.3. Method 4: Kissinger–Akahira–Sunose (KAS)
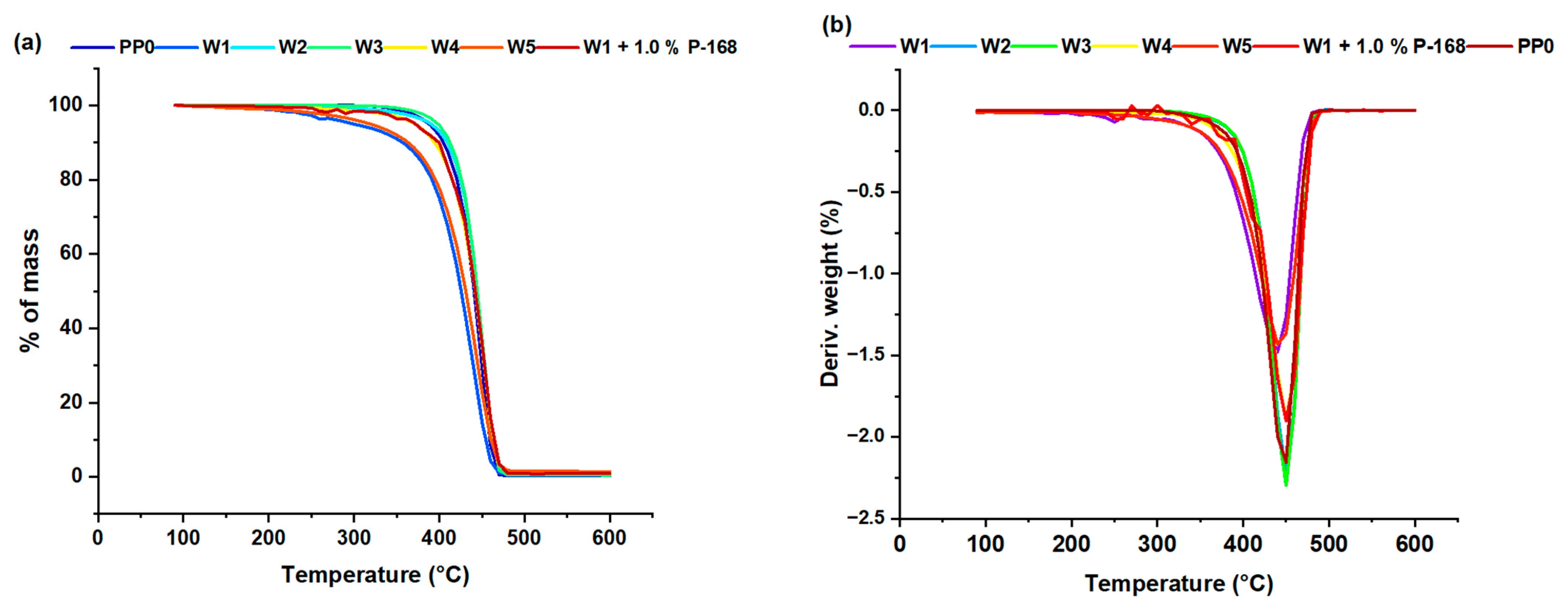
3. Results
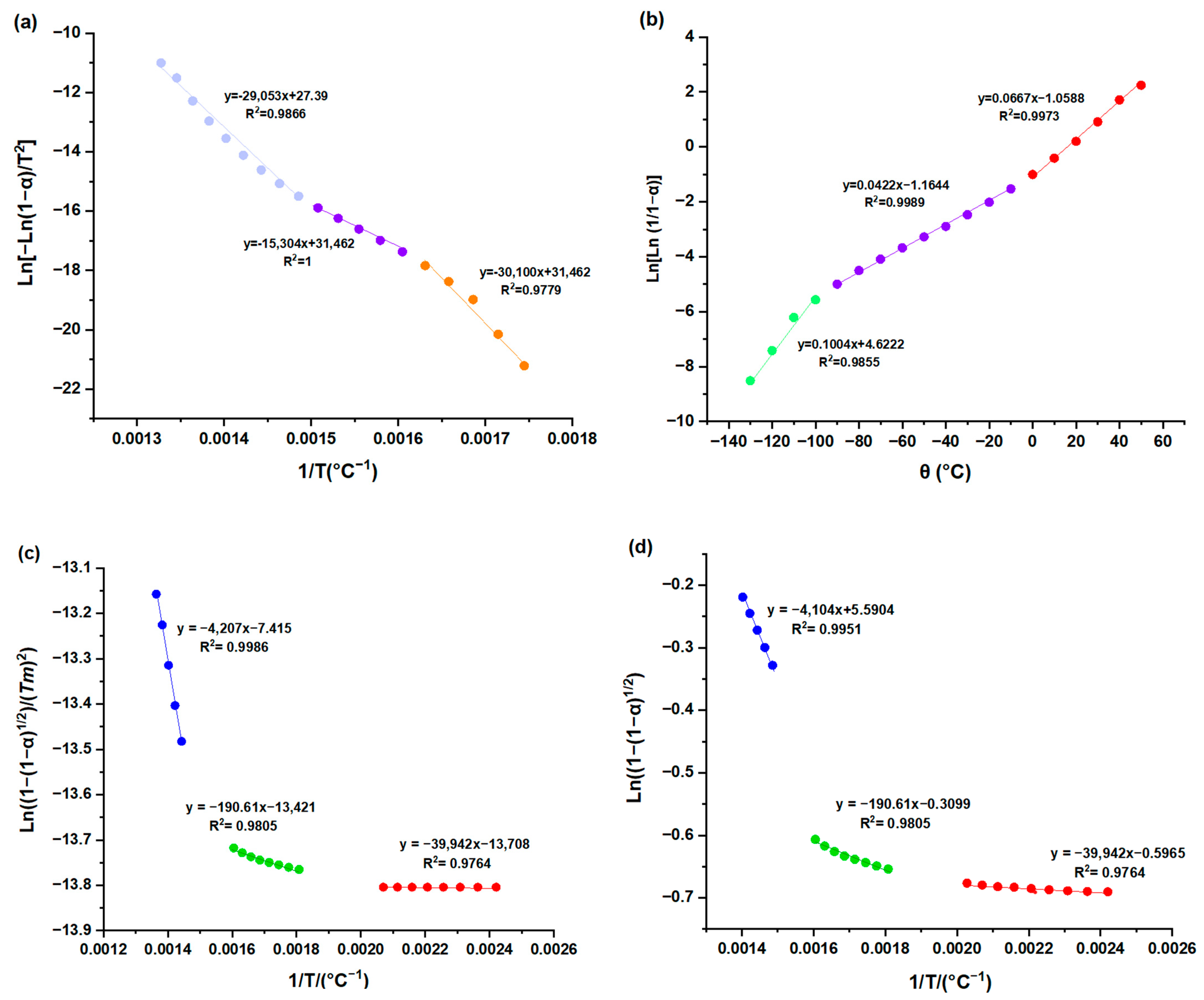
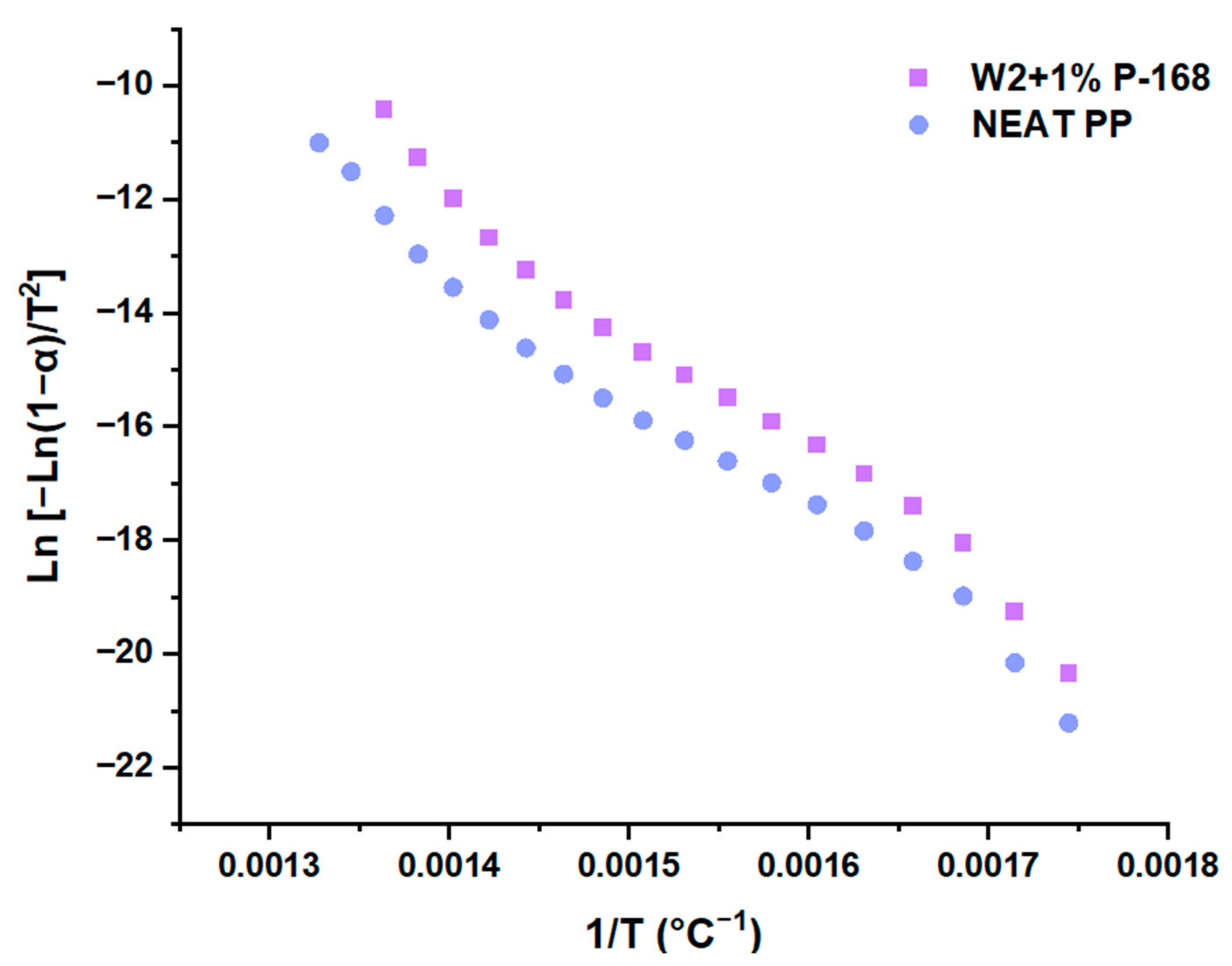
3.1. Kinetic Parameters
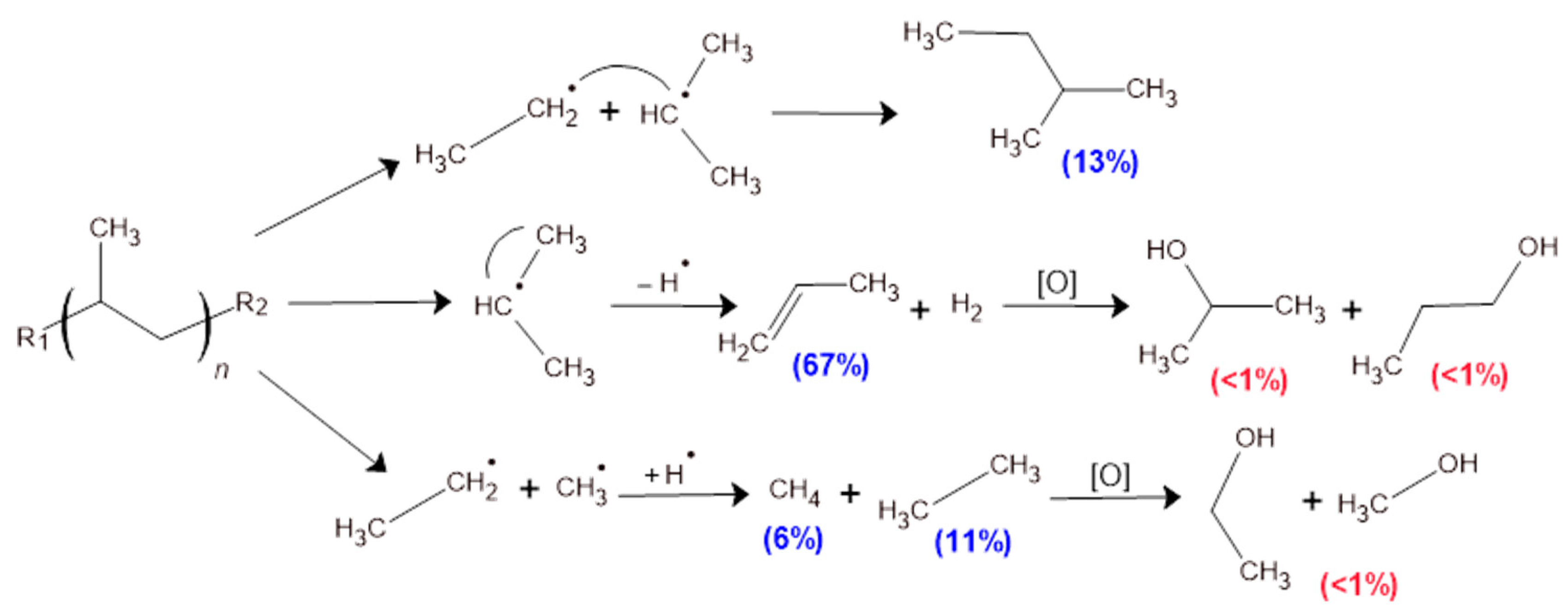
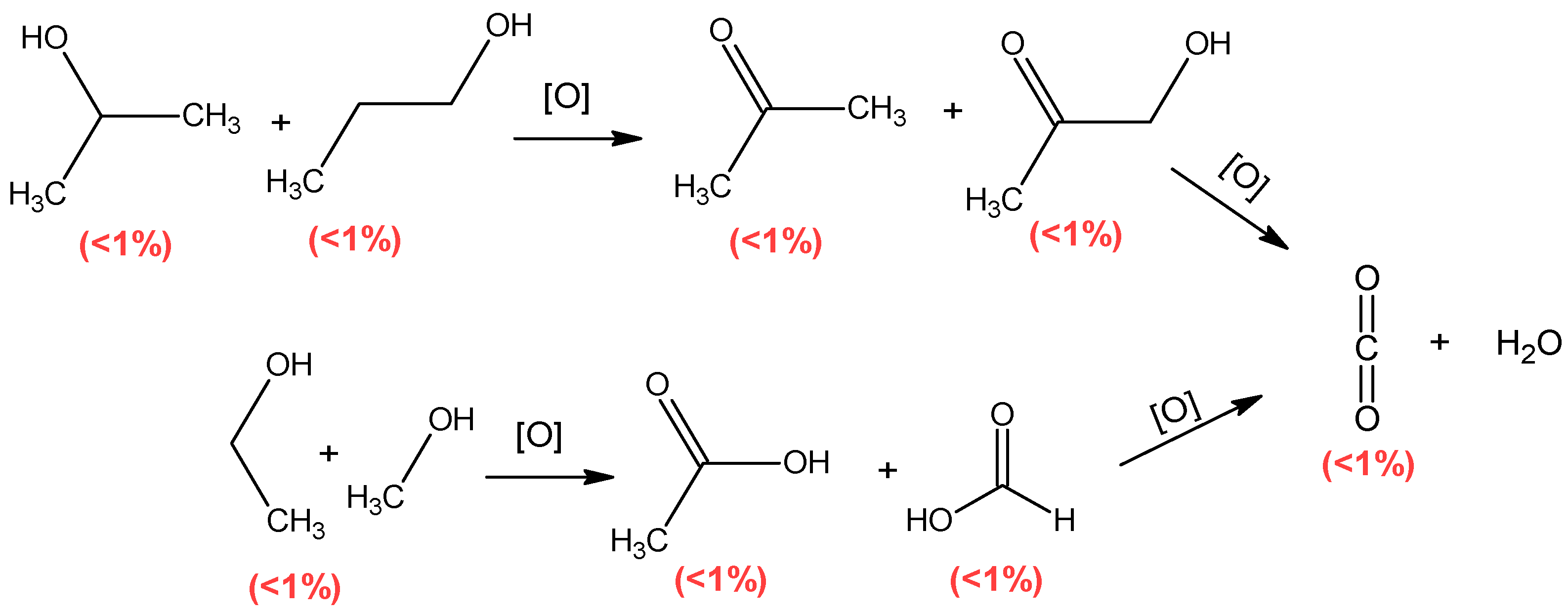
Mechanism of Pyrolysis
3.2. Thermodynamic Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zweifel, H. Stabilization of Polymeric Materials; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Pospíšil, J.; Klemchuk, P.P. Oxidation Inhibition in Organic Materials; CRC Press: Boca Raton, FL, USA, 1990; Volumes 1 and 2. [Google Scholar]
- Scott, G. Atmospheric Oxidation and Antioxidants; Elsevier Science Publishers: Amsterdam, The Netherlands, 1993; Volume 1, ISBN 9780444896179. [Google Scholar] [CrossRef]
- Rabek, J.F. (Ed.) Photostabilization of Polymers: Priciples and Application; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Kleinhans, K.; Demets, R.; Dewulf, J.; Ragaert, K.; De Meester, S. Non-household end-use plastics: The ‘forgotten’plastics for the circular economy. Curr. Opin. Chem. Eng. 2021, 32, 100680. [Google Scholar] [CrossRef]
- Brems, A.; Dewil, R.; Baeyens, J.; Zhang, R. Gasification of plastic waste as waste-to-energy or waste-to-syngas recovery route. Solid Waste A Renew. Resour. 2013, 5, 241–263. [Google Scholar] [CrossRef]
- Al-Salem, S.M.; Lettieri, P.; Baeyens, J. Recycling and recovery routes of plastic solid waste (PSW): A review. Waste Manag. 2009, 29, 2625–2643. [Google Scholar] [CrossRef] [PubMed]
- Pfaendner, R. Restabilization–30 years of research for quality improvement of recycled plastics review. Polym. Degrad. Stab. 2022, 203, 110082. [Google Scholar] [CrossRef]
- Alotaibi, M.; Aldhafeeri, T.; Barry, C. The Impact of Reprocessing with a Quad Screw Extruder on the Degradation of Polypropylene. Polymers 2022, 14, 2661. [Google Scholar] [CrossRef]
- Blázquez-Blázquez, E.; Díez-Rodríguez, T.M.; Pérez, E.; Cerrada, M.L. Recycling of metallocene isotactic polypropylene: Importance of antioxidants. J. Therm. Anal. Calorim. 2022, 147, 13363–13374. [Google Scholar] [CrossRef]
