3.1. N-I…O=C Halogen Bond
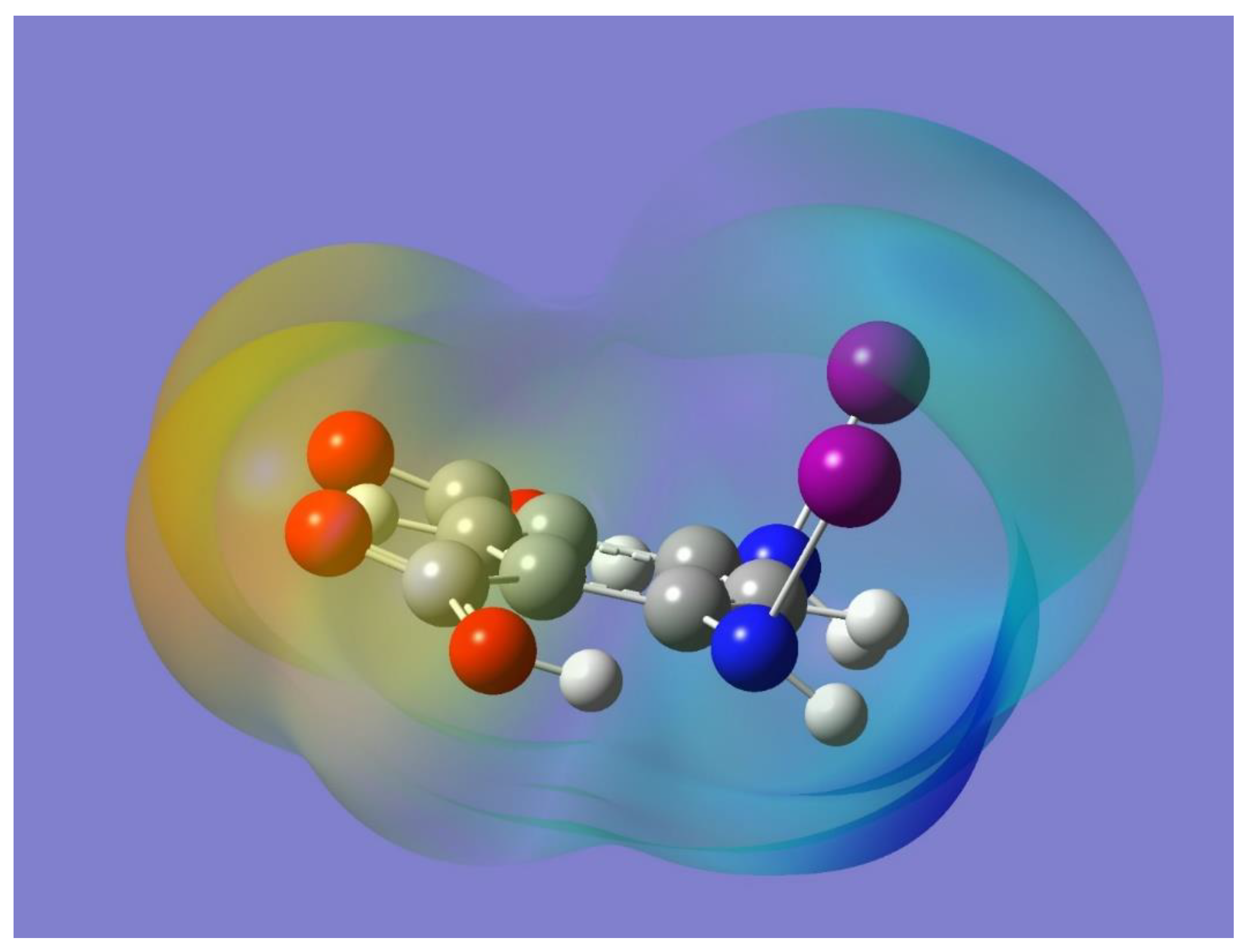
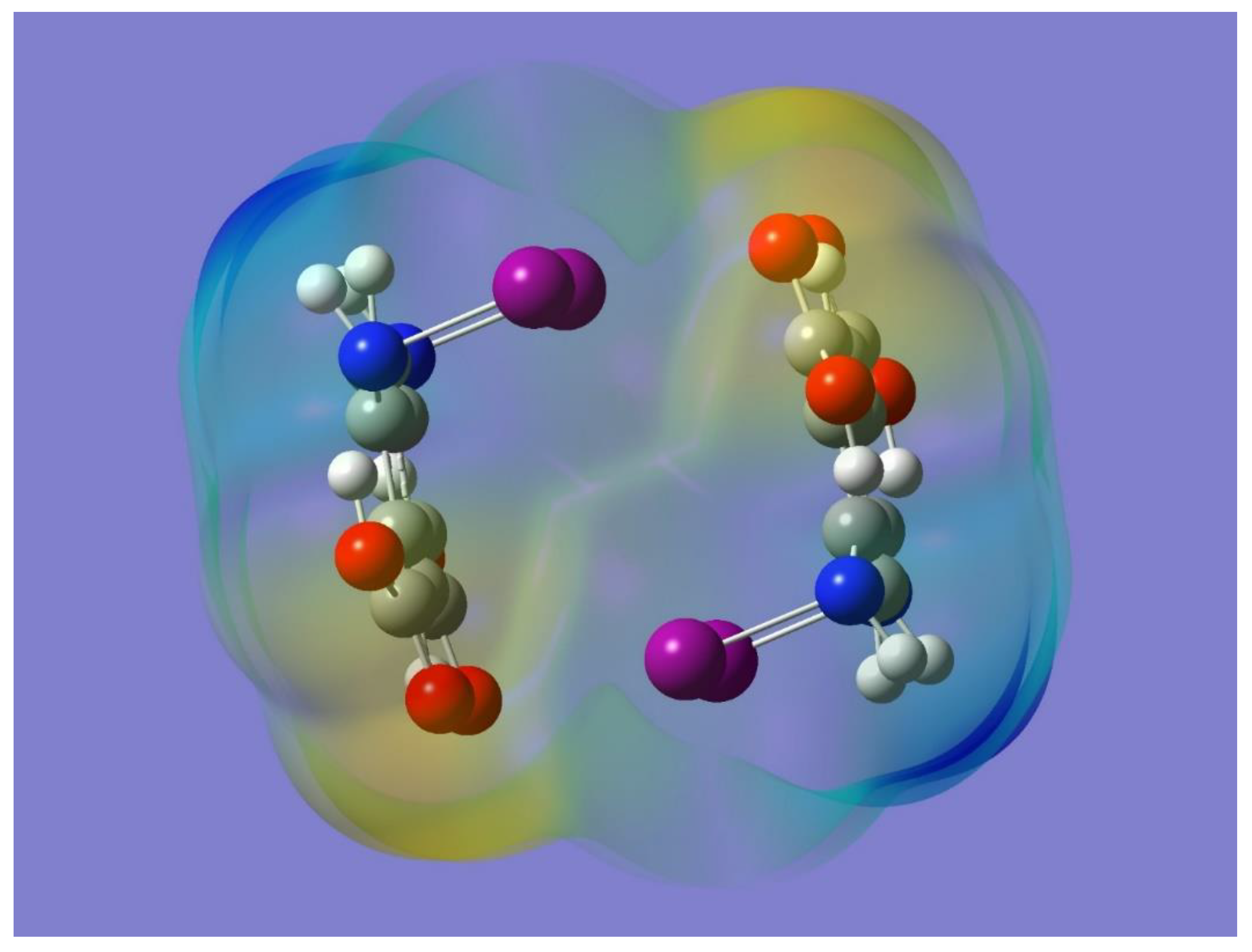
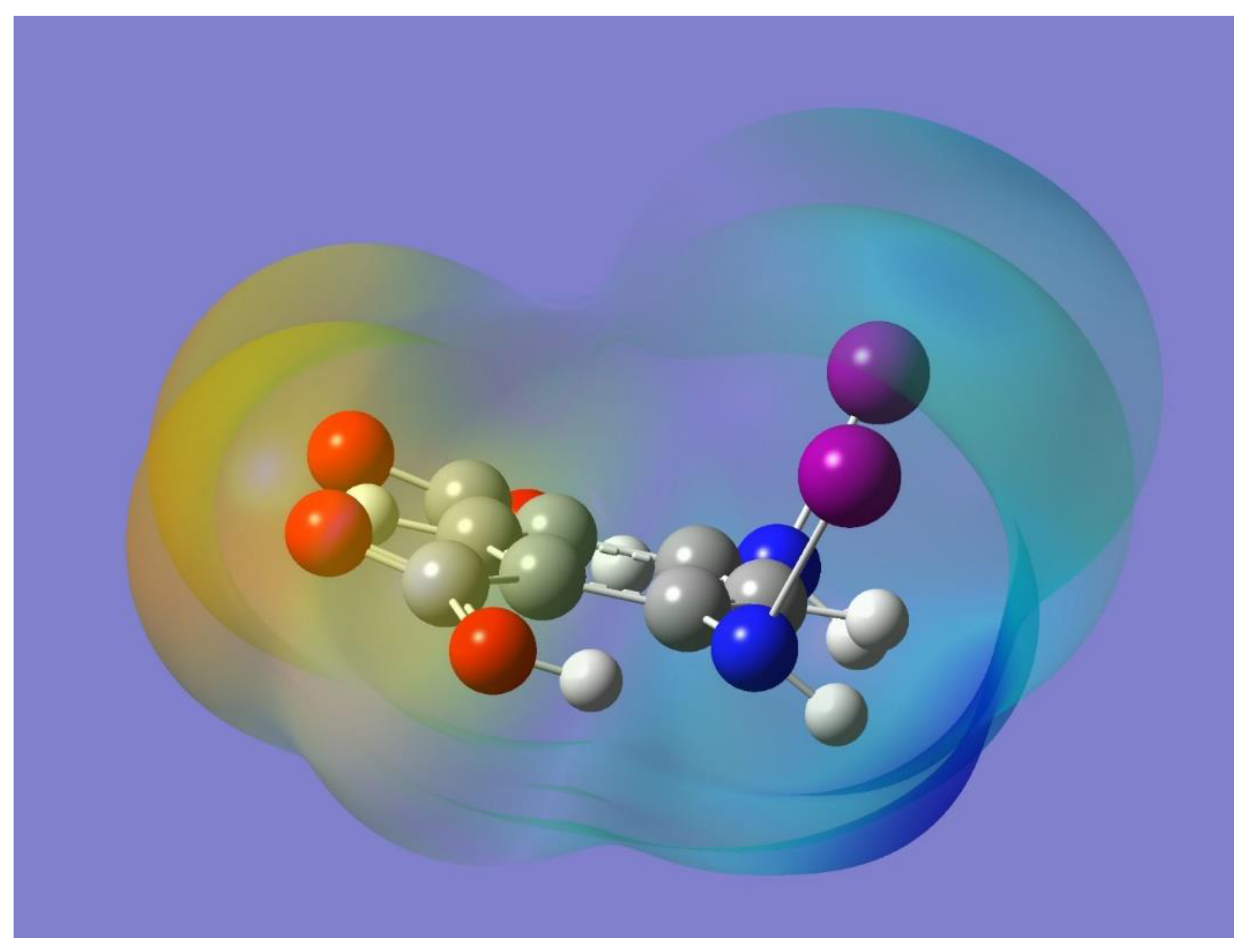
The potential for halogen bonding was first examined by optimizing the monomer and then by considering its electrostatic potential. The optimized geometry of the monomer is displayed in
Figure 1. Incidentally, a monomer with the halogen atoms facing in opposite directions with respect to the organic framework was also optimized and found to be a stable minimum with no imaginary frequencies. The two optimized monomers were essentially isoenergetic, although the one used in this work is the one that allows for the intermolecular halogen bonding. The orientation of each O-H bond is conveniently chosen trans to the C=O group in the pertinent carboxy group to allow for intramolecular O-H…N hydrogen bonding with the nitrogen atom of the adjacent NHI group. The intramolecular hydrogen bonding should polarize the corresponding N-I bond, which would then enhance a potential intermolecular N-I…O=C halogen bond interaction. Frequency calculations resulted in no imaginary frequencies, and hence demonstrated that the selected optimized monomer is indeed a minimum on the potential energy surface of the molecule. It has been shown that a halogen atom covalently bonded to a more electronegative element tends to exhibit an asymmetric distribution of electron density with less electron density on the region opposite to the covalent bond than on the region orthogonal to it. The former region has been termed the halogen σ-hole and it usually has a positive electrostatic potential [
43]. The presence of the σ-hole on each of the N-I bonds in the optimized monomer can be appreciated in
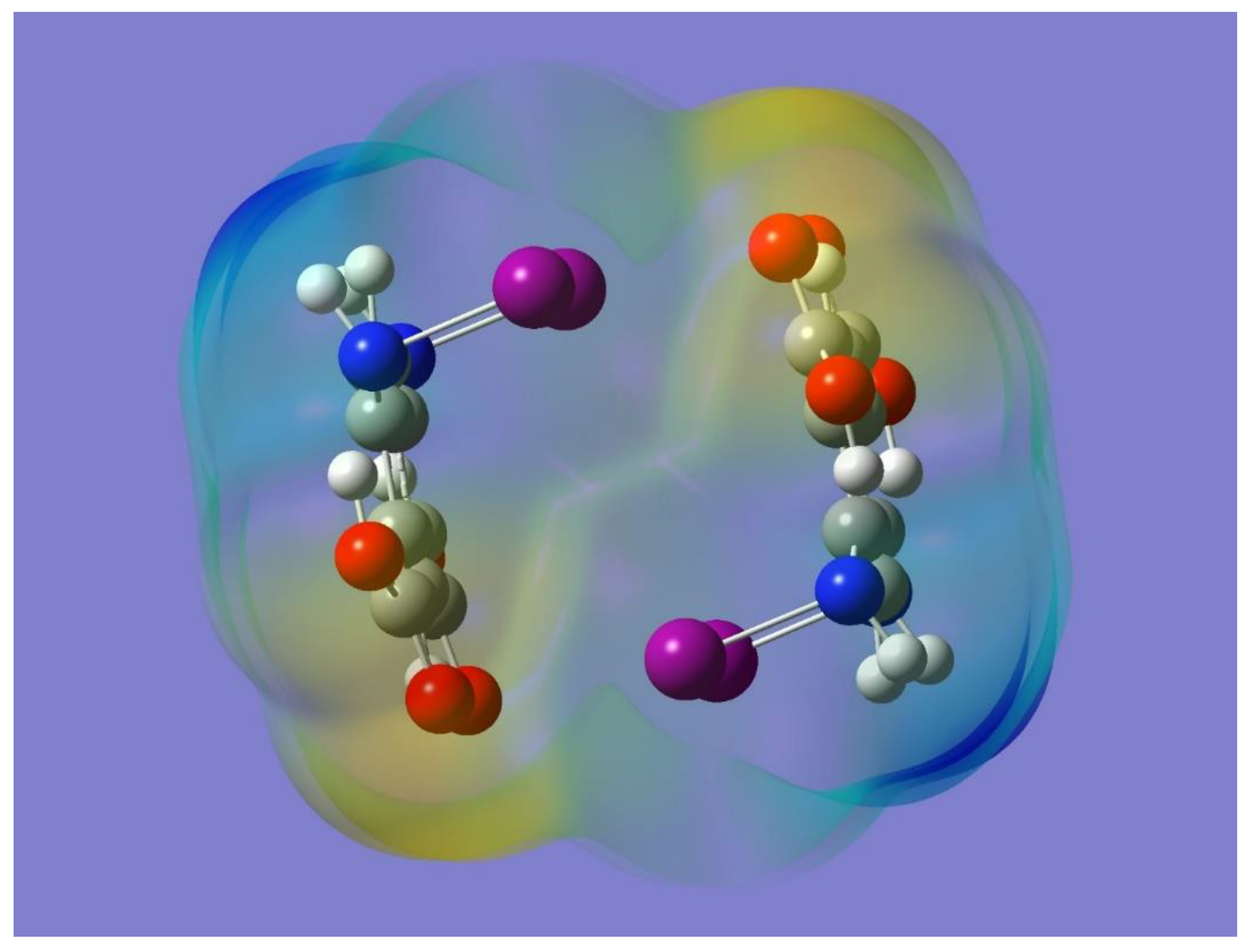
Figure 2 as a light blue region that indicates a corresponding positive electrostatic potential. Also apparent in
Figure 2 is the negative electrostatic potential around the oxygen atoms (yellow-red region). It is worth noting that the presence and relative locations of the complementary (positive and negative) electrostatic potentials in the monomer can indeed facilitate the formation of halogen bonding, N-I…O=C, via intermolecular interactions of two or more monomers.
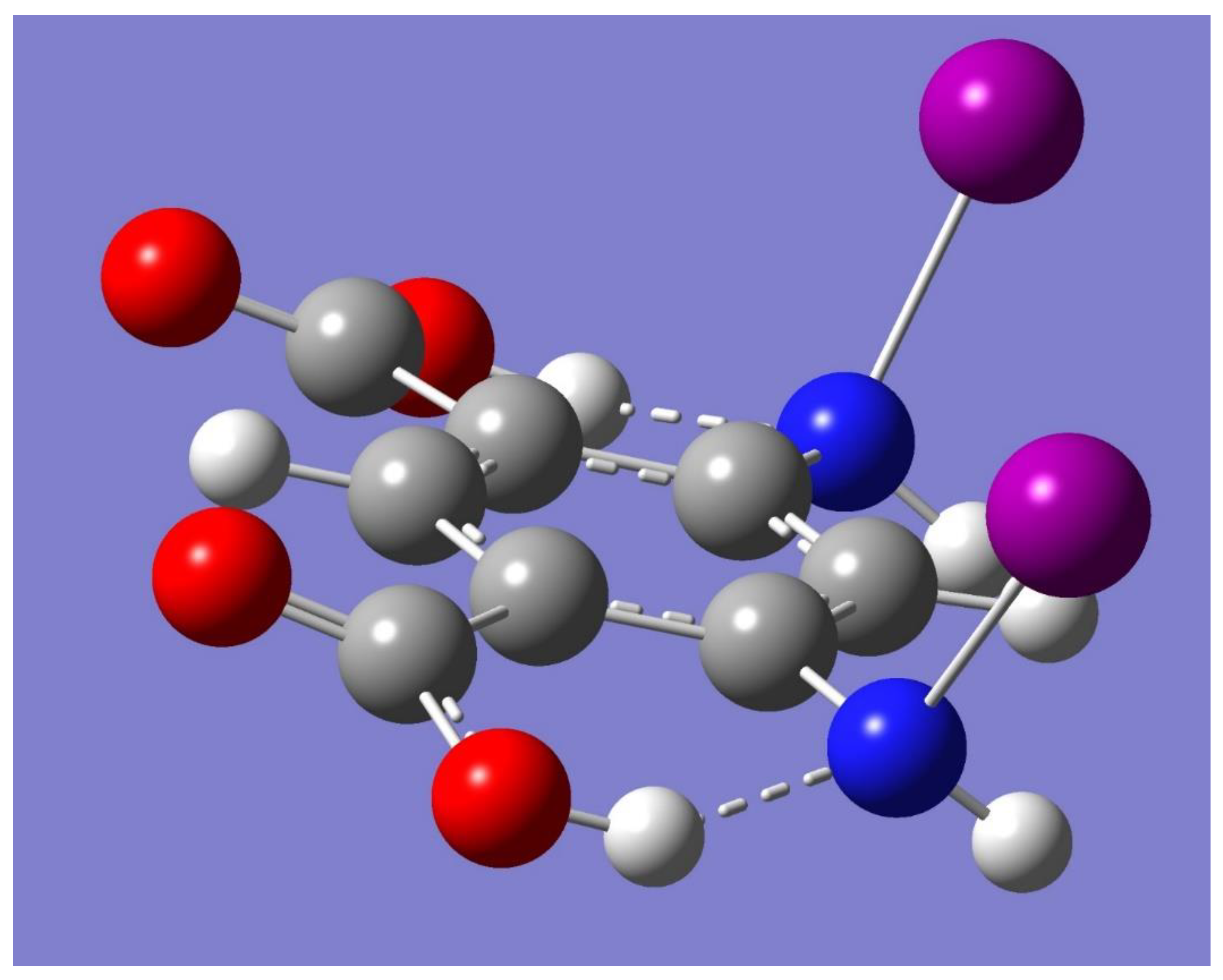
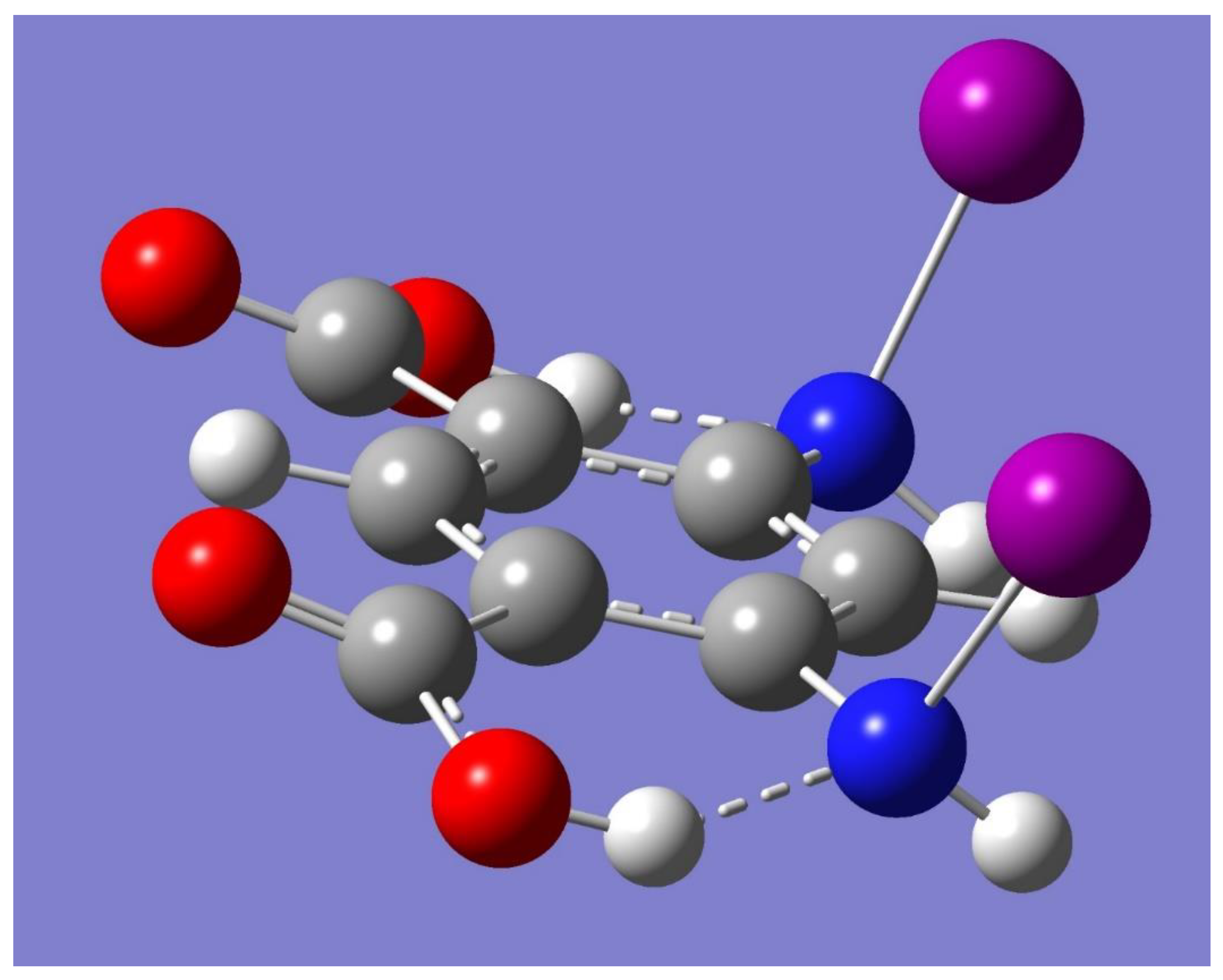
