
Malonaldehyde-like Systems: BeF2 Clusters—A Subtle Balance between Hydrogen Bonds, Beryllium Bonds, and Resonance


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
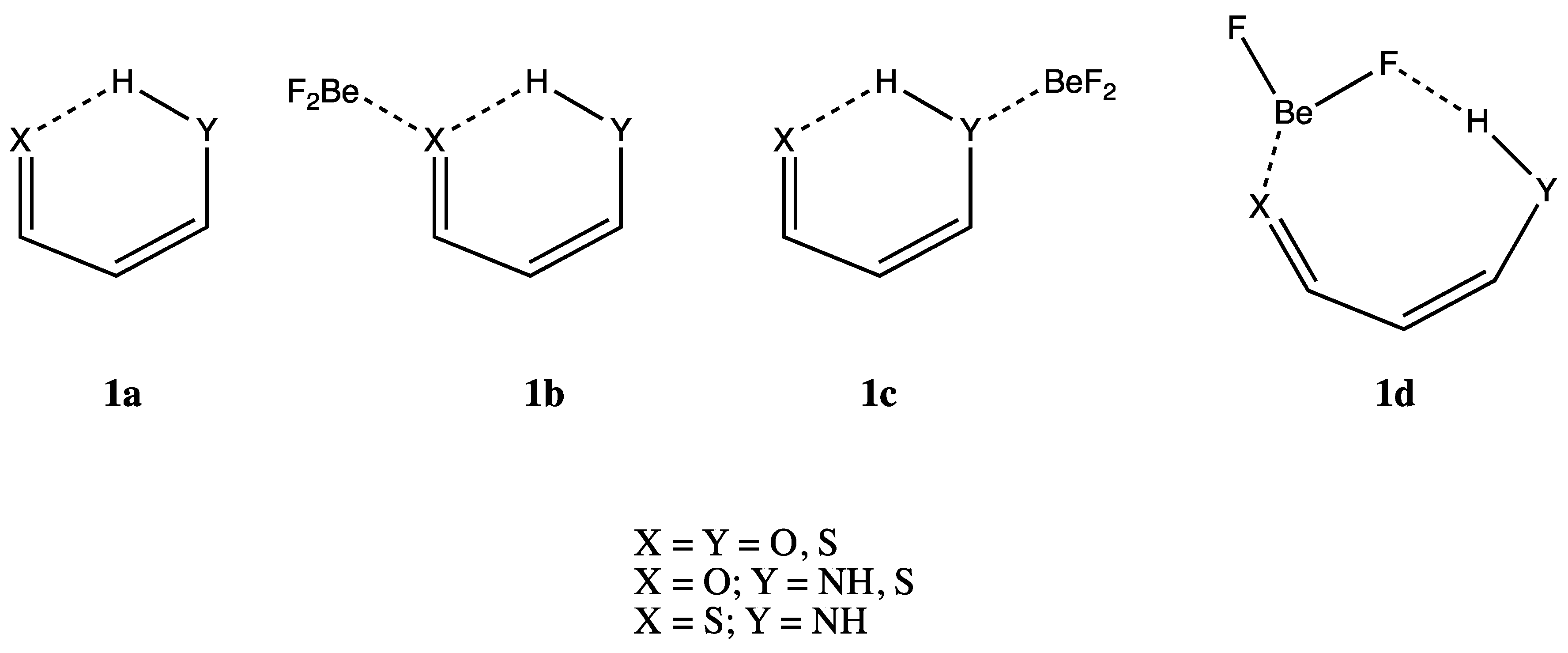
:1. Introduction
2. Computational Details
3. Results and Discussion