- Schweighuber, A.; Felgel-Farnholz, A.; Bögl, T.; Fischer, J.; Buchberger, W. Investigations on the influence of multiple extrusion on the degradation of polyolefins. Polym. Degrad. Stab. 2021, 192, 109689. [Google Scholar] [CrossRef]
- Schyns, Z.O.; Shaver, M.P. Mechanical recycling of packaging plastics: A review. Macromol. Rapid Commun. 2021, 42, 2000415. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, D.S.; Saragih, R.H.P. Impact of repetitive recycling on optical properties of virgin and recycled polypropylene blends based on material value conservation paradigm. Mater. Sci. Forum 2021, 1020, 192–198. [Google Scholar] [CrossRef]
- Saikrishnan, S.; Jubinville, D.; Tzoganakis, C.; Mekonnen, T.H. Thermo-mechanical degradation of polypropylene (PP) and low-density polyethylene (LDPE) blends exposed to simulated recycling. Polym. Degrad. Stab. 2020, 182, 109390. [Google Scholar] [CrossRef]
- Lindqvist, K.; Andersson, M.; Boss, A.; Oxfall, H. Thermal and mechanical properties of blends containing PP and recycled XLPE cable waste. J. Polym. Environ. 2019, 27, 386–394. [Google Scholar] [CrossRef]
- Hernández-Fernández, J.; Castro-Suares, J.; Toloza, C. Iron Oxide Powder as Responsible for the Generation of Industrial Polypropylene Waste and as a Co-Catalyst for the Pyrolysis of Non-Additive Resins. Int. J. Mol. Sci. 2022, 23, 11708. [Google Scholar] [CrossRef] [PubMed]
- La Mantia, F.P.; Morreale, M.; Botta, L.; Mistretta, M.C.; Ceraulo, M.; Scaffaro, R. Degradation of polymer blends: A brief review. Polym. Degrad. Stab. 2017, 145, 79–92. [Google Scholar] [CrossRef]
- Hamskog, M.; Klügel, M.; Forsström, D.; Terselius, B.; Gijsman, P. The effect of base stabilization on the recyclability of polypropylene as studied by multi-cell imaging chemiluminescence and microcalorimetry. Polym. Degrad. Stab. 2004, 86, 557–566. [Google Scholar] [CrossRef]
- Hamskog, M.; Kluegel, M.; Forsstroem, D.; Terselius, B.; Gijsman, P. The effect of adding virgin material or extra stabilizer on the recyclability of polypropylene as studied by multi-cell imaging chemiluminescence and microcalorimetry. Polym. Degrad. Stab. 2006, 91, 429–436. [Google Scholar] [CrossRef]
- Ranjan, V.P.; Goel, S. Recyclability of polypropylene after exposure to four different environmental conditions. Resour. Conserv. Recycl. 2021, 169, 105494. [Google Scholar] [CrossRef]