A halogen-bonded driven cyclic dimer can be built by conveniently placing two monomers facing each other with the N-I bonds in one monomer interacting with the carbonyl oxygen atoms in the other monomer. The resulting optimized geometry has C
2h symmetry, and it is displayed in
Figure 3. Frequency calculations show, however, a small imaginary frequency (−9 cm
−1). A new initial guess for geometry optimization was created by perturbing the small negative frequency. The newly optimized geometry is very similar to the C
2h dimer, but with a reduced C
i symmetry instead, and with no imaginary frequencies. Moreover, the C
i monomer has virtually the same electronic energy as its C
2h counterpart. A halogen-bonded driven trimer was built in a manner similar to the cyclic dimer. The resulting optimized geometry has C
3h symmetry, and it is displayed in
Figure 4. Frequency calculations confirm that the optimized geometry is a minimum on the corresponding potential energy surface, with no imaginary frequencies.
Some key geometrical parameters for the optimized monomer, dimer, and trimer systems are listed in
Table 1. It should be noted that the parameters listed in
Table 1 for the dimer of C
i symmetry represent average values, and these average values are very close to the corresponding geometrical values of the C
2h dimer. More specifically, in the dimer of C
i symmetry, there are two distinct pairs of N-I…O=C halogen bond interactions, with NI…O=C distances of 2.962 Å and of 3.080 Å, respectively. The NI…O=C angles are 162.2° and 154.7° for the former and latter halogen bonds, respectively. Similarly, two distinct pairs of COH…N hydrogen bond interactions are present, with COH…N distances of 1.771 Å and of 1.775 Å, respectively. The CO-H…N angles are 147.2° and 146.9° for the former and latter hydrogen bonds, respectively. Lastly, two sets of covalent N-I (2.130 and 2.128 Å) and C=O (1.218 and 1.216 Å) bonds are found in the C
i dimer.
An inspection of
Table 1 reveals that dimer formation brings about an elongation, relative to the monomer, of the two covalent bonds N-I and C=O. These changes are consistent with attractive N-I…O=C interactions that can be considered halogen bonded interactions, given that the interaction distances are smaller than the sum (3.50 Å) of the van der Waals radii of the iodine (1.98 Å) and oxygen (1.52 Å) atoms [