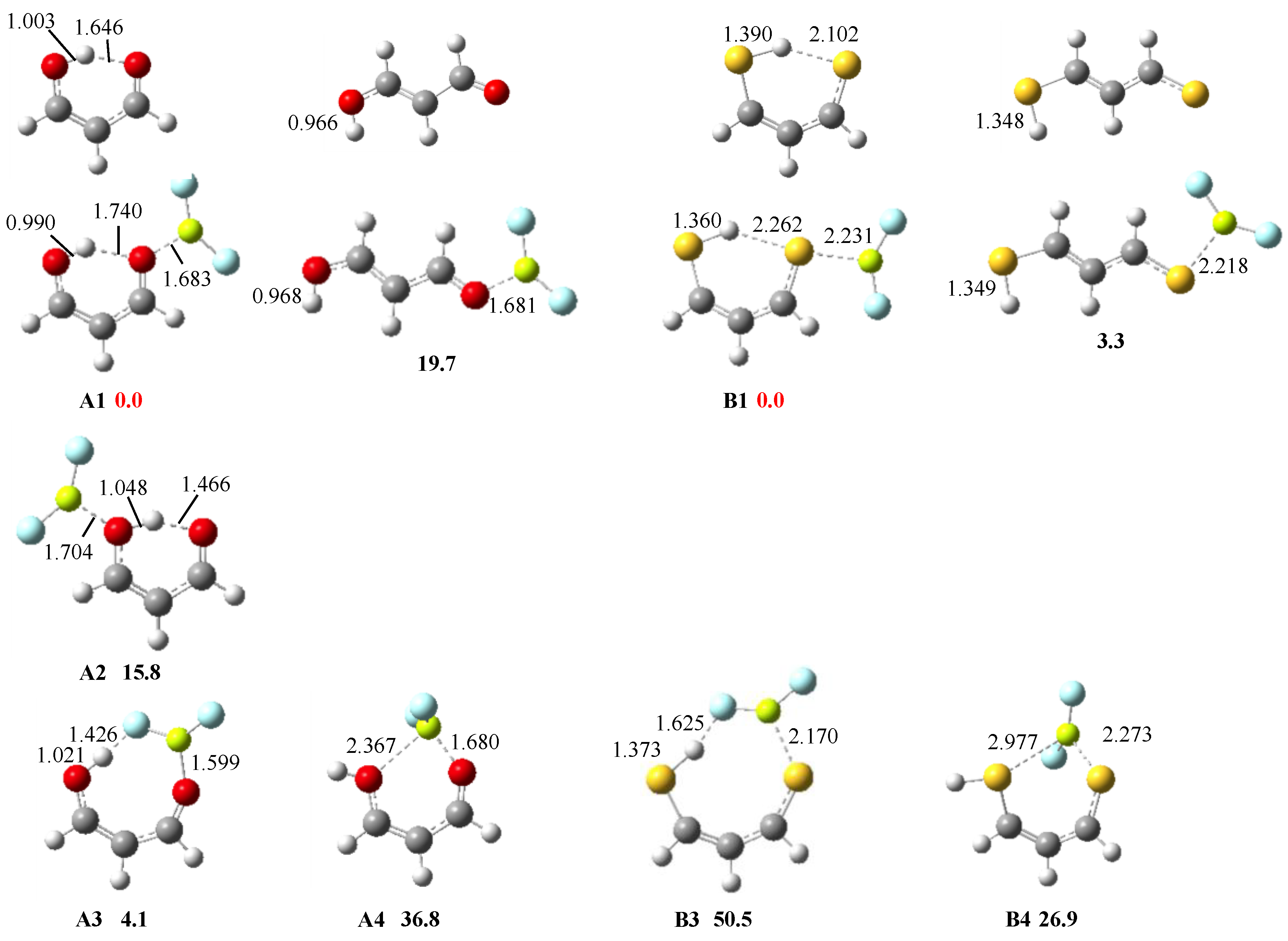
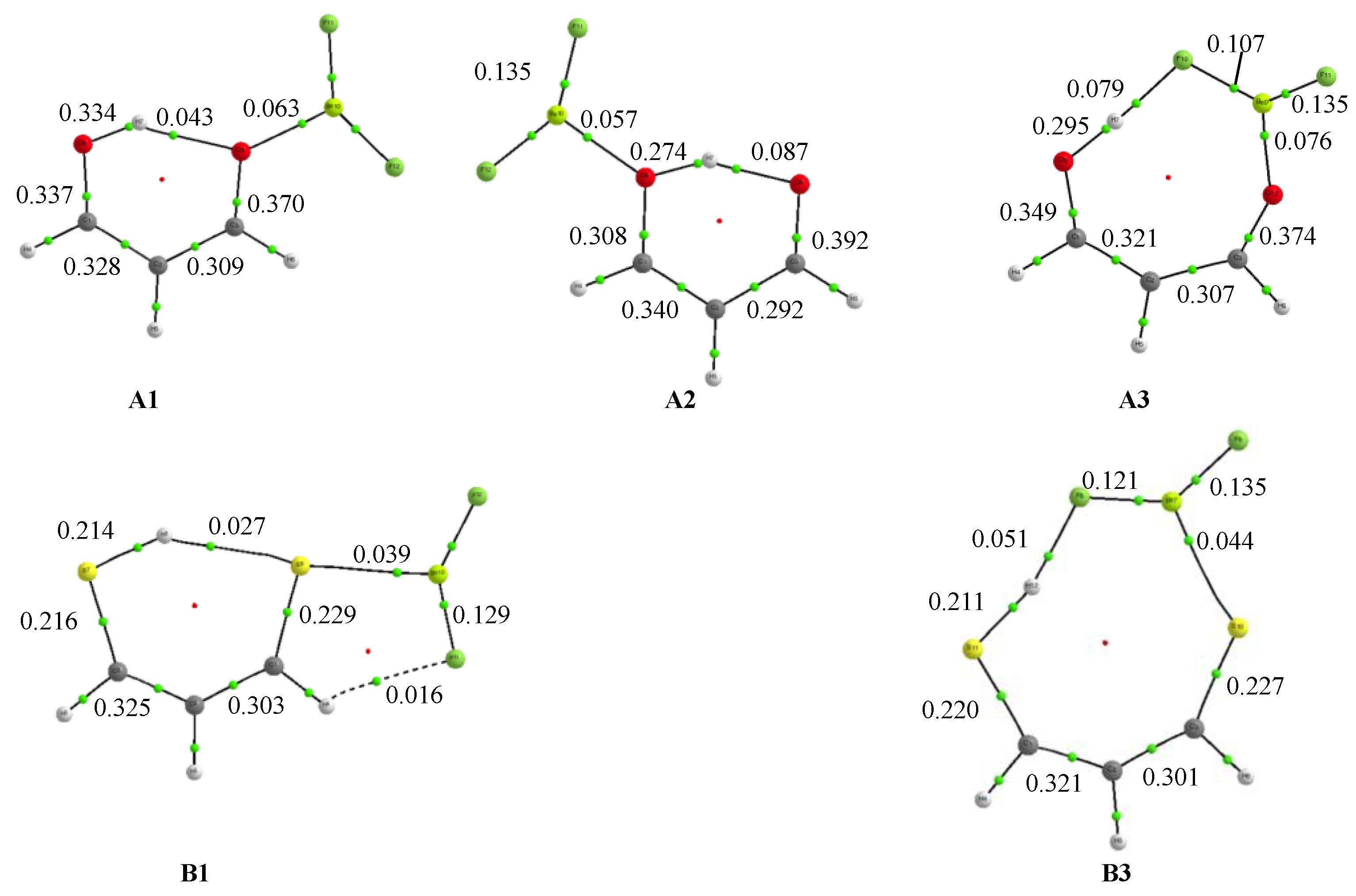
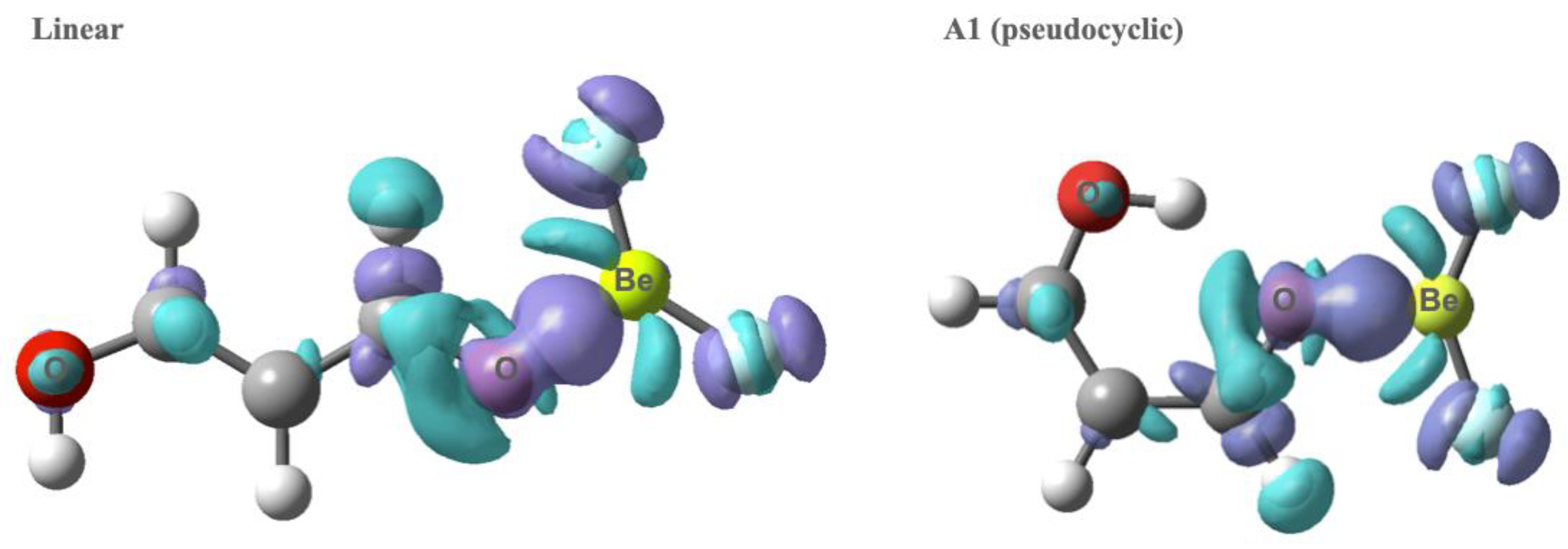
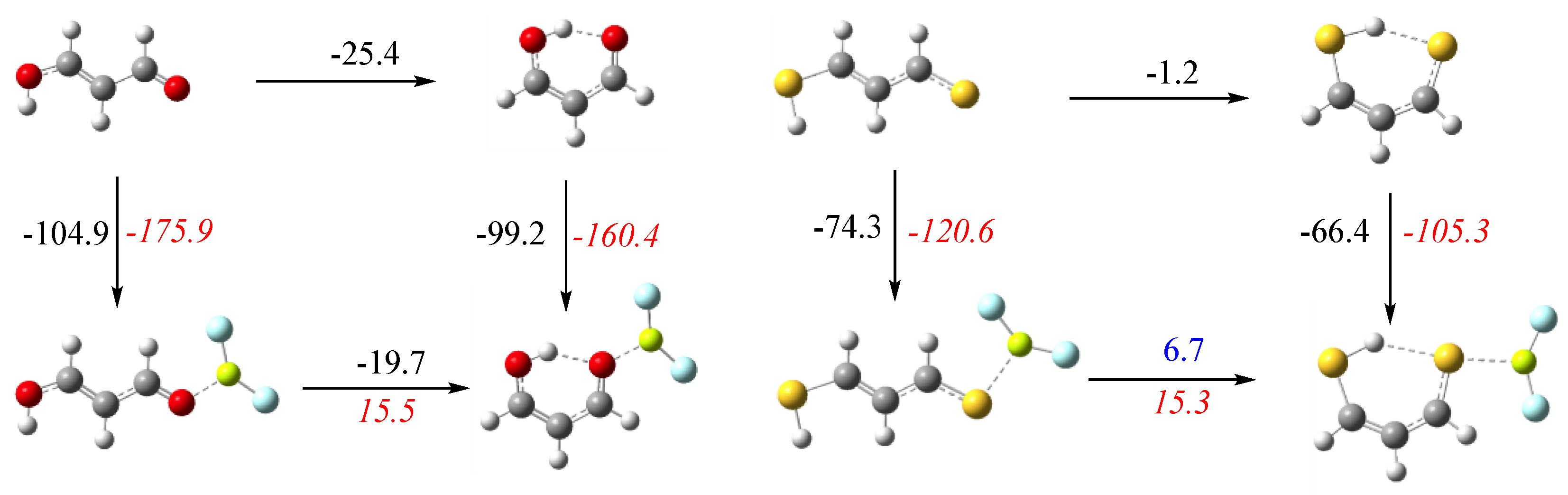