- Knight, J.B.; Calvert, P.D.; Billingham, N.C. Localization of oxidation in polypropylene. Polymer 1985, 26, 1713–1718. [Google Scholar] [CrossRef]
- Billingham, N.C.; Calvert, P.D.; Knight, J.B. Application of ultraviolet microscopy to oxidation of polyolefin. In Proc. IUPAC, IUPAC, Macromol. Symp. 1982.
- Cicchetti, O.; De Simone, R.; Gratani, F. Titanium-catalysed-inhibited autoxidation of polypropylene and of its models. Eur. Polym. J. 1973, 9, 1205–1229. [Google Scholar] [CrossRef]
- Kresta, J.; Majer, J.; Veselý, K. Reactions of low molecular weight polypropylene induced by titanium compounds. J. Polym. Sci. Part C Polym. Symp. 1968, 22, 329–338. [Google Scholar] [CrossRef]
- Allen, N.S.; Fatinikun, K.O.; Henman, T.J. Thermal and photochemical oxidation of polypropylene. Influence of residual catalyst levels in unstabilised diluent and gas phase polymers. Eur. Polym. J. 1983, 19, 551–554. [Google Scholar] [CrossRef]
- Gijsman, P.; Fiorio, R. Long term thermo-oxidative degradation and stabilization of polypropylene (PP) and the implications for its recyclability. Polym. Degrad. Stab. 2023, 208, 110260. [Google Scholar] [CrossRef]
- Goss, B.G.; Nakatani, H.; George, G.A.; Terano, M. Catalyst residue effects on the heterogeneous oxidation of polypropylene. Polym. Degrad. Stab. 2003, 82, 119–126. [Google Scholar] [CrossRef]
- Ahlblad, G.; Gijsman, P.; Terselius, B.; Jansson, A.; Möller, K. Thermo-oxidative stability of PP waste films studied by imaging chemiluminescence. Polym. Degrad. Stab. 2001, 73, 15–22. [Google Scholar] [CrossRef]
- Scheirs, J.; Delatycki, O.; Bigger, S.W.; Billingham, N.C. Staining techniques for detecting localized oxidation in high density polyethylene powders and films. Polym. Int. 1991, 26, 187–193. [Google Scholar] [CrossRef]
- Celina, M.; George, G.A. A heterogeneous model for the thermal oxidation of solid polypropylene from chemiluminescence analysis. Polym. Degrad. Stab. 1993, 40, 323–335. [Google Scholar] [CrossRef]
- Hernández-Fernández, J.; Cano, H.; Aldas, M. Impact of Traces of Hydrogen Sulfide on the Efficiency of Ziegler–Natta Catalyst on the Final Properties of Polypropylene. Polymers 2022, 14, 3910. [Google Scholar] [CrossRef] [PubMed]