44]. The directional character of the halogen bond in a cyclic dimer is seen in the relatively large interaction angle of about 158°. Dimer formation strengthens the intramolecular hydrogen bonds, as seen in the sizeable shortening of the intramolecular COH…N hydrogen bond distances. The enhanced hydrogen bond interactions may be traced back to a concerted resonance-assisted cooperativity involving the halogen and the hydrogen bonds. Indeed, a cyclic network of alternating halogen and hydrogen bonds can be observed on either side of the dimer: [NI…O=C-OH…NI…O=C-OH…NI]. This sequence of interactions results in resonance-assisted cooperativity, reflected in an increase in the O-H bond length and a decrease in the C-O bond length, in addition to the shortening of the COH…N distances (
Table 1).
Table 1 also shows that, upon trimer formation, cooperativity brings about an enhancement of the NI…O=C halogen bonds in addition to further strengthening the COH…N hydrogen bonds. Particularly striking is the sizeable decrease in the halogen bond distance (about 9% reduction) and widening of the corresponding angle (about 7% wider) relative to the dimer.
In addition to geometrical parameters, interaction energies provide a direct measure of both the strength and the cooperativity of the intermolecular interactions holding the dimer and trimer systems together.
Table 2 lists the interaction energies corrected for basis set superposition error, BSSE. An inspection of
Table 2 shows that the interaction energies for both dimers (C
i and C
2h) are virtually the same. Cooperativity is made apparent in an important increase in the interaction energy in the trimer relative to the dimer. More specifically, and on a per-halogen bond interaction basis, the magnitude of the interaction energy increases more than 36% from the dimer to the trimer. Also shown in
Table 2 are the intermolecular halogen bond distances and the corresponding bond critical point electron densities, ρ
NI…O=C, calculated within the framework of the quantum theory of atoms in molecules, QTAIM [
35,
36,
37]. In particular, the existence of a bond critical point (that is, the point where the gradient of the density is zero) provides evidence of the existence of a bond, whereas its absence indicates the lack of bonding. Moreover, the magnitude of the electron density at the bond critical point directly correlates with the strength of the bond. The more than 50% increase in ρ