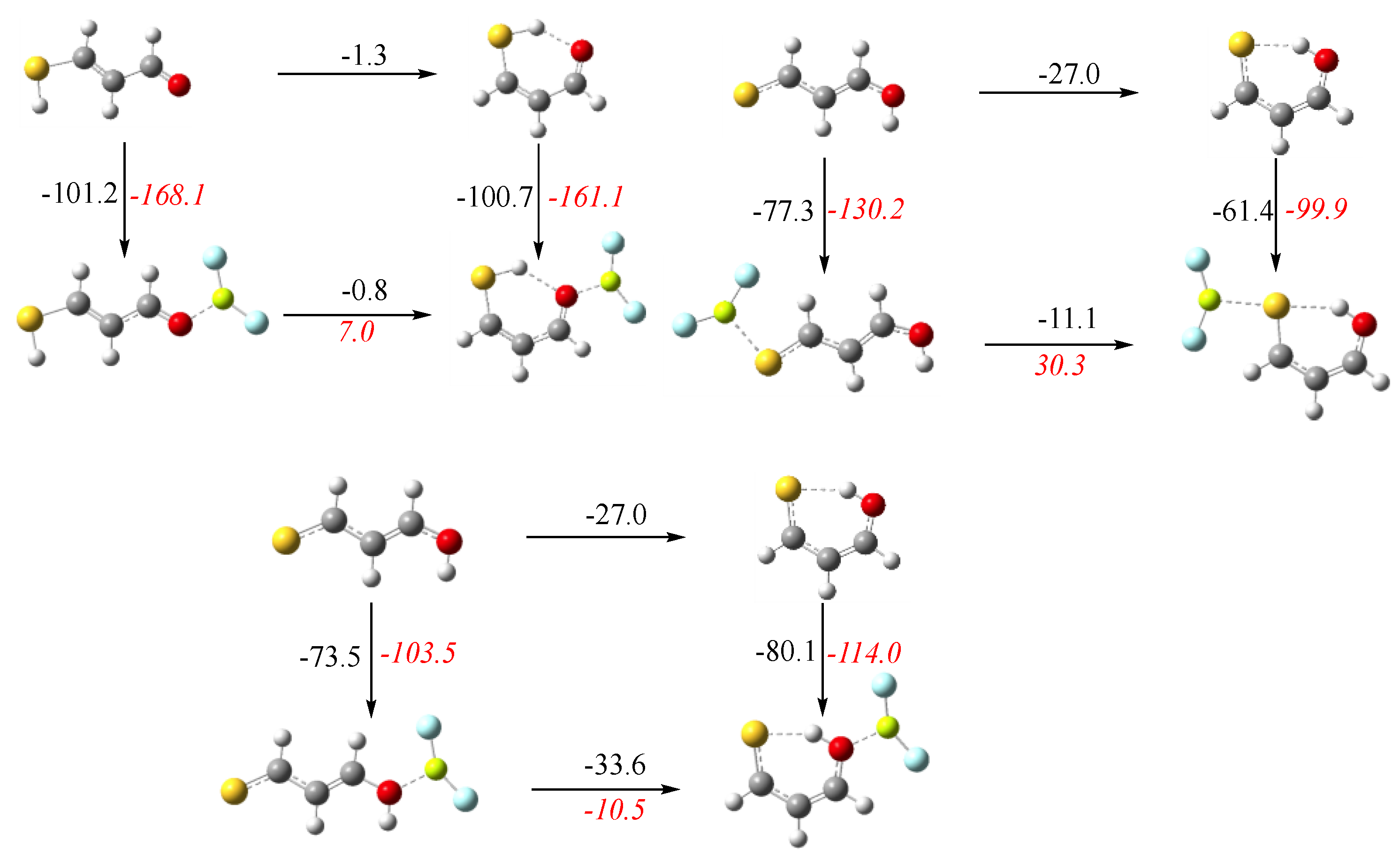
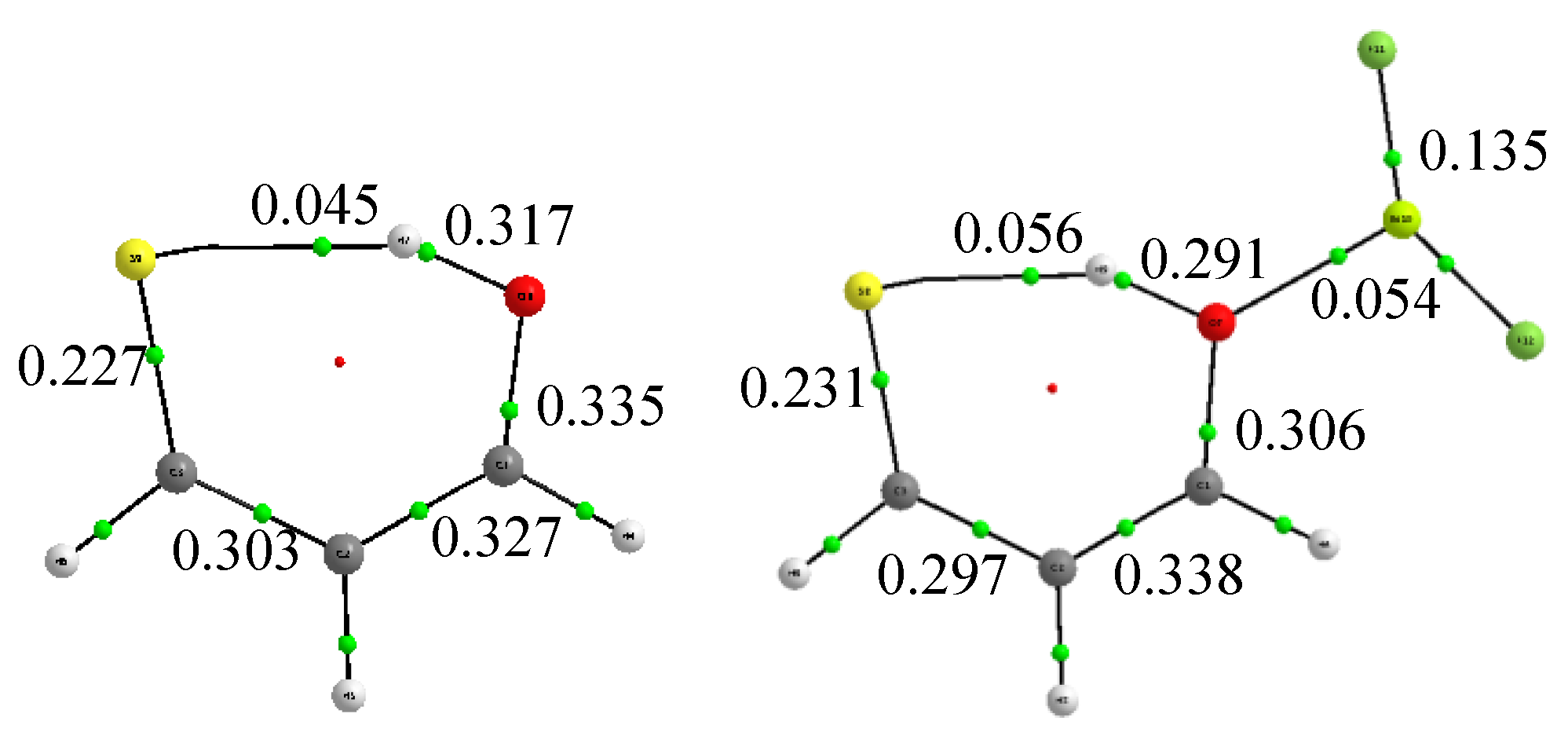
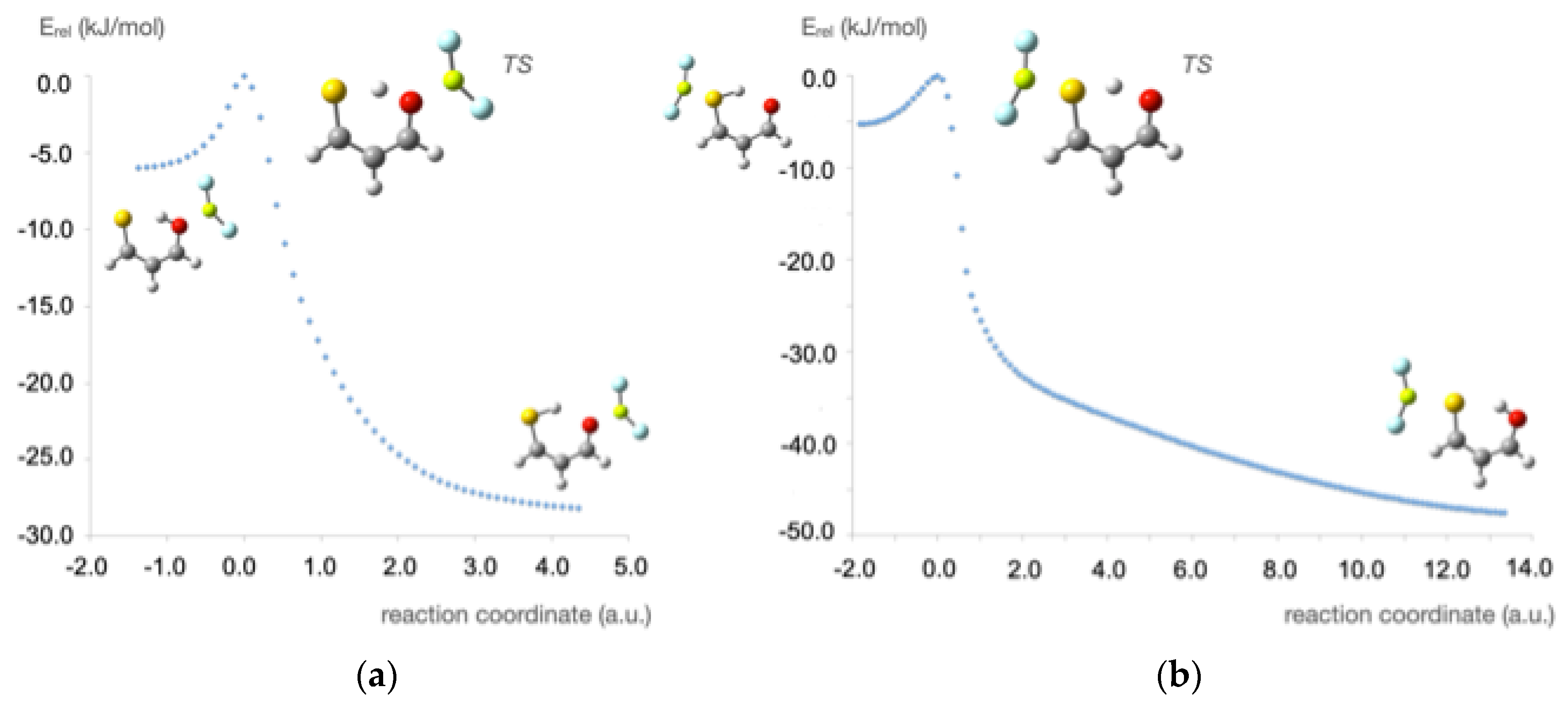