- Ablblad, G.; Stenberg, B.; Terselius, B.; Reitberger, T. Imaging chemiluminescence instrument for the study of heterogeneous oxidation effects in polymers. Polym. Test. 1997, 16, 59–73. [Google Scholar] [CrossRef]
- Eriksson, P.; Reitberger, T.; Ahlblad, G.; Stenberg, B. Oxidation fronts in polypropylene as studied by imaging chemiluminescence. Polym. Degrad. Stab. 2001, 73, 177–183. [Google Scholar] [CrossRef]
- Nakatani, H.; Shibata, H.; Miyazaki, K.; Yonezawa, T.; Takeda, H.; Azuma, Y.; Watanabe, S. Studies on heterogeneous degradation of polypropylene/talc composite: Effect of iron impurity on the degradation behavior. J. Appl. Polym. Sci. 2010, 115, 167–173. [Google Scholar] [CrossRef]
- Drake, W.O.; Pauquet, J.-R.; Todesco, R.V. Polypropylene the Way Ahead, Madrid, Spain; PRI: London, UK, 1989. [Google Scholar]
- Richters, P. Initiation process in the oxidation of polypropylene. Macromolecules 1970, 3, 262–264. [Google Scholar] [CrossRef]
- Billingham, N.C. Localization of oxidation in polypropylene. In Makromolekulare Chemie. Macromolecular Symposia; Hüthig & Wepf Verlag: Basel, Switzerland, 1989; Volume 28, pp. 145–163. [Google Scholar]
- Blakey, I.; Billingham, N.; George, G.A. Use of 9,10-diphenylanthracene as a contrast agent in chemiluminescence imaging: The observation of spreading of oxidative degradation in thin polypropylene films. Polym. Degrad. Stab. 2007, 92, 2102–2109. [Google Scholar] [CrossRef]
- Hernández-Fernández, J.; Guerra, Y.; Espinosa, E. Development and Application of a Principal Component Analysis Model to Quantify the Green Ethylene Content in Virgin Impact Copolymer Resins During Their Synthesis on an Industrial Scale. J. Polym. Environ. 2022, 30, 4800–4808. [Google Scholar] [CrossRef]
- Hernández-Fernández, J.; Vivas-Reyes, R.; Toloza, C.A.T. Experimental Study of the Impact of Trace Amounts of Acetylene and Methylacetylene on the Synthesis, Mechanical and Thermal Properties of Polypropylene. Int. J. Mol. Sci. 2022, 23, 12148. [Google Scholar] [CrossRef] [PubMed]
- Pavon, C.; Aldas, M.; Hernández-Fernández, J.; López-Martínez, J. Comparative characterization of gum rosins for their use as sustainable additives in polymeric matrices. J. Appl. Polym. Sci. 2022, 139, 51734. [Google Scholar] [CrossRef]