NI…O=C in the trimer compared with the dimer confirms the significant cooperative strengthening of the N-I…O=C interaction already evinced by the changes in both geometrical parameters and interaction energies.
3.2. Na+ Binding by Dimer
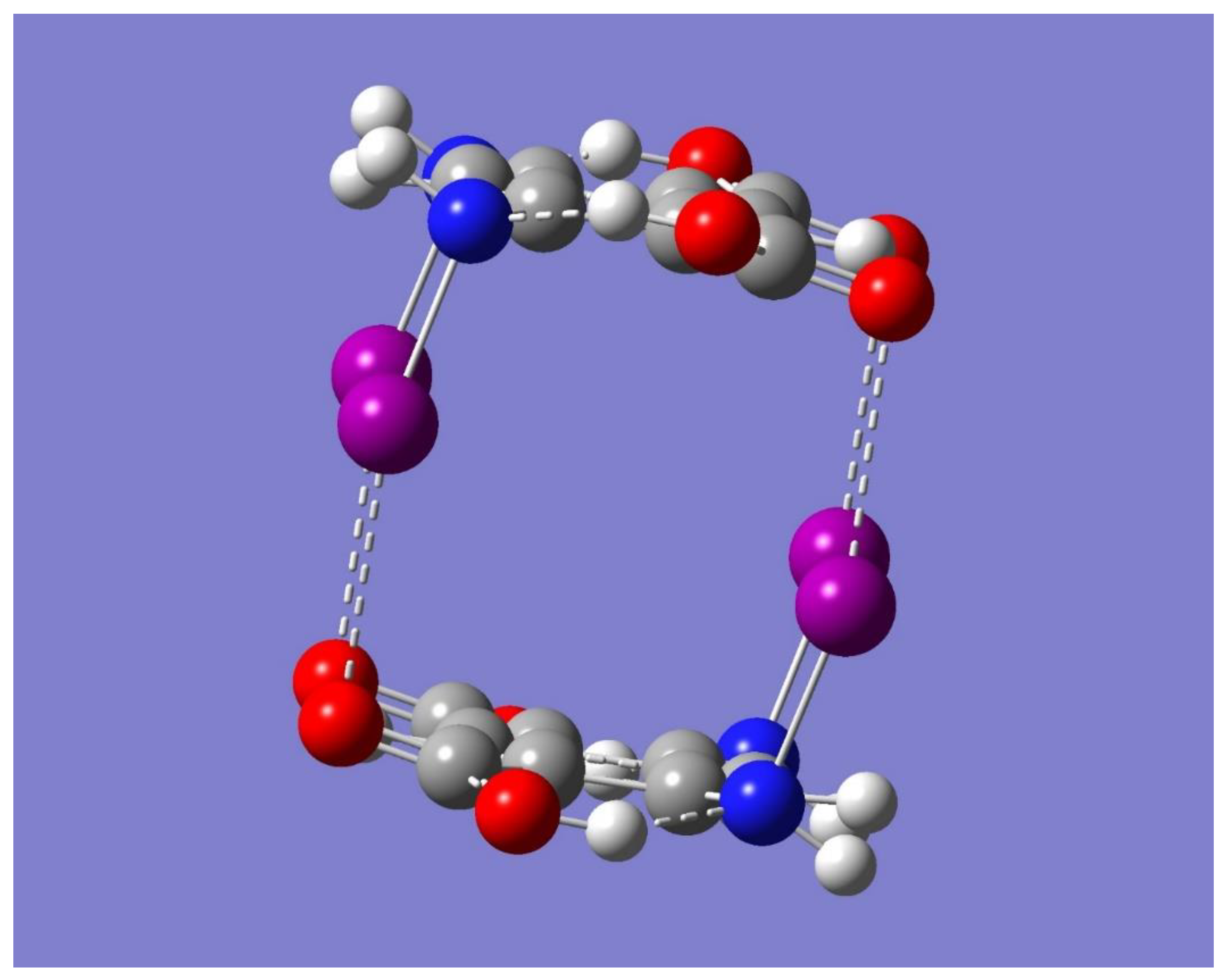
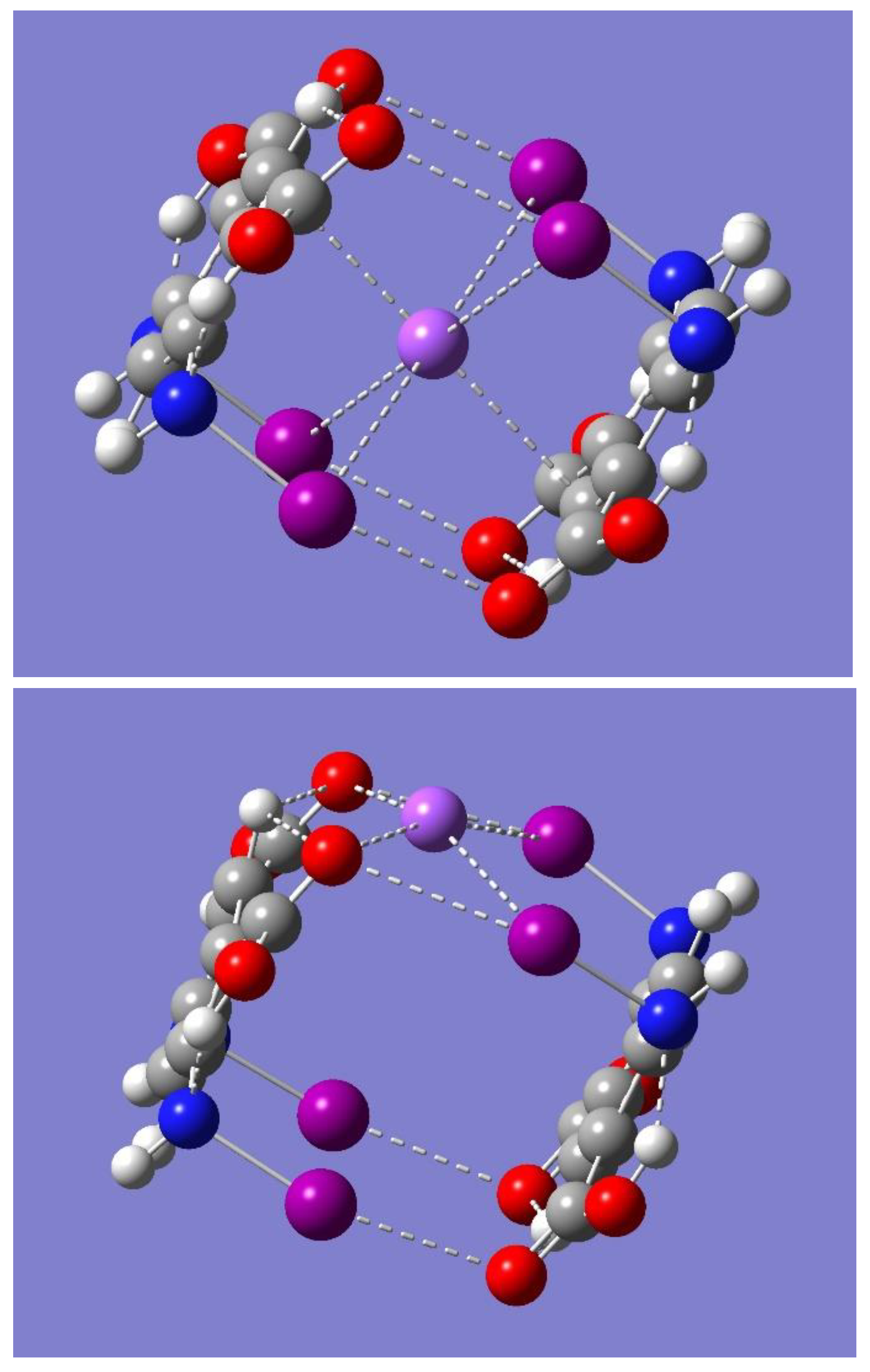
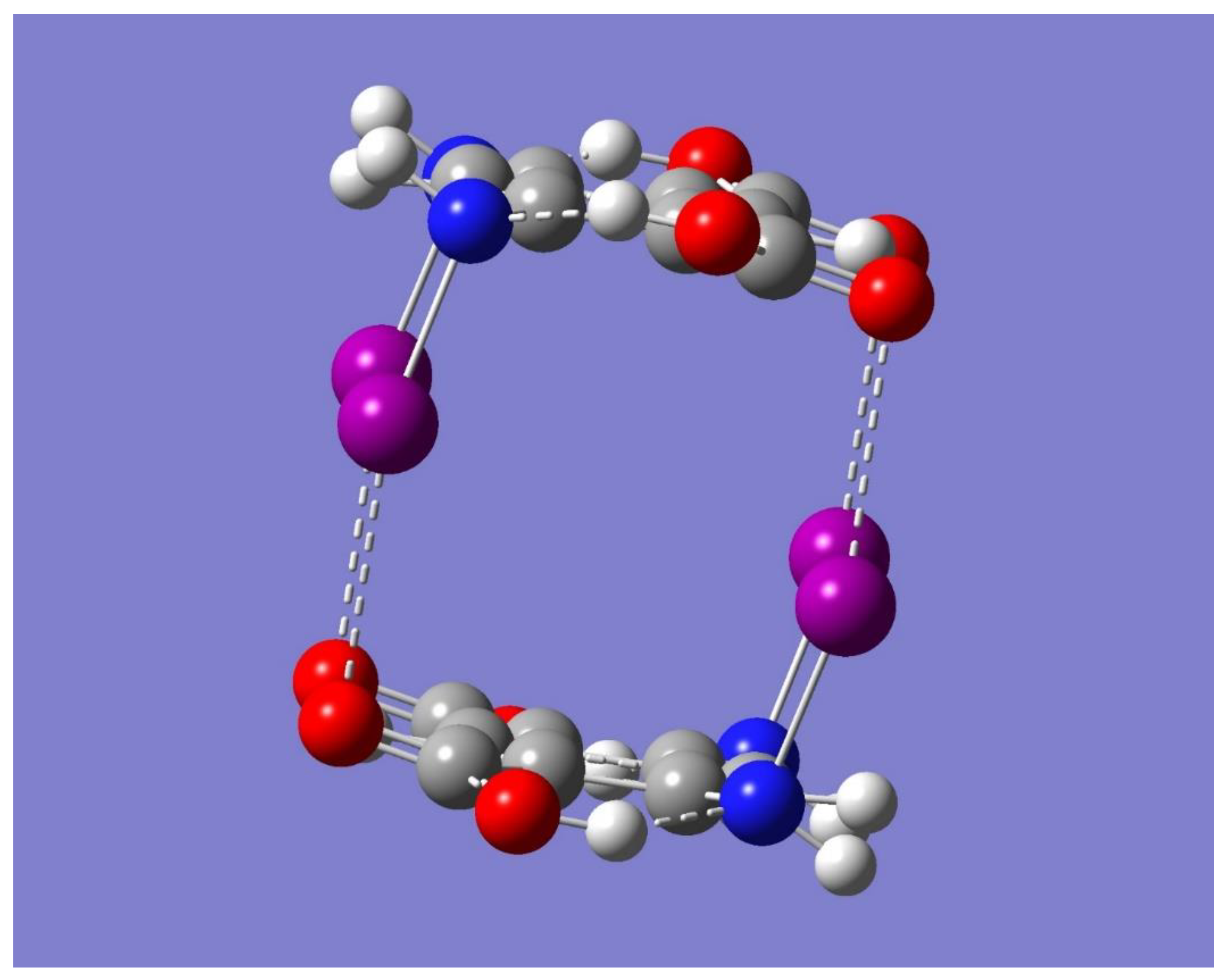
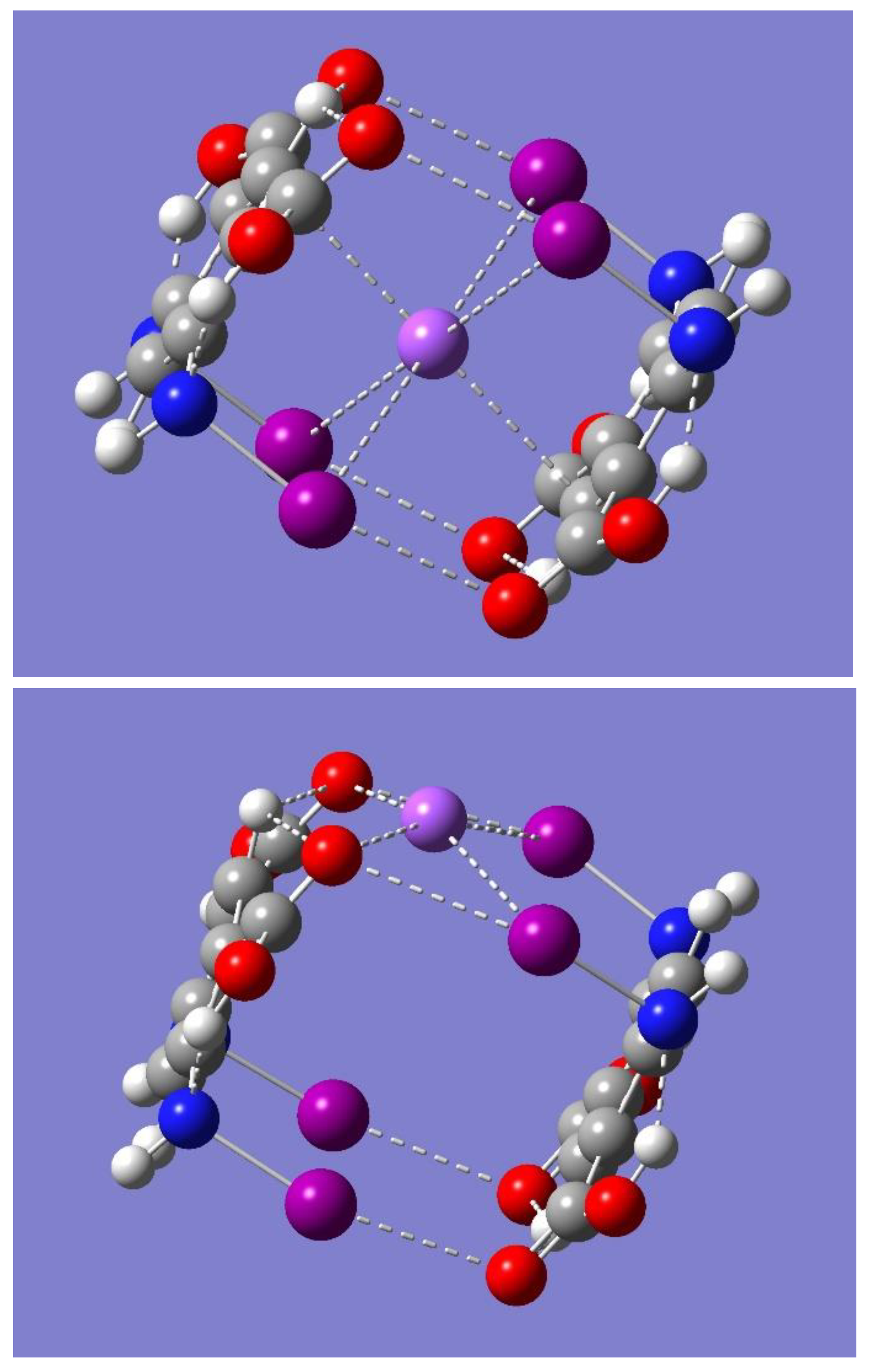
The potential for Na
+ binding by the dimer was examined by considering the electrostatic potential of the dimer using the optimized dimer of symmetry C
2h. Inspection of
Figure 5 reveals a couple of regions of negative electrostatic potential. One region can be identified as the yellow-red color, mostly near the oxygen atoms, while the other as the light purple color around the iodine atoms, mostly perpendicular to the respective N-I…O=C interaction lines. These regions are located on the sides of the dimer and to some extent in the cavity of the cyclic structure. Accordingly, the dimer should be able to bind a Na
+ on either side of the dimer or in the middle of the dimer cavity. To test these hypotheses, a full geometry optimization was first performed starting with the metal ion on the center of the dimer cavity. The result was the binding of Na
+ in a complex geometry of C
2h symmetry. It is worth noting that the same complex geometry occurred regardless of the neutral dimer (C
i or C
2h symmetry) used as the host. Next, a full geometry optimization with a Na
+ on each side of the dimer (that is, two Na
+ ions) resulted in a minimum energy structure of C
2h symmetry, confirming the ability of the dimer to bind a metal ion on the sides based on their corresponding negative electrostatic potentials. Lastly, a geometry optimization using the optimized dimer with the two Na
+ ions was carried after removing one of the Na
+. The successful geometry optimization resulted in a complex geometry of C
s symmetry. Frequency calculations demonstrated that all of the metal-complexed optimized geometries are indeed minimum energy structures with no imaginary frequencies. The optimized geometries for the Dimer-Na
+ and Dimer-2Na
+ complexes are displayed in
Figure 6, while
Table 3 lists some relevant geometrical parameters.