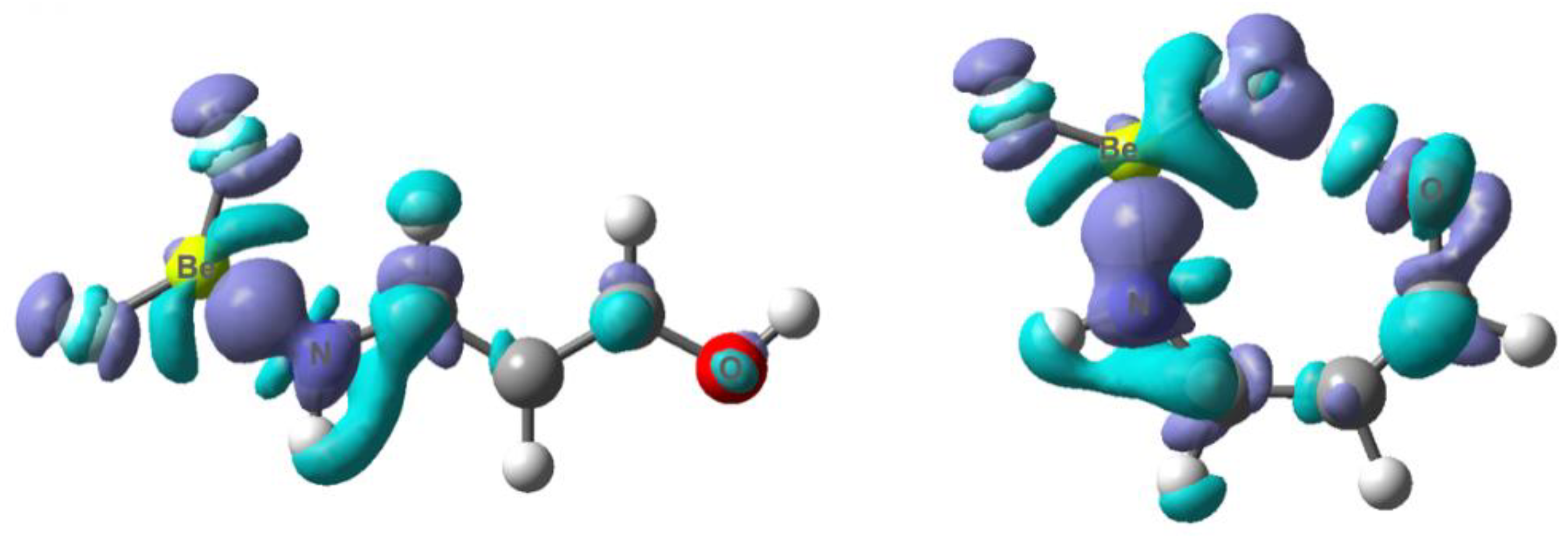
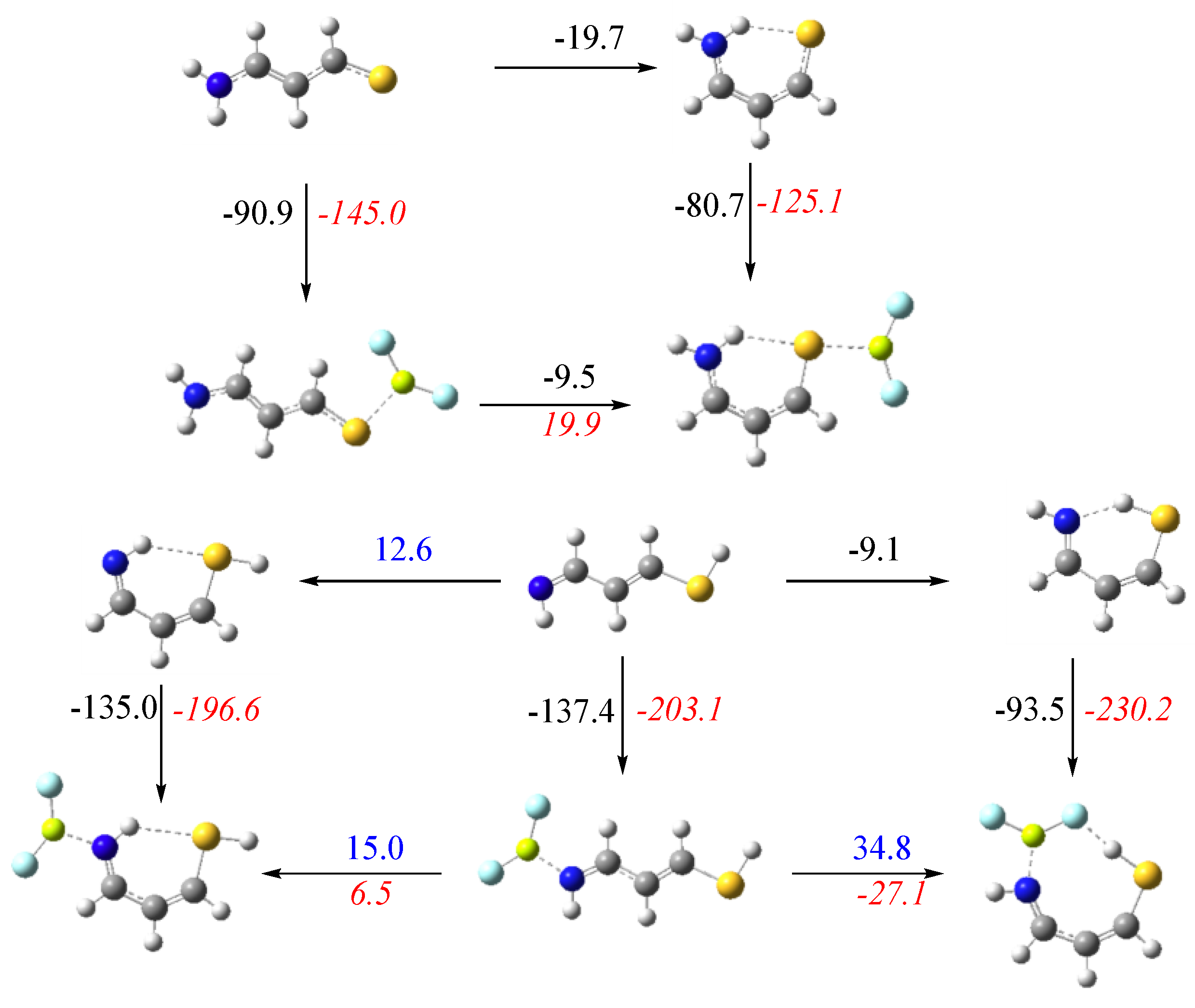
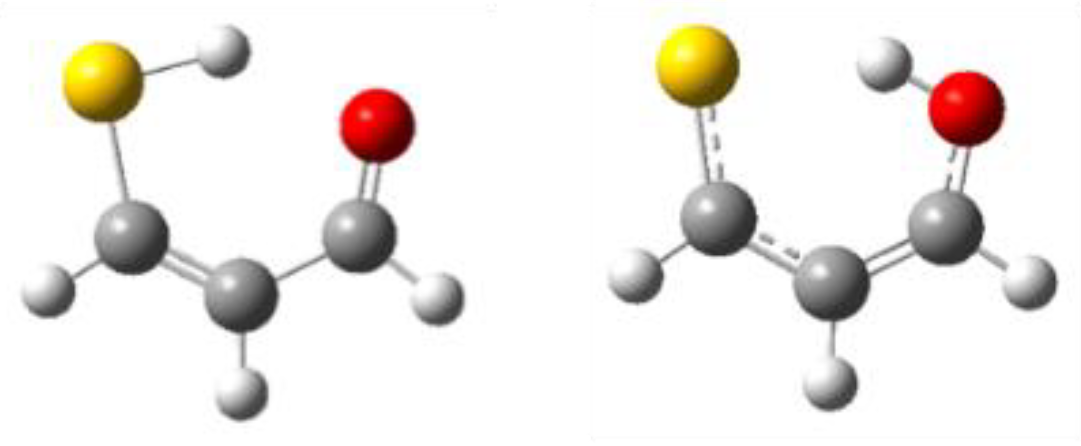