- Hernández-Fernández, J.; Lopez-Martinez, J.; Barceló, D. Development and validation of a methodology for quantifying parts-per-billion levels of arsine and phosphine in nitrogen, hydrogen and liquefied petroleum gas using a variable pressure sampler coupled to gas chromatography-mass spectrometry. J. Chromatogr. A 2021, 1637, 461833. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Tang, Z.; Yao, D.; Yang, H.; Shao, J.; Chen, H. Thermal behavior, kinetics and gas evolution characteristics for the co-pyrolysis of real-world plastic and tyre wastes. J. Clean. Prod. 2020, 260, 121102. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Burnham, A.K.; Criado, J.M.; Pérez-Maqueda, L.A.; Popescu, C.; Sbirrazzuoli, N. ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis data. Thermochim. Acta 2011, 520, 1–19. [Google Scholar] [CrossRef]
- Coats, A.W.; Redfern, J.P. Kinetic Parameters from Thermogravimetric Data. Nature 1964, 201, 68–69. [Google Scholar] [CrossRef]
- Horowitz, H.H.; Metzger, G. A New Analysis of Thermogravimetric Traces. Anal. Chem. 1963, 35, 1464–1468. [Google Scholar] [CrossRef]
- Meng, X.; Yang, R. How formaldehyde affects the thermo-oxidative and photo-oxidative mechanism of polypropylene: A DFT/TD-DFT study. Polym. Degrad. Stab. 2022, 205, 110131. [Google Scholar] [CrossRef]
- Nguyen, H.M.; Tang, H.-Y.; Huang, W.-F.; Lin, M. Mechanisms for reactions of trimethylaluminum with molecular oxygen and water. Comput. Theor. Chem. 2014, 1035, 39–43. [Google Scholar] [CrossRef]
- Naumkin, F.Y. Flat-structural Motives in Small Alumino−Carbon Clusters CnAlm (n = 2−3, m = 2−8). J. Phys. Chem. A 2008, 112, 4660–4668. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Narro, G.; Royston, N.J.; Billsborough, K.L.; Phan, A.N. Kinetic modelling of mixed plastic waste pyrolysis. Chem. Thermodyn. Therm. Anal. 2023, 9, 100105. [Google Scholar] [CrossRef]
- Dubdub, I.; Al-Yaari, M. Pyrolysis of Mixed Plastic Waste: I. Kinet. Study. Mater. 2020, 13, 4912. [Google Scholar] [CrossRef]