Inspection of both
Table 3 and
Figure 6 suggests that the binding of a single Na
+ within the cavity of the cyclic dimer is driven primarily by the attractive interactions of the metal ion with the four iodine atoms and with both aromatic rings. The electron-rich region, resulting from the halogen atom lone pairs not involved in the halogen bonds around the center of symmetry of the dimer, helps stabilize and therefore bind electron-deficient species such as Na
+. The aromatic rings also provide electron-rich regions to help bind the metal ion. Specifically, the smallest separation between Na
+ and one of the CH groups in a benzene ring is readily visible in
Figure 6, while the corresponding distance, HC…Na
+, is listed in
Table 3. It is important to note that the halogen bonds were strengthened rather than weakened following the complexation of Na
+ at the center of symmetry of the dimer. Supporting evidence to this effect is found in a decrease in the halogen bond distances (3.007 Å to 2.836 Å) and a slight widening of the angles (158.9° to 162.5°).
When the binding of Na+ occurs on the side of the dimer, the metal ion sits on top of the plane formed by the iodine and oxygen atoms participating in one of the halogen-bonding networks. Here, the Na+ binding is driven mostly by the attractive interactions of the metal ion with the two carbonyl oxygen atoms, the two iodine atoms, and one of the aromatic rings. The enhanced contribution of the carbonyl oxygen atoms to bind Na+, with respect to the binding inside the dimer cavity, is seen in a substantial decrease (35% reduction) in the Na+…O=C distances. Moreover, the dihedral angle between the planes formed by the metal ion and the halogen-bonded network, Na+…I…I…O, changes from -73.8° (Na+ inside) to 13.1° (Na+ outside), which is consistent with the larger role played by the carbonyl oxygen atoms in binding the metal ion. Although in this complex the metal ion interacts with only one of the aromatic rings and just two iodine atoms, these interactions appear somewhat stronger, as suggested by a reduction of 5% and 2% in the HC…Na+ and NI…Na+ distances, respectively. When compared with the free C2h dimer, the halogen bond network that interacts with the metal ion is weakened upon Na+ complexation, as seen in an increase in the halogen bond distances (3.007 Å to 3.157 Å) and a narrowing of the angles (158.9° to 147.2°). In contrast, the halogen bond network that is farther away from Na+ becomes somewhat stronger, as indicated by shorter halogen bond distances, 2.847 Å, and wider angles, 164.4°.
The binding of two metal ions by the cyclic dimer involves both halogen-bonded networks, each binding one Na
+ respectively. Both Na
+ ions are symmetrically equivalent within an overall C
2h symmetry. As in the binding of a single Na
+ on the outer side of the dimer, each metal ion sits on top of the plane formed by the iodine and oxygen atoms participating in the appropriate halogen-bonded network, as seen in
Figure 6. Similarly, each Na
+ binding is driven mostly by the attractive interactions of the Na
+ with two carbonyl oxygen atoms, an aromatic ring, and two iodine atoms. Inspection of
Table 3 reveals, however, that the interactions with the ring and the iodine atoms are weaker compared with the binding of a single Na
+ counterpart. For example, both the NI…Na
+ and the HC…Na
+ distances are, respectively, 0.098 Å and 0.133 Å longer than those found in the binding of a single Na
+ on the complex side. Additionally, the larger value of the dihedral angle (an increase of 9.8°), Na
+…I…I…O, demonstrates a larger displacement of Na
+ from the pertinent halogen-bond plane which results in the longer NI…Na
+ and HC…Na
+ separations. The moving away of each Na
+ from its respective halogen-bond plane is a manifestation of the moving away of the two Na
+ ions from each other to reach an equilibrium distance, Na
+…Na
+, of 6.748 Å, in response to their repulsive electrostatic interactions.
One direct measure of the strength of a metal ion binding is provided by the corresponding binding energy, calculated as the negative of the complex interaction energy, with the metal ion taken as one unit in the complex and the dimer as the other unit in the complex. The BSSE-corrected binding energies listed in
Table 4 demonstrate that the binding of a single Na
+ is energetically favorable. The binding is even more favorable when Na
+ is found on the outside rather than in the interior of the halogen-bonded dimer. The bond critical point electron densities shown also in
Table 4 serve to reveal more specifically the role of the aromatic rings, the iodine atoms, and the carbonyl oxygen atoms in the binding of Na
+. In particular, a critical point is found along each of the four NI…Na
+ bond paths in the Dimer-Na
+ complex of C
2h symmetry. Additionally, the interaction of Na
+ with each aromatic ring is demonstrated with the presence of a related critical point along the line connecting Na
+ with the closest CH group in the aromatic ring. No critical points are found, however, between Na
+ and any other regions on either aromatic ring. Likewise, no critical points are found in any of the C=O…Na
+ interaction lines, which indicate that the binding of Na
+ is driven mainly by the two aromatic rings and the four iodine atoms. In contrast, the Dimer-Na
+ complex of C
s symmetry exhibits a critical point along each C=O…Na
+ interaction line, which demonstrates the important role of the carbonyl oxygen atoms to bind the metal ion in this complex. Additionally, a critical point is also found along each of the two NI…Na
+ interaction lines, and one critical point with the closer aromatic ring. The corresponding electron density values ρ
NI…Na+ and ρ
HC…Na+ appear larger in this complex than in that of C
2h symmetry. Overall, the combined effect of all these stabilization factors, especially the important contribution of the carbonyl oxygen atoms, as shown in their ρ
C=O…Na+ values, helps explain the larger binding energy in the Dimer-Na
+ complex of C
s symmetry.
As shown in
Table 4, bringing together the two monomers and the two Na
+ ions upon formation of the Dimer-2Na
+ system is energetically favorable, and results in a binding energy comparable to those in the Dimer-Na
+ systems. This result is particularly important given the repulsive interaction between the two positively charged metal ions. Indeed, the calculated repulsive interaction for the two isolated Na
+ ions, at the distance they have in the Dimer-2Na
+ complex, is 49.12 kcal/mol.
Table 4 shows also that the electron densities on the bond critical points along the C=O…Na
+ and the HC…Na
+ interaction lines are comparable to those in the system with only one Na
+. However, the importance of the Na
+ interaction with the halogen atoms in stabilizing the complex appears diminished to the point that no critical point is actually found along any of the NI…Na
+ interaction lines.
The impact on the relative strength of the NI…O=C halogen bonds upon Na+ binding can be seen in the changes in the corresponding critical point electron densities, ρNI…O=C. For example, the halogen bond in the Dimer-Na+ of C2h symmetry exhibits a significant 34% increase in ρNI…O=C relative, for example, to the optimized dimer of C2h symmetry in the absence of the metal ion. In contrast, binding two Na+, as in the Dimer-2Na+, weakens each of the halogen bond, as seen in a respective 11% decrease in ρNI…O=C. The two sets of halogen bonds in the Dimer-Na+ of Cs symmetry are impacted differently by the presence of Na+. Accordingly, the two halogen bonds closer to the metal ion are weakened (15% decrease in ρNI…O=C) while the two halogen bonds farther from the metal ion, each with a ρNI…O=C value of 0.0189 a.u., are actually strengthened (29% increase in ρNI…O=C).