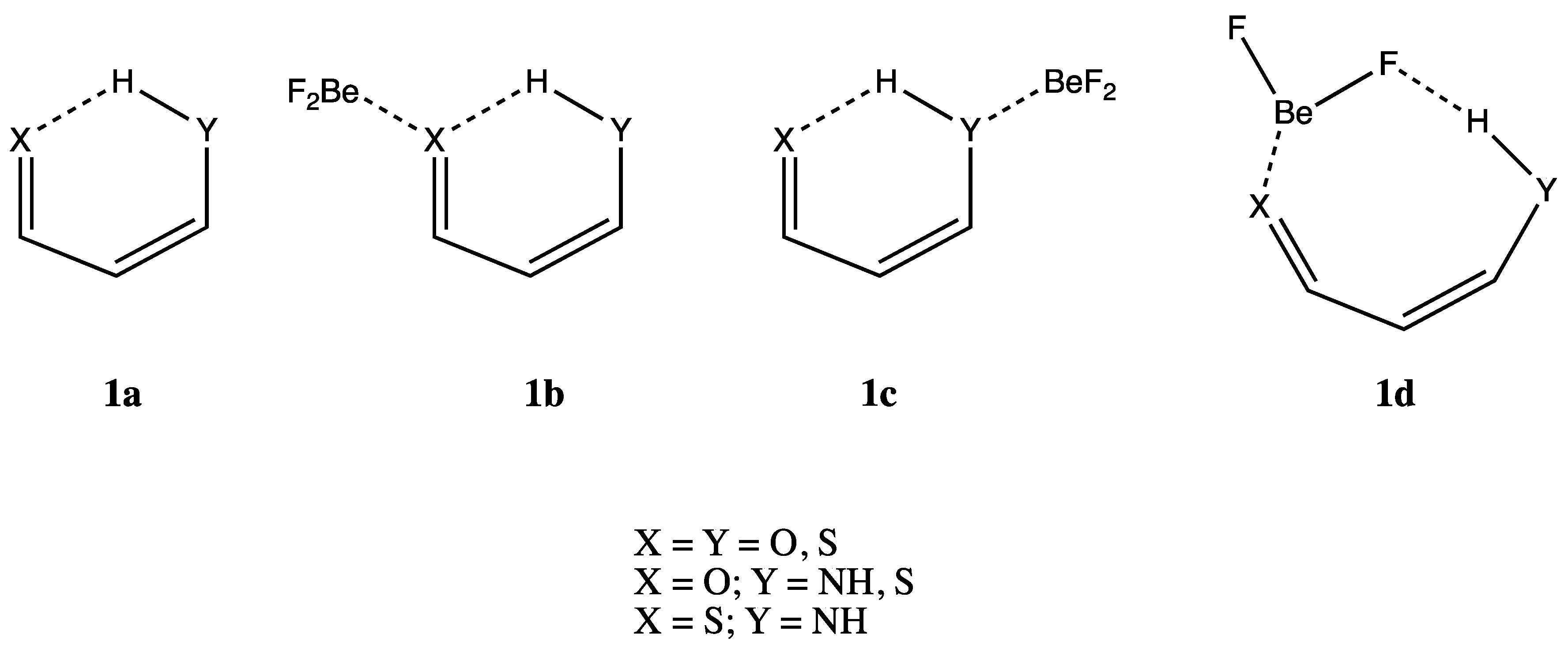
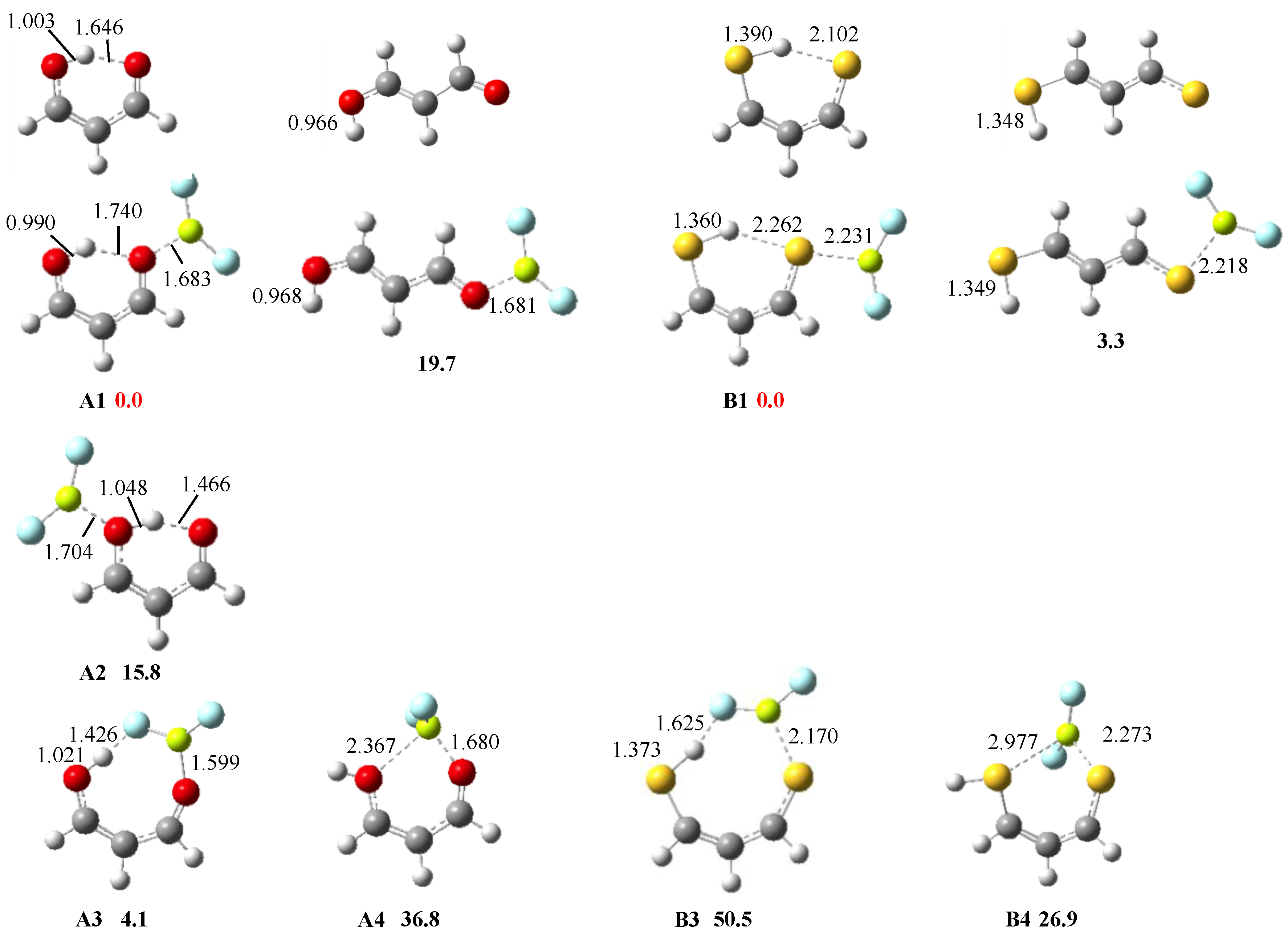
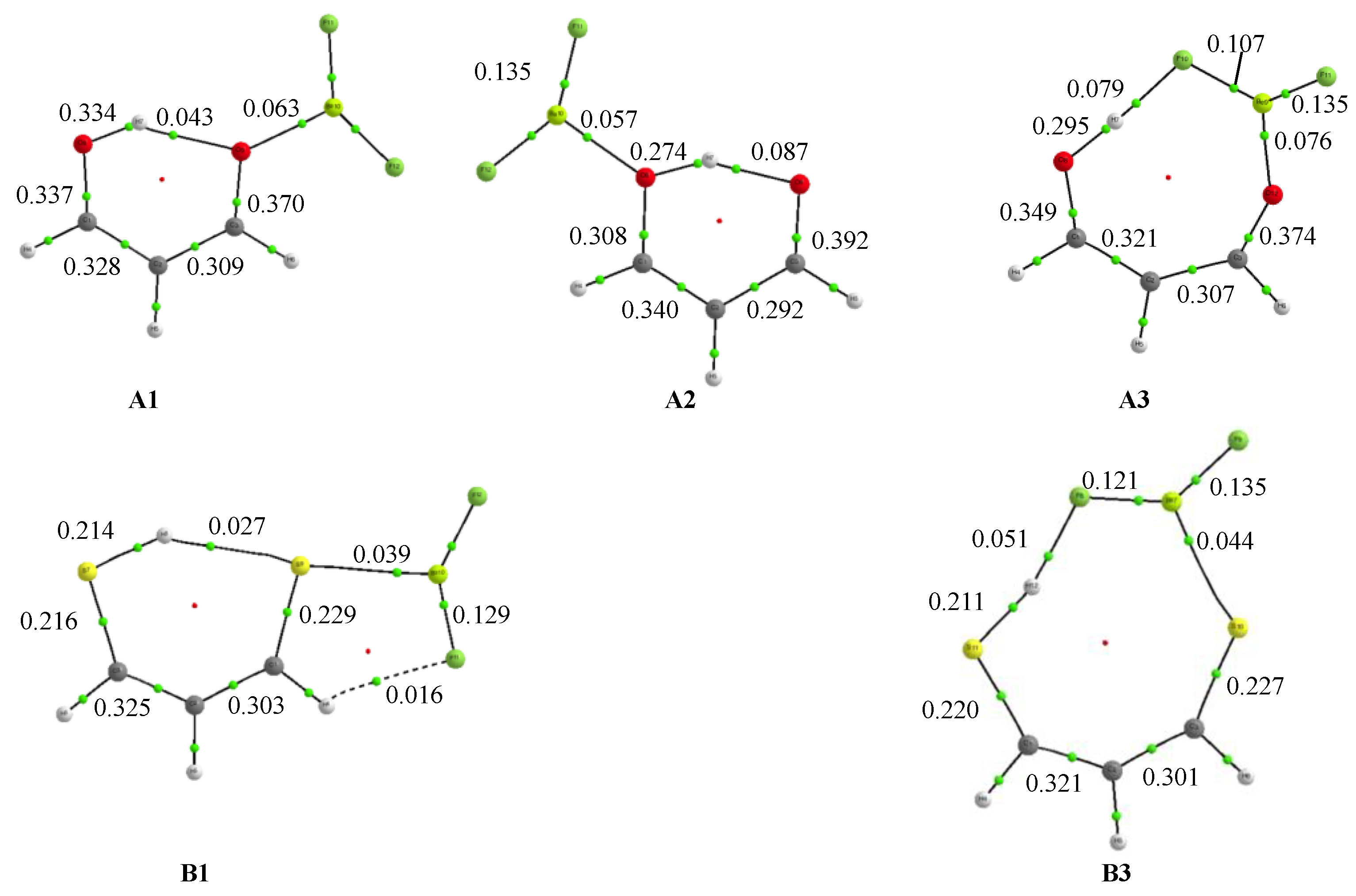
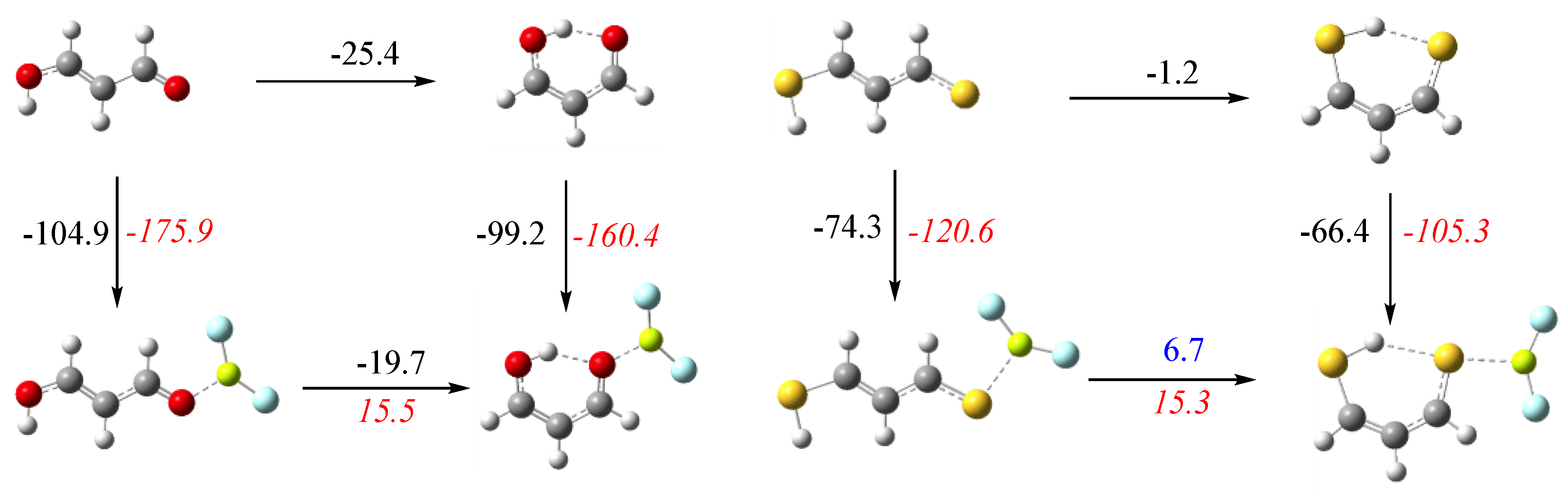