- Gul, H.; Shah, A.U.H.A.; Gul, S.; Arjomandi, J.; Bilal, S. Study on the thermal decomposition kinetics and calculation of activation energy of degradation of poly (o-toluidine) using thermogravimetric analysis. Iran. J. Chem. Chem. Eng. (IJCCE) 2018, 37, 193–204. [Google Scholar]
- Cai, J.; Bi, L. Precision of the Coats and Redfern Method for the Determination of the Activation Energy without Neglecting the Low-Temperature End of the Temperature Integral. Energy Fuels 2008, 22, 2172–2174. [Google Scholar] [CrossRef]
- Arrhenius, S. Über die Dissociationswärme und den Einfluss der Temperatur auf den Dissociationsgrad der El-ektrolyte. Z. Für Phys. Chem. 1889, 4U, 96–116. [Google Scholar] [CrossRef]
- Palmay, P.; Pillajo, L.; Andrade, M.; Medina, C.; Barzallo, D. Kinetic Analysis of Thermal Degradation of Recycled Polypropylene and Polystyrene Mixtures Using Regenerated Catalyst from Fluidized Catalytic Cracking Process (FCC). Polymers 2023, 15, 2035. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Amount (%W/W) |
|---|---|
| White mineral oil (petroleum) | ≤75% |
| Magnesium chloride–titanium tetrachloride complex | ≤30% |
| Organic ester | <7% |
| Diethyl phthalate | <5% |
| Isopentane | <8% |
| Titanium tetrachloride | ≤0.65% |
| Phthalic anhydride | ≤0.6% |
| Chlorobenzene | ≤0.5% |
| 2-Chloro-1-methylbenzene | ≤0.04% |
| Iron chloride | ≤0.004% |
| Aluminum chloride | ≤0.005% |
| Moisture, wt% | Volatile Matter, wt% | Ti ppm | Al ppm | Cl ppm | Fe ppm | |
|---|---|---|---|---|---|---|
| PPO | 0.18 | 99.49 | 0.98 | 8.53 | 13.37 | 4.13 |
| W1 | 0.33 | 99.49 | 0.98 | 320.23 | 201.03 | 99.11 |
| W2 | 0.21 | 99.49 | 0.98 | 171.12 | 81.71 | 51.34 |
| W3 | 0.18 | 99.49 | 0.98 | 5.15 | 58.75 | 7.03 |
| W4 | 0.22 | 99.49 | 0.98 | 5.15 | 105.23 | 44.51 |
| W5 | 0.35 | 99.49 | 0.98 | 410.13 | 205.33 | 100.13 |
| Sample Name | Stages Mechanism | Horowitz and Metz (HM) | Ea/kJ | Coats and Redfern (CR) | Ea/kJ | A |
|---|---|---|---|---|---|---|
| PPO | 1 | 412.73 | 250.26 | 4.22 × 1017 | ||
| 2 | 173.48 | 127.24 | 6.01 × 106 | |||
| 3 | 274.19 | 241.56 | 6.76 × 1015 | |||
| W1 | 1 | 0.82 | 27.09 | 1.90 × 107 | ||
| 2 | 2.88 | 59.66 | 9.40 × 104 | |||
| 3 | 70.30 | 155.74 | 4.00 × 109 | |||
| W2 | 1 | 65.36 | 32.09 | 2.49 × 107 | ||
| 2 | 155.39 | 70.86 | 1.85 × 104 | |||