3.3. Na+ Binding by Trimer
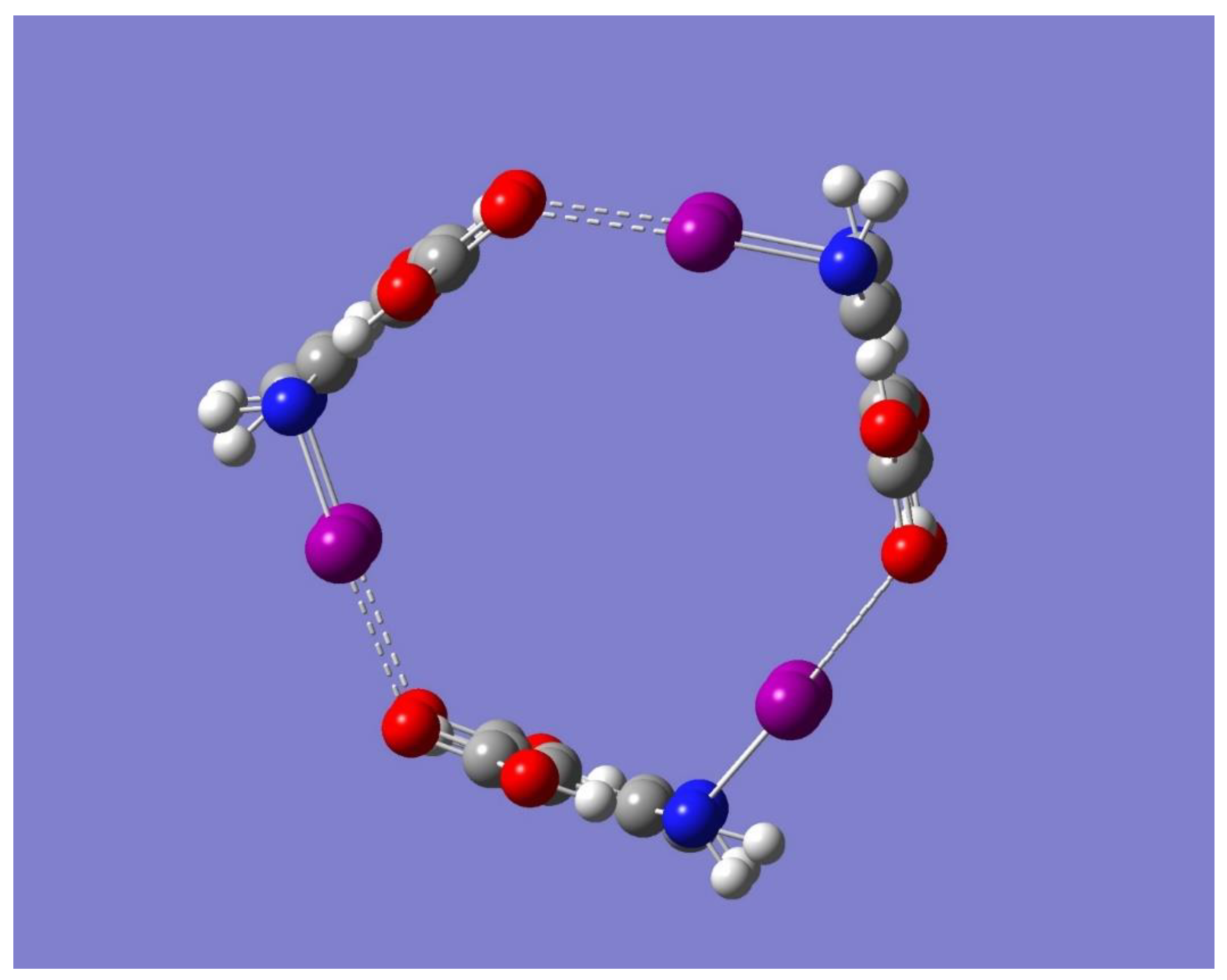
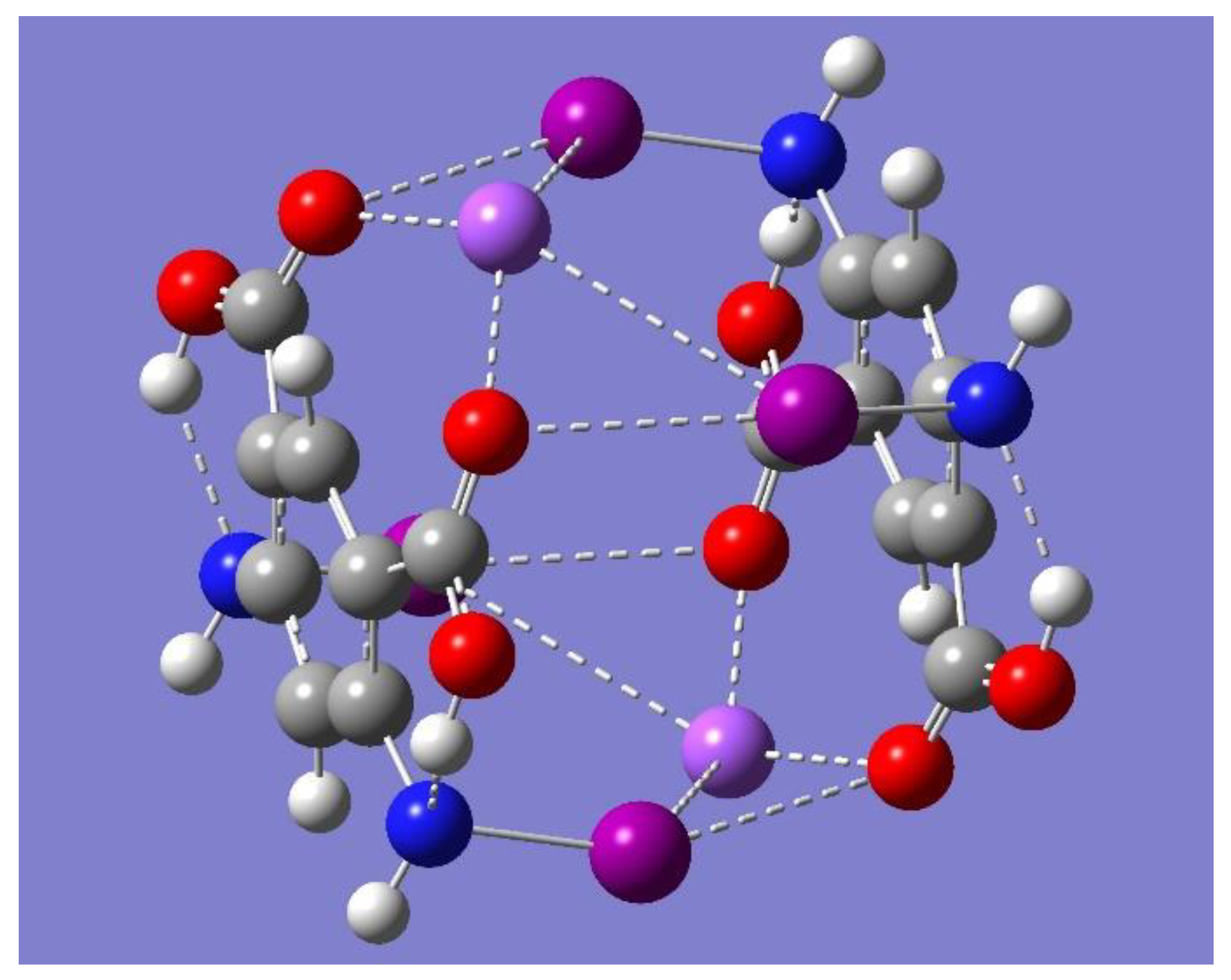
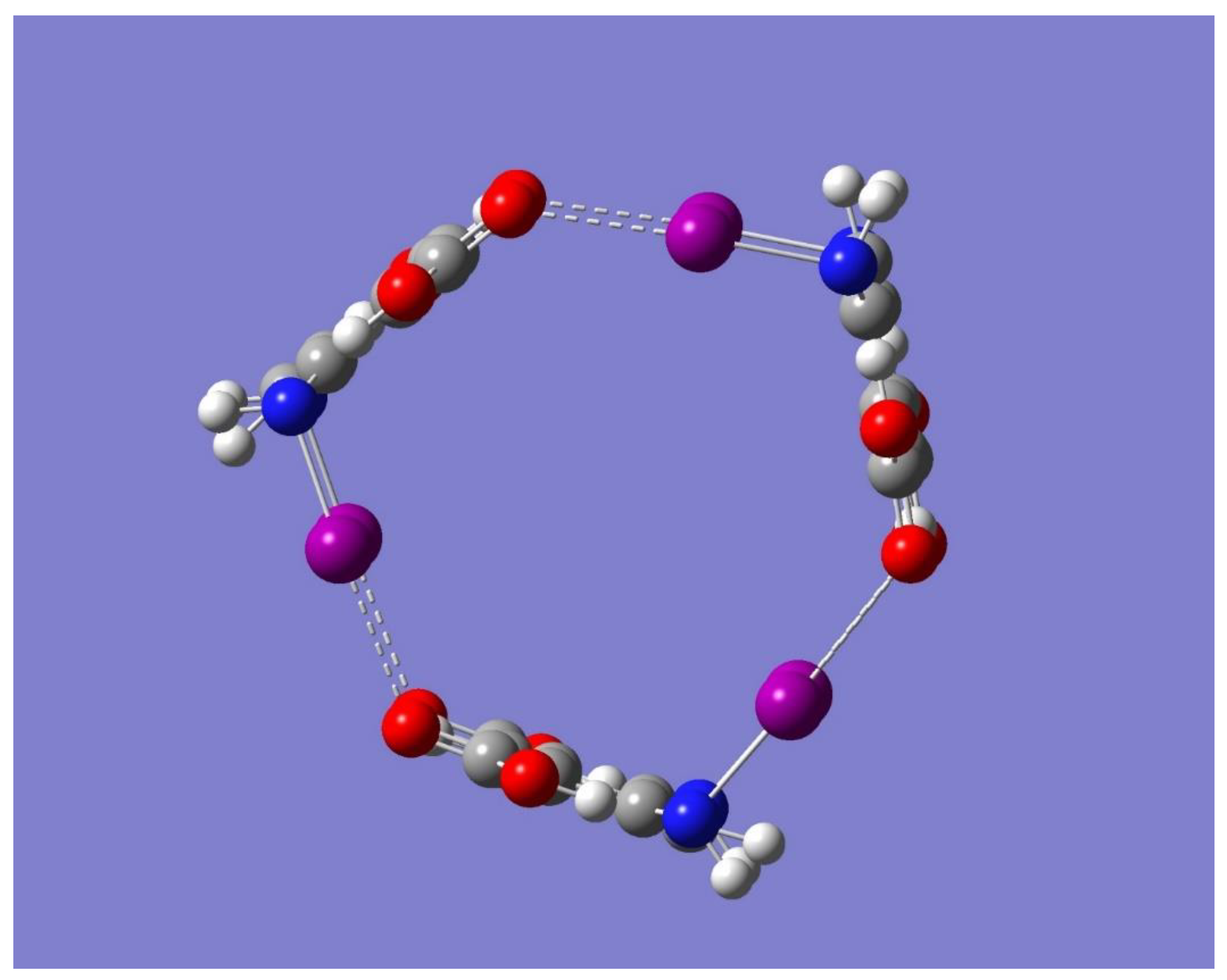
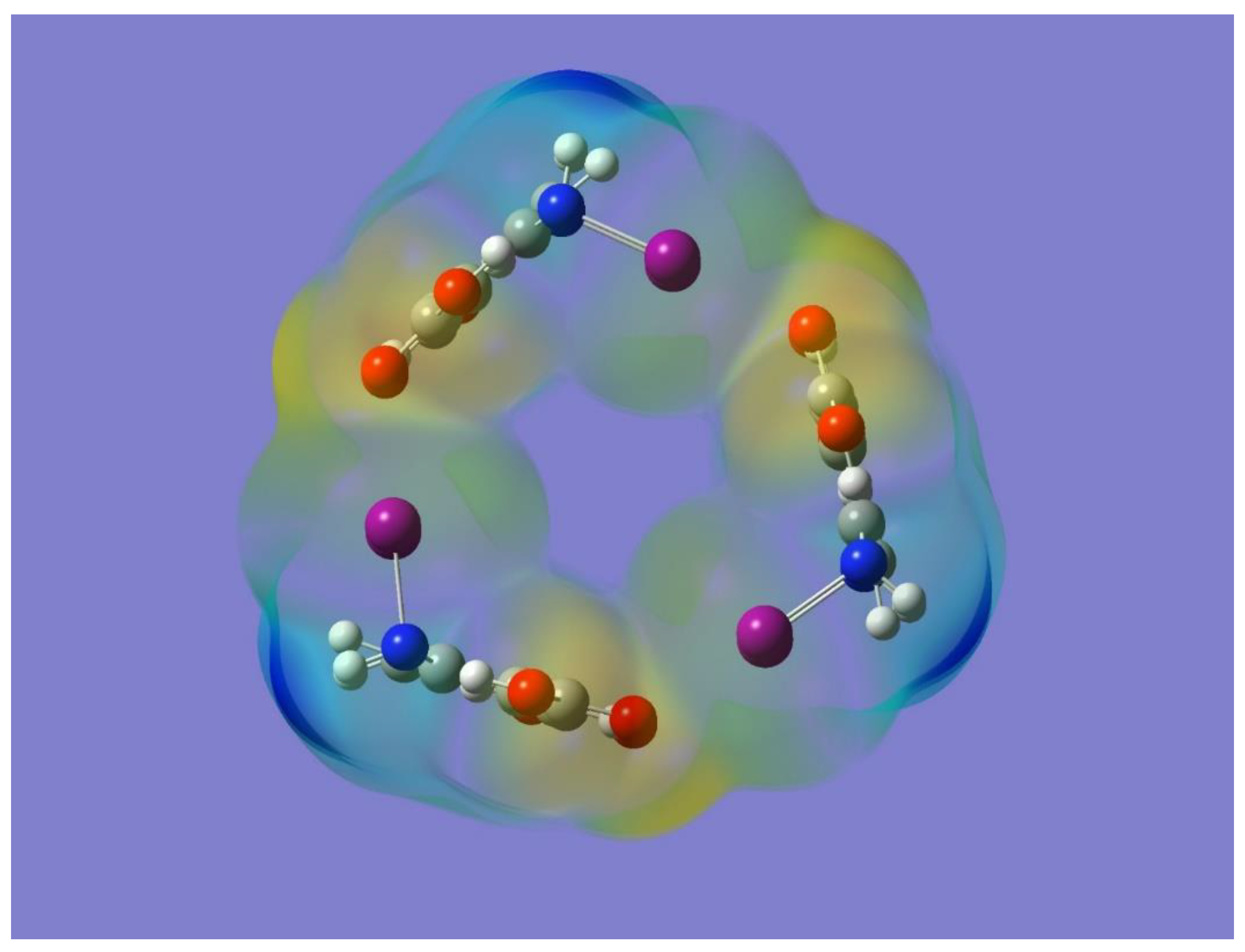
The electrostatic potential of the optimized cyclic trimer of symmetry C
3h shown in
Figure 7 shows the potential of this trimer to bind an electron-deficient species such as Na
+ mostly on the sides or edges of the trimer. One striking result from
Figure 7 is, however, the lack of any negative electrostatic potential that can attract Na
+ in the central cavity of the molecule. In fact, a full geometry optimization, followed by frequency calculations performed with the metal ion on the point of symmetry of the trimer, resulted in a structure with three imaginary frequencies. On the other hand, a geometry optimization with Na
+ on one side of the cyclic trimer was confirmed to be a minimum energy structure, with frequency calculations showing no imaginary frequencies. Similarly, a geometry optimization with one Na