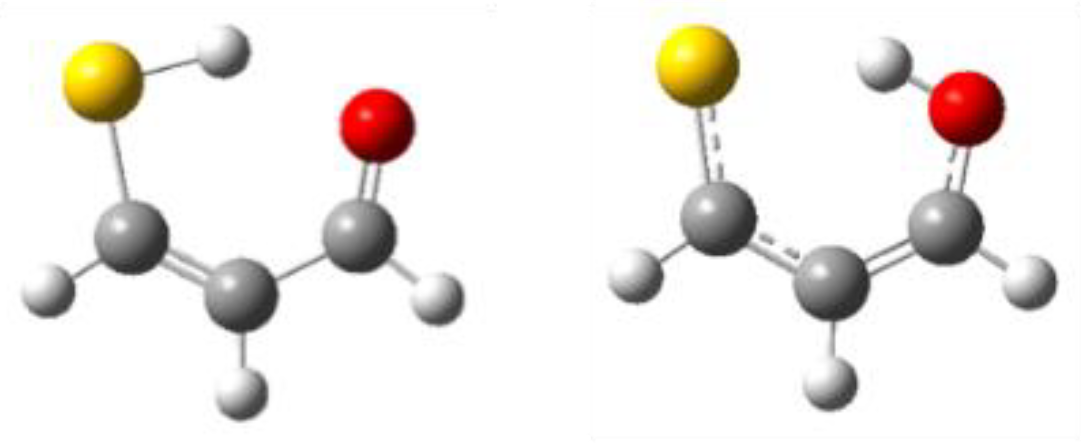
3.1. X = Y = O, S
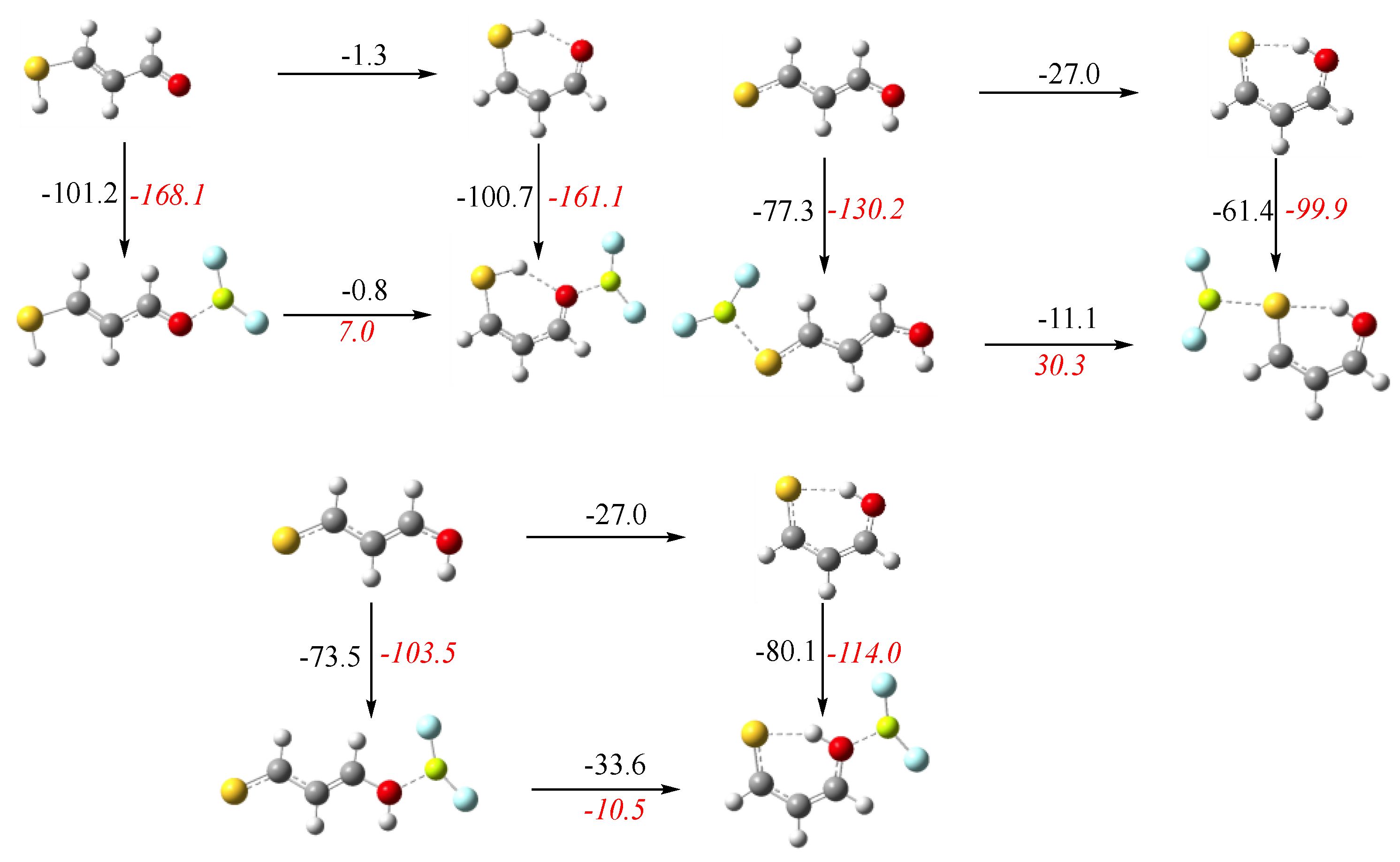
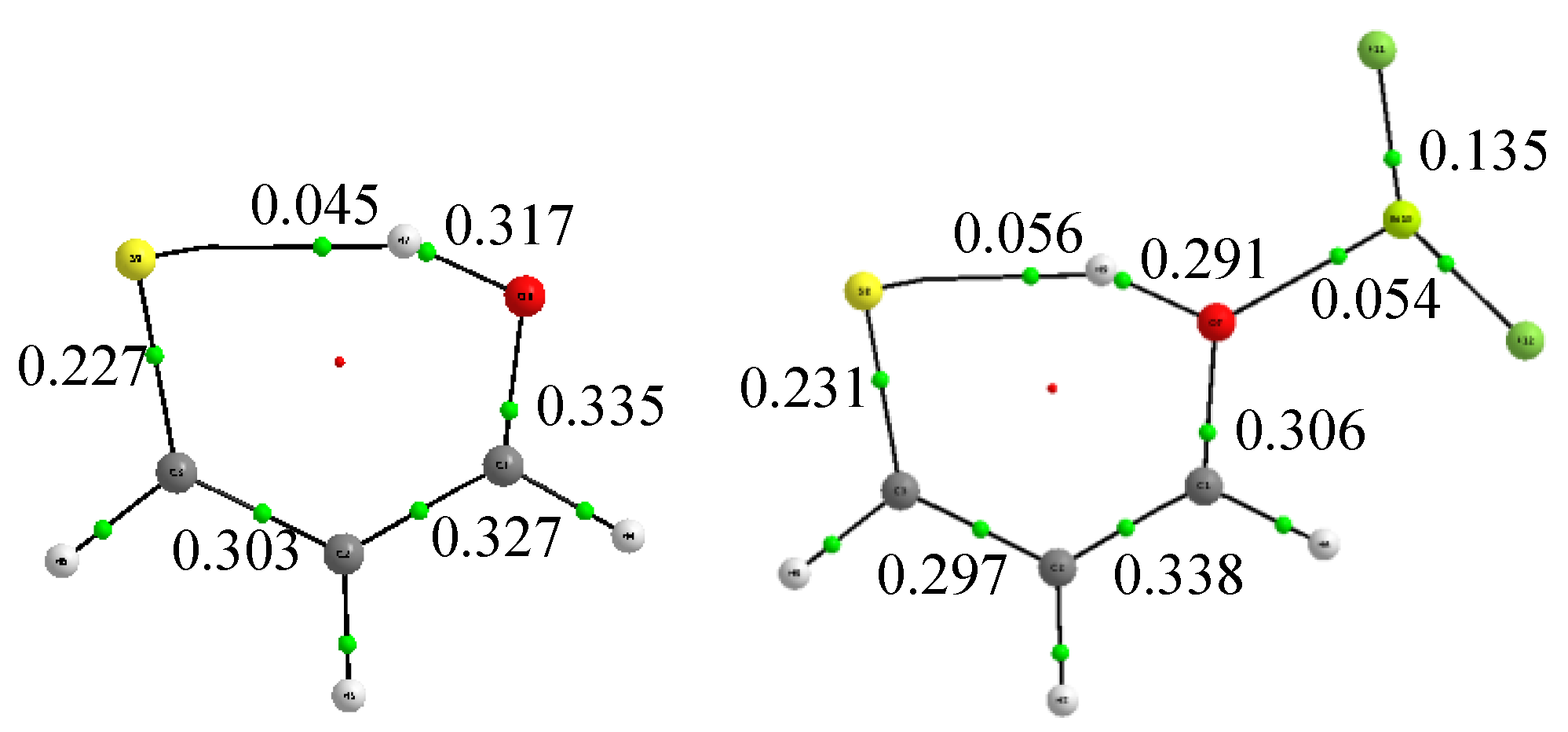