| 3 | 310.37 | 289.65 | 1.20 × 1020 | |||
| W3 | 1 | 224.45 | 143.68 | 2.48 × 108 | ||
| 2 | 174.71 | 222.86 | 6.25 × 1014 | |||
| 3 | 272.55 | 345.18 | 1.16 × 1024 | |||
| W4 | 1 | 147.99 | 42.84 | 1.50 × 106 | ||
| 2 | 60.02 | 86.46 | 1.24 × 104 | |||
| 3 | 143.88 | 267.21 | 2.47 × 1018 | |||
| W5 | 1 | 106.47 | 26.40 | 1.37 × 107 | ||
| 2 | 57.14 | 70.48 | 5.00 × 103 | |||
| 3 | 128.67 | 212.09 | 2.67 × 1014 |
| Sample Name | Stages Mechanism | Flynn–Wall–Ozawa (FWO) | Kissinger–Akahira–Sunose (KAS) | ||||
|---|---|---|---|---|---|---|---|
| Equation | Ea/kJ | A | Equation | Ea/kJ | A | ||
| PPO | 1 | 10.73 | 1.38×105 | 2.97 | 2.92 × 107 | ||
| 2 | 10.27 | 5.66×105 | 10.80 | 7.14 × 106 | |||
| 3 | 61.72 | 2.13×1012 | 885.41 | 6.66 × 1060 | |||
| W1 | 1 | 1.16 | 1.15×105 | 1.22 | 7.63 × 107 | ||
| 2 | 21.83 | 3.04×106 | 28.37 | 5.14 × 105 | |||
| 3 | 26.14 | 6.83×106 | 34.12 | 1.89 × 105 | |||
| W2 | 1 | 3.05 | 6.83×106 | 3.20 | 3.10 × 107 | ||
| 2 | 2.74 | 1.17×105 | 2.86 | 3.43 × 107 | |||
| 3 | 2.65 | 1.15×105 | 2.81 | 3.43 × 107 | |||
| W3 | 1 | 0.09 | 1.53×105 | 0.16 | 1.03 × 108 | ||
| 2 | 13.23 | 5.07×105 | 22.82 | 1.71 × 106 | |||
| 3 | 50.77 | 4.69×108 | 52.49 | 1.04 × 104 | |||
| W4 | 1 | 0.47 | 1.39×105 | 0.49 | 8.44 × 107 | ||
| 2 | 15.90 | 8.88×105 | 23.29 | 1.40 × 106 | |||
| 3 | 29.89 | 1.28×107 | 43.54 | 4.67 × 104 | |||
| W5 | 1 | 1.25 | 1.14×105 | 1.32 | 7.63 × 107 | ||
| 2 | 19.33 | 1.85×106 | 25.61 | 7.48 × 107 | |||
| 3 | 42.19 | 1.03×108 | 34.98 | 1.71 × 105 | |||
| Sample Name | Activation Energies | Elemental Composition of Catalyst Residues | ||||||
|---|---|---|---|---|---|---|---|---|
| * Ea/kJ | ** Ea/kJ | *** Ea/kJ | **** Ea/kJ | Ti/ppm | Al/ppm | Cl/ppm | Fe/ppm | |
| PPO | 412.73 | 250.26 | 10.73 | 2.97 | 0.98 | 8.53 | 13.37 | 4.13 |
| 173.48 | 127.24 | 10.27 | 10.80 | |||||
| 274.19 | 241.56 | 61.72 | 885.41 | |||||
| W1 | 0.82 | 27.09 | 1.16 | 1.22 | 0.98 | 320.23 | 201.03 | 99.11 |
| 2.88 | 59.66 | 21.83 | 28.37 | |||||
| 70.3 | 155.74 | 26.14 | 34.12 | |||||
| W2 | 65.36 | 32.09 | 3.05 | 3.20 | 0.98 | 171.12 | 81.71 | 51.34 |
| 155.39 | 70.86 | 2.74 | 2.86 | |||||
| 310.37 | 289.65 | 2.65 | 2.81 | |||||
| W3 | 224.45 | 143.68 | 0.09 | 0.16 | 0.98 | 5.15 | 58.75 | 7.03 |