+ on each side of the trimer also resulted in a minimum energy structure with no imaginary frequencies. The symmetry of the optimized Trimer-Na
+ complex is C
s, while the optimized Trimer-3Na
+ complex retains the symmetry of the trimer, C
3h. It is worth noting that a geometry optimization of a Trimer-2Na
+ complex, using the optimized Trimer-3Na
+ geometry as an initial guess but removing one of the Na
+, resulted in a geometry that retains the overall shape of the complex. However, this optimized Trimer-2Na
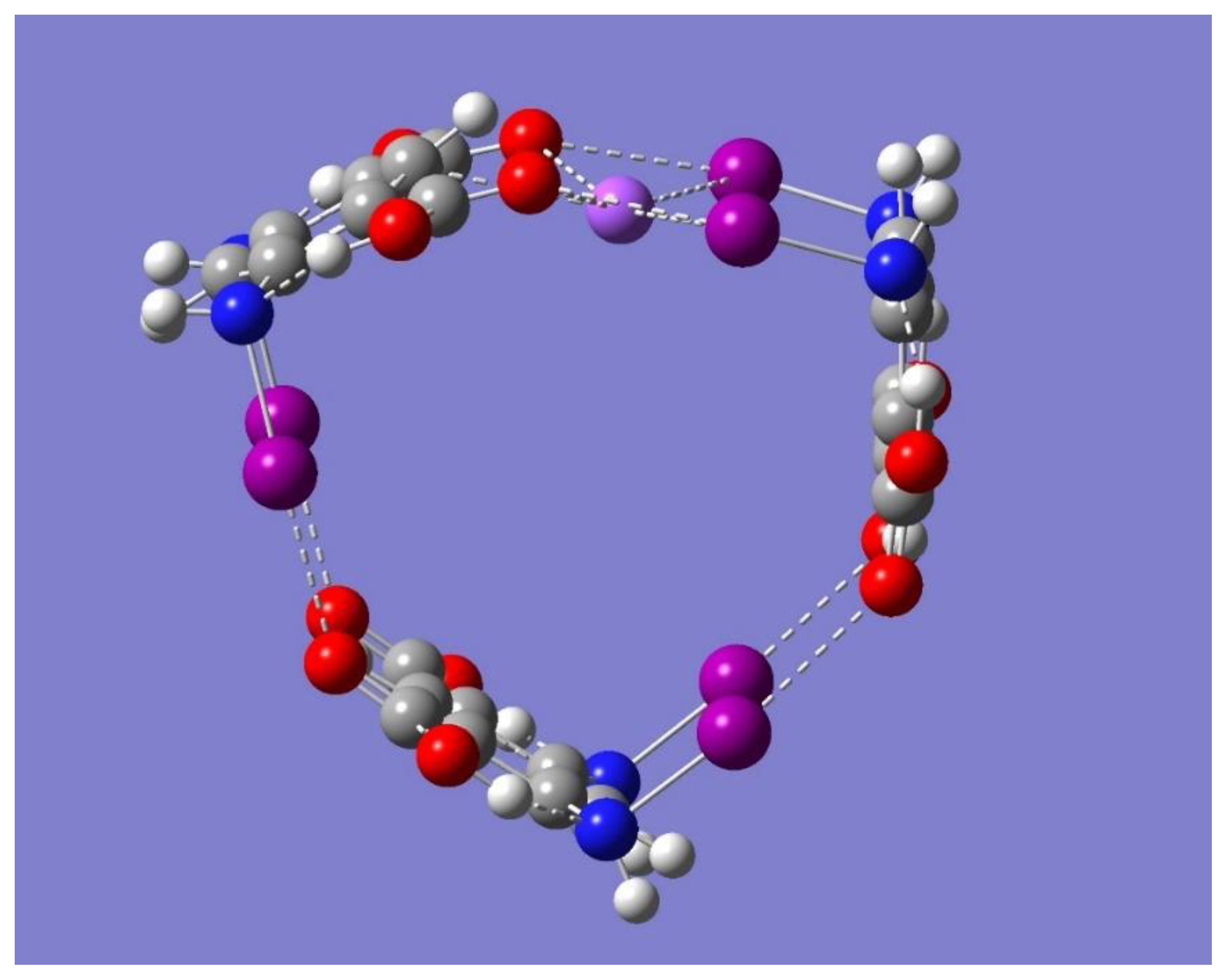
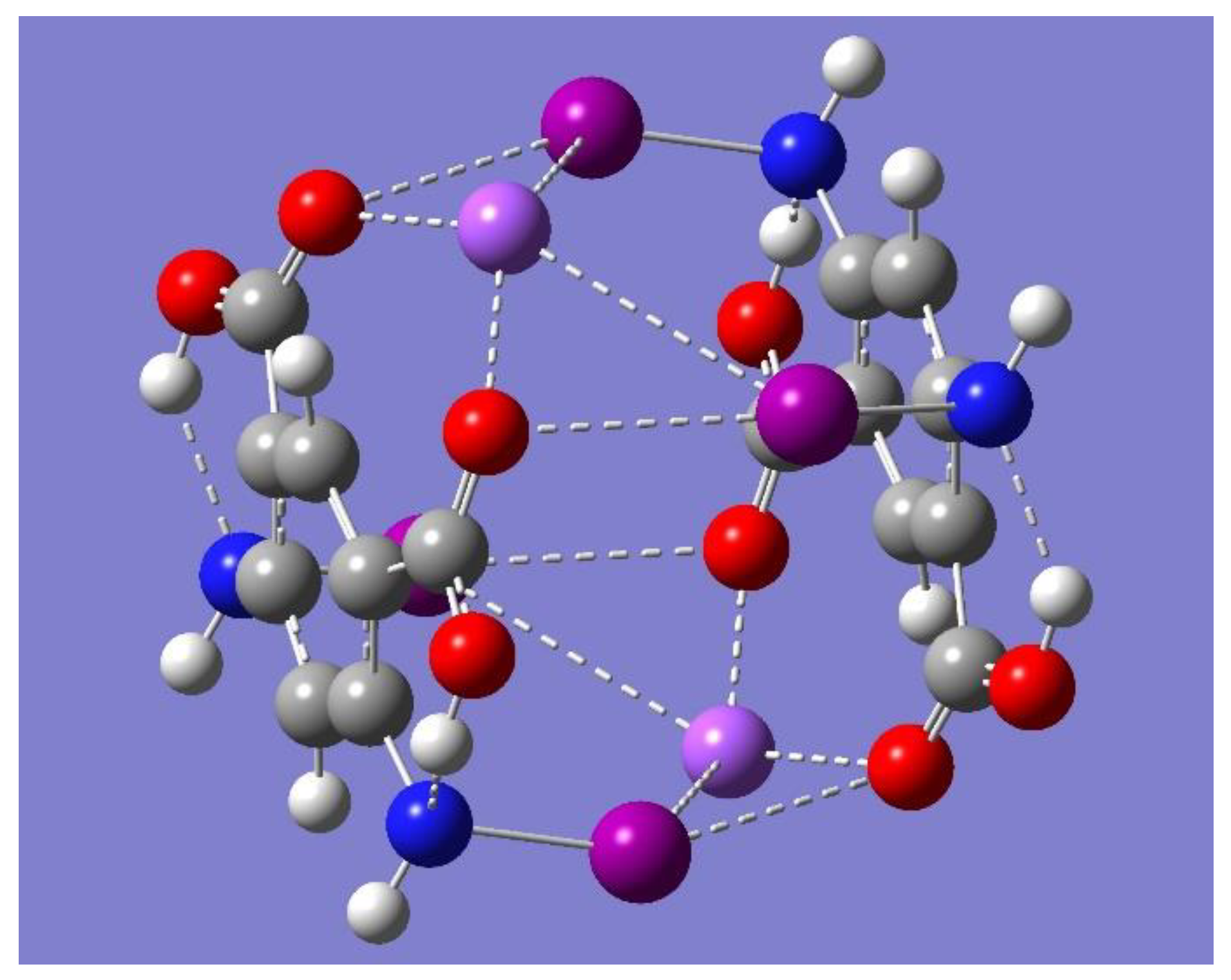
+ complex was shown to be a saddle point of first order (frequency calculations resulted in one imaginary frequency). Thus, the focus of the discussion is on the two minimum energy structures shown in
Figure 8 which have no imaginary frequencies, Trimer-Na
+ and Trimer-3Na
+, and whose relevant geometrical parameters are listed in
Table 5.