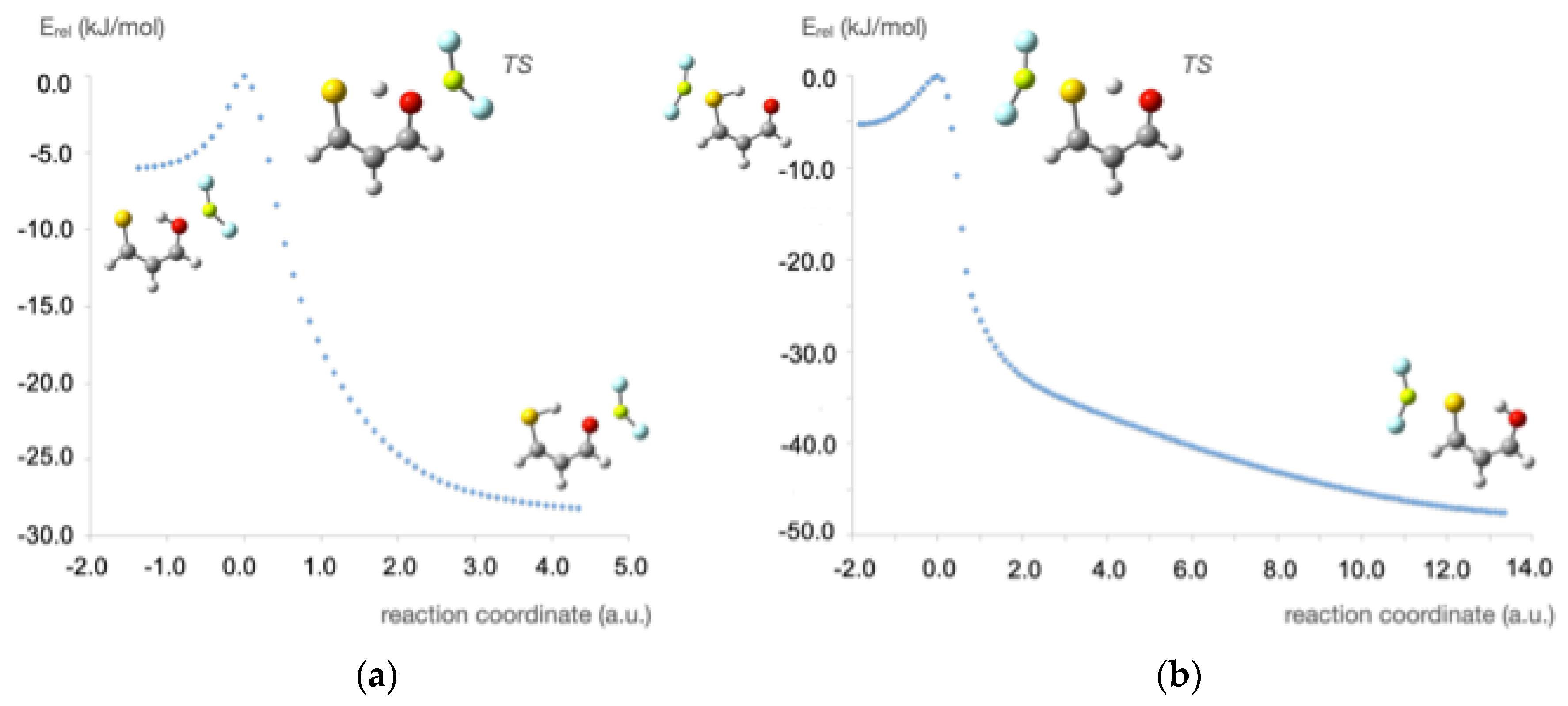
3.2. X = O, Y = S
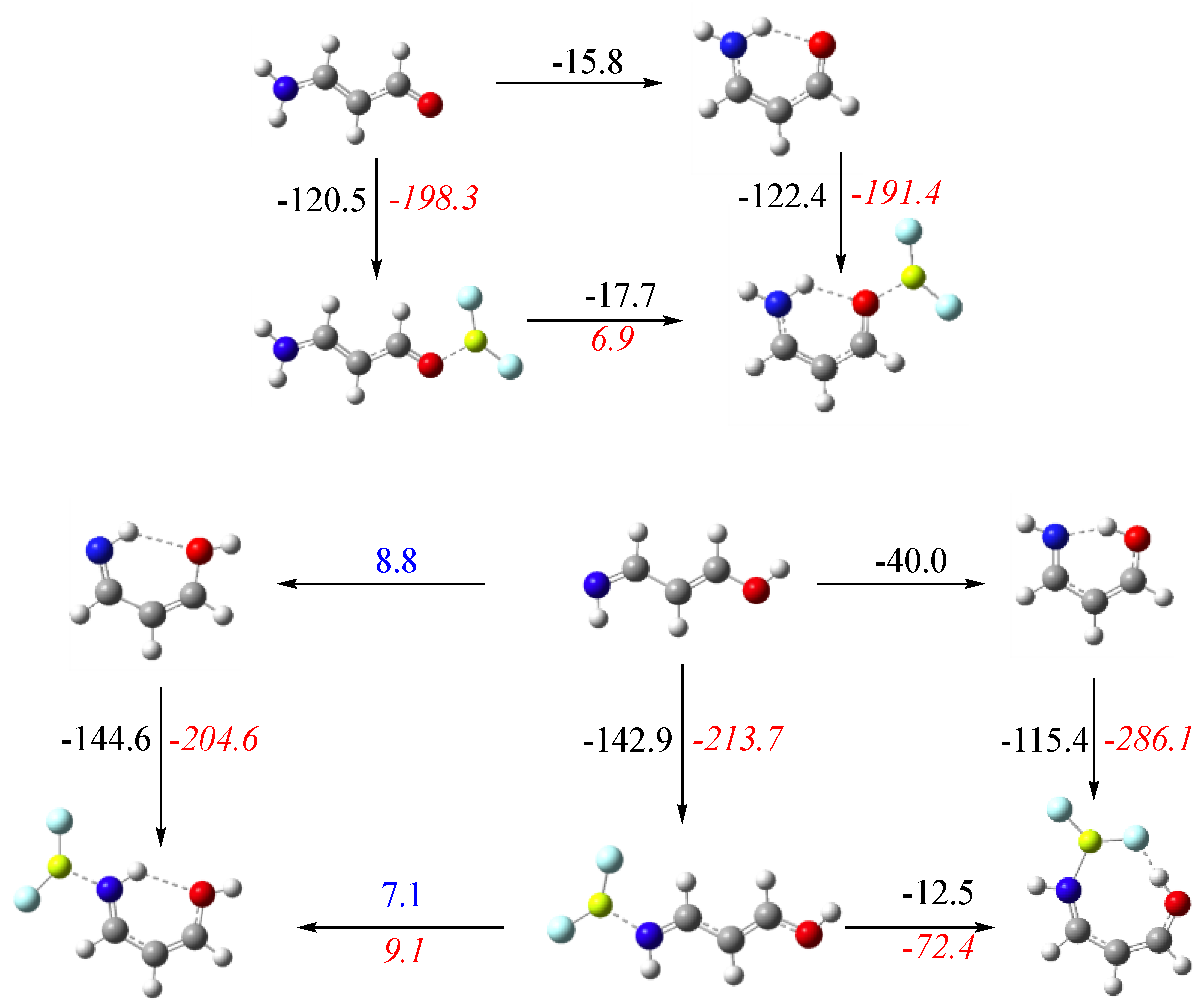
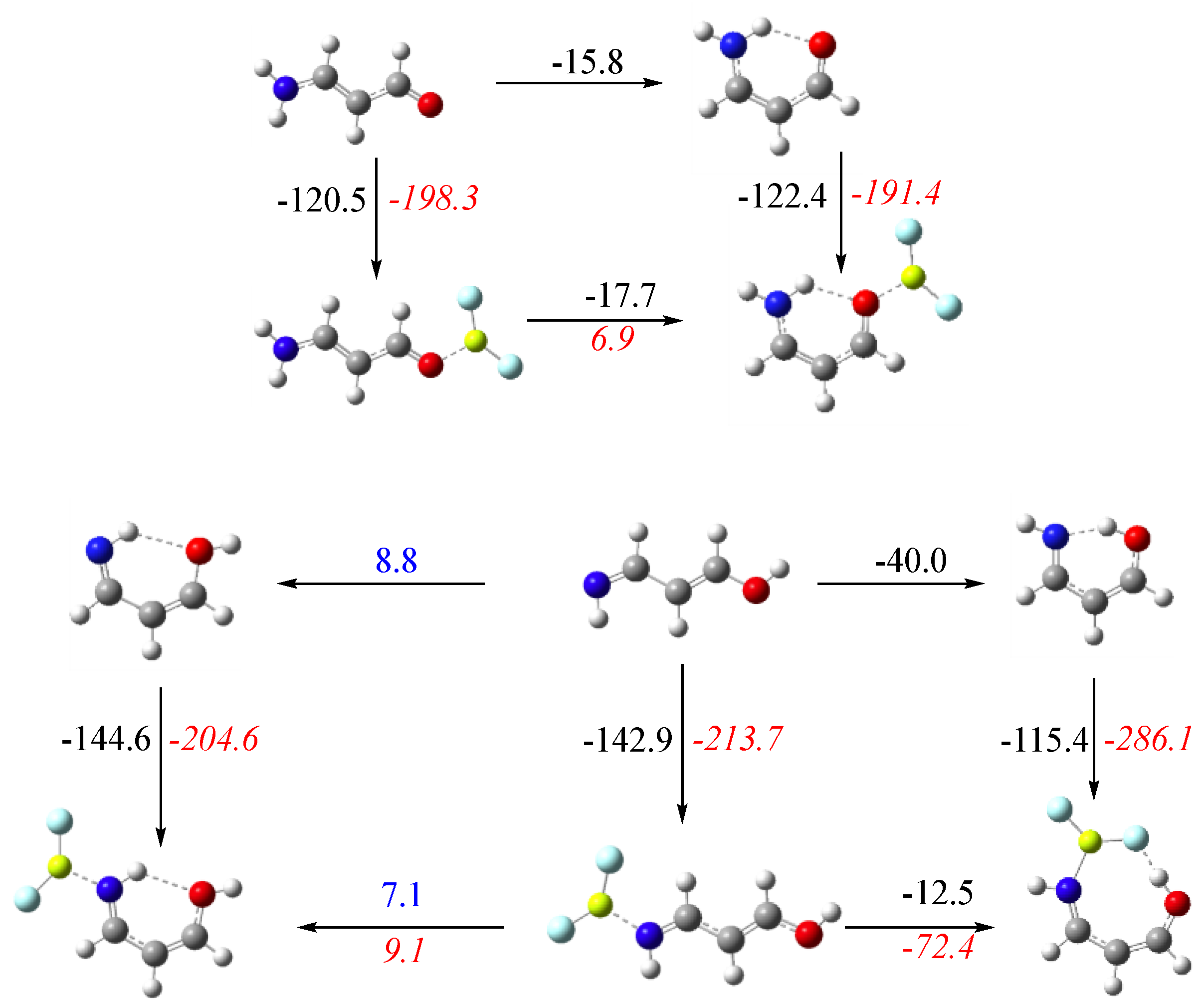