| 174.71 | 222.86 | 13.23 | 22.82 | |||||
| 272.55 | 345.18 | 50.77 | 52.49 | |||||
| W4 | 147.99 | 42.84 | 0.47 | 0.49 | 0.98 | 5.15 | 105.23 | 44.51 |
| 60.02 | 86.46 | 15.90 | 23.29 | |||||
| 143.88 | 267.21 | 29.89 | 43.54 | |||||
| W5 | 106.47 | 26.4 | 1.25 | 1.32 | 0.98 | 410.13 | 205.33 | 100.13 |
| 57.14 | 70.48 | 19.33 | 25.61 | |||||
| 128.67 | 212.09 | 42.19 | 34.98 | |||||
| Compounds | PP0 | Waste 1 | Waste 2 | Waste 3 | Waste 4 | Waste 5 |
|---|---|---|---|---|---|---|
| Alkanes | 30.52 | 15.54 | 26.89 | 32.34 | 25.1 | 16.85 |
| Alkenes | 67.63 | 55.15 | 49.85 | 66.32 | 49.39 | 52.05 |
| Alkynes | 0.93 | 3.88 | 2.84 | 0.79 | 4.29 | 3.75 |
| Alcohols | 0 | 9.7 | 7.44 | 0 | 8.58 | 10.78 |
| Ketones | 0 | 5.25 | 4.7 | 0 | 5.1 | 5.89 |
| Acids | 0 | 5.29 | 3.95 | 0 | 4.18 | 4.96 |
| Permanent | 0.82 | 5.25 | 4.32 | 0.83 | 3.42 | 5.79 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández Fernández, J.A.; Ortega-Toro, R.; Fuentes, E.A.E. A Study on the Early Degradation of the Non-Additive Polypropylene–Polyethylene Composite Sampled between the Polymerization Reactor and the Deactivation-Degassing Tank. J. Compos. Sci. 2024, 8, 311. https://doi.org/10.3390/jcs8080311
Hernández Fernández JA, Ortega-Toro R, Fuentes EAE. A Study on the Early Degradation of the Non-Additive Polypropylene–Polyethylene Composite Sampled between the Polymerization Reactor and the Deactivation-Degassing Tank. Journal of Composites Science. 2024; 8(8):311. https://doi.org/10.3390/jcs8080311
Chicago/Turabian StyleHernández Fernández, Joaquín Alejandro, Rodrigo Ortega-Toro, and Eduardo Antonio Espinosa Fuentes. 2024. "A Study on the Early Degradation of the Non-Additive Polypropylene–Polyethylene Composite Sampled between the Polymerization Reactor and the Deactivation-Degassing Tank" Journal of Composites Science 8, no. 8: 311. https://doi.org/10.3390/jcs8080311
APA StyleHernández Fernández, J. A., Ortega-Toro, R., & Fuentes, E. A. E. (2024). A Study on the Early Degradation of the Non-Additive Polypropylene–Polyethylene Composite Sampled between the Polymerization Reactor and the Deactivation-Degassing Tank. Journal of Composites Science, 8(8), 311. https://doi.org/10.3390/jcs8080311