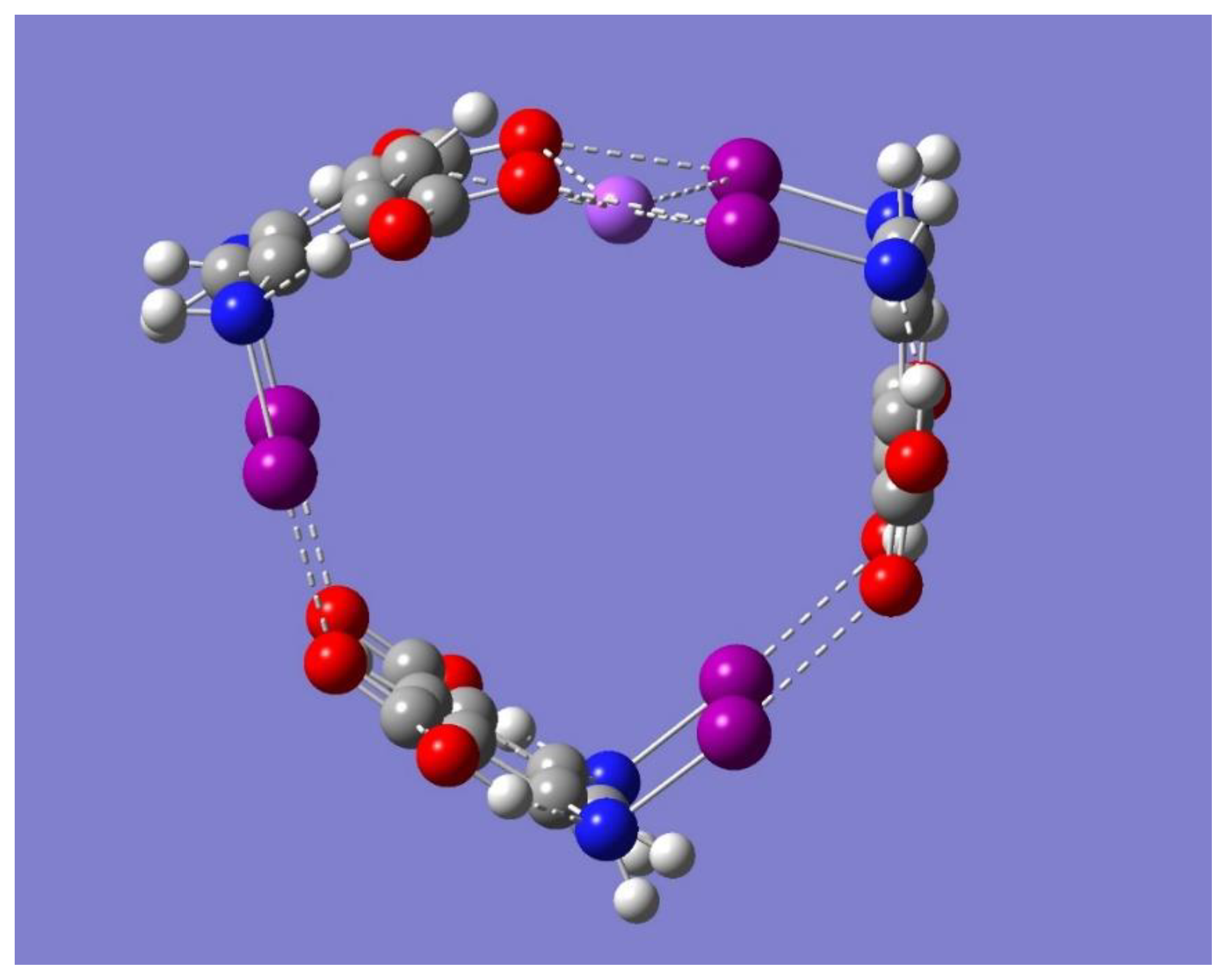
Figure 6 and
Figure 8 show that the binding in Trimer-Na
+ can be conveniently compared with the corresponding binding in Dimer-Na
+ of symmetry C
s, given that, in each case, the binding of Na
+ occurs primarily with one of the halogen-bonded networks. One interesting difference is that the metal ion in the Trimer-Na
+ complex is found toward the interior of the complex, in contrast to the corresponding Dimer-Na
+ complex, where the metal ion is found toward the outside of the complex. This difference is further confirmed by the opposite signs of the dihedral angle, Na
+…I…I…O: 13.1° and −14.4° for the Dimer-Na
+ and Trimer-Na
+, respectively (
Table 3 and
Table 5). Moreover, geometrical parameters suggest an enhanced role of the NI…Na
+ interactions for Na
+ binding by the trimer. For example, an inspection of
Table 3 and
Table 5 shows, relative to the Dimer-Na
+, a sizeable decrease in the NI…Na
+ distances concomitant with respectively minor increases in the C=O… Na
+ and HC…Na
+ distances. Compared with the free C
3h trimer, the halogen bond network that interacts with the metal ion is weakened upon Na
+ complexation, as indicated by longer halogen bond distances (2.746 Å to 2.919 Å) and smaller angles (169.5° to 156.0°). The impact of Na
+ complexation on the other two halogen-bonded networks can be conveniently described by considering the monomer that is farthest away from Na
+ in its dual role as the halogen-bond donor or as the halogen-bond acceptor. In the former case, it is found that the associated NI…Na
+ distances are longer (2.823 Å), while the opposite is true in the latter case (2.655 Å). These results indicate an enhanced halogen-bond donor ability of the monomer that interacts with Na
+ through the carbonyl oxygen atoms, C=O. The increase in the halogen-bond strength with the farthest (from Na
+) monomer is offset, on the other hand, by a concurrent reduced halogen-bond acceptor ability of the monomer that interacts with Na
+ through the iodine atoms.
The binding of three metal ions by the cyclic trimer involves all three halogen-bonded networks, each binding one Na
+. The three Na
+ ions are equivalent within an overall C
3h symmetry. Unlike the binding of a single Na
+, which occurs towards the inner side of the trimer, each of the three metal ions sits now on top of the plane formed by the iodine and oxygen atoms participating in the pertinent halogen-bonding network, as displayed in
Figure 8. The sign change in Na
+…I…I…O, (from −14.4° to 14.1°), results in relatively large Na
+…Na
+ separations of 7.327 Å, which help minimize the repulsive electrostatic interactions among the three metal ions. As with the binding of a single Na
+, the binding of each Na
+ in the Trimer-3Na
+ complex is driven mostly by the attractive interactions of the Na
+ with the corresponding halogen-bonded network through two carbonyl oxygen atoms, an aromatic ring, and two iodine atoms. Inspection of
Table 5 shows, however, that the interactions with the ring and the iodine atoms appear weaker (longer distances) compared with the binding of a single Na
+ counterpart, while the C=O…Na
+ interactions appear stronger (shorter distances).
The BSSE-corrected metal ion binding energies listed in
Table 6 demonstrate that the binding of a single Na
+ by the trimer is energetically favorable. Moreover, the binding energy is actually larger than that in either dimer (C
s or C
2h symmetry,
Table 4). The bond critical point electron densities also shown in
Table 6 help to better appreciate the role of the aromatic rings, the iodine atoms, and the carbonyl oxygen atoms in the binding of Na
+. In particular, a critical point is found along each of the associated NI…Na
+ and C=O…Na
+ interaction lines in the Trimer-Na
+ complex. Additionally, the interaction of Na
+ with the closest aromatic ring is demonstrated with the presence of a related critical point along the line connecting Na
+ with the closest carbon atom of the aromatic ring. The values of the critical point electron densities, ρ
C=O…Na+ and ρ
C…Na+, in the Trimer-Na
+ complex are comparable to those in the Dimer-Na
+ complex of symmetry C
s. However, the ρ
NI…Na+ values are much larger in the Trimer-Na
+ complex. It should be noted that the changes in the critical point electron density values consistently mirror the interrelated changes in the geometrical parameters, namely a sizeable decrease in the NI…Na
+ distances concomitant with respectively minor changes in the C=O…Na
+ and HC…Na
+ distances.
As shown in
Table 6, bringing together the three monomers and the three Na
+ ions upon formation of the Trimer-3Na
+ system is still an energetically favorable process. Compared with Trimer-Na
+, the binding energy in Trimer-3Na
+ is, nonetheless, substantially smaller. This result is not surprising, given the large repulsive electrostatic interaction among the three positively charged metal ions. Indeed, the calculated molar repulsive energy for the three Na
+ ions is 135.92 kcal/mol. An inspection of
Table 6 shows that the electron densities on the bond critical points ρ
HC…Na+ and ρ
NI…Na+ are reduced, while those for ρ
C=O…Na+ are increased relative to the Trimer-Na
+ complex.
The critical point electron density values in
Table 6 and
Table 4 indicate that the HC…Na
+ interactions are enhanced, while the C=O…Na
+ interactions weakened in the Trimer-3Na
+ complex relative to the Dimer-2Na
+ complex. One interaction that appears heightened in the Trimer–3Na
+ is the NI…Na
+ interaction, as the existence of a corresponding bond critical point demonstrates. The presence of bond critical points along each of the NI…Na
+ interactions denotes the enhanced role of these interactions in the binding of each Na
+ in the Trimer-3Na
+ complex. In contrast, no such critical points were found the Dimer-2Na
+ complex.
The impact on the relative strength of the NI…O=C halogen bonds upon Na
+ binding by the trimer can be appreciated in the changes of the corresponding critical point electron densities, ρ
NI…O=C. For example, an inspection of
Table 2 and
Table 6 shows that each of the halogen bonds interacting with Na
+ in the Trimer-Na
+ exhibits a significant 23% decrease in ρ
NI…O=C relative to the trimer optimized in the absence of the metal ion. The two sets of halogen bonds in the Trimer-Na
+ not directly involved in binding the metal ion are impacted differently by the presence of Na
+. One set of halogen bonds has ρ
NI…O=C values of 0.0188 a.u. (16% decrease in ρ
NI…O=C), while the other set has ρ
NI…O=C values of 0.0263 a.u. (18% increase in ρ
NI…O=C). Each of the halogen bonds in the Trimer-3Na
+ complex exhibits a substantial 40% decrease in ρ
NI…O=C. As expected, the changes in the critical point electron density values are in accordance with the interrelated changes in the relevant geometrical parameters.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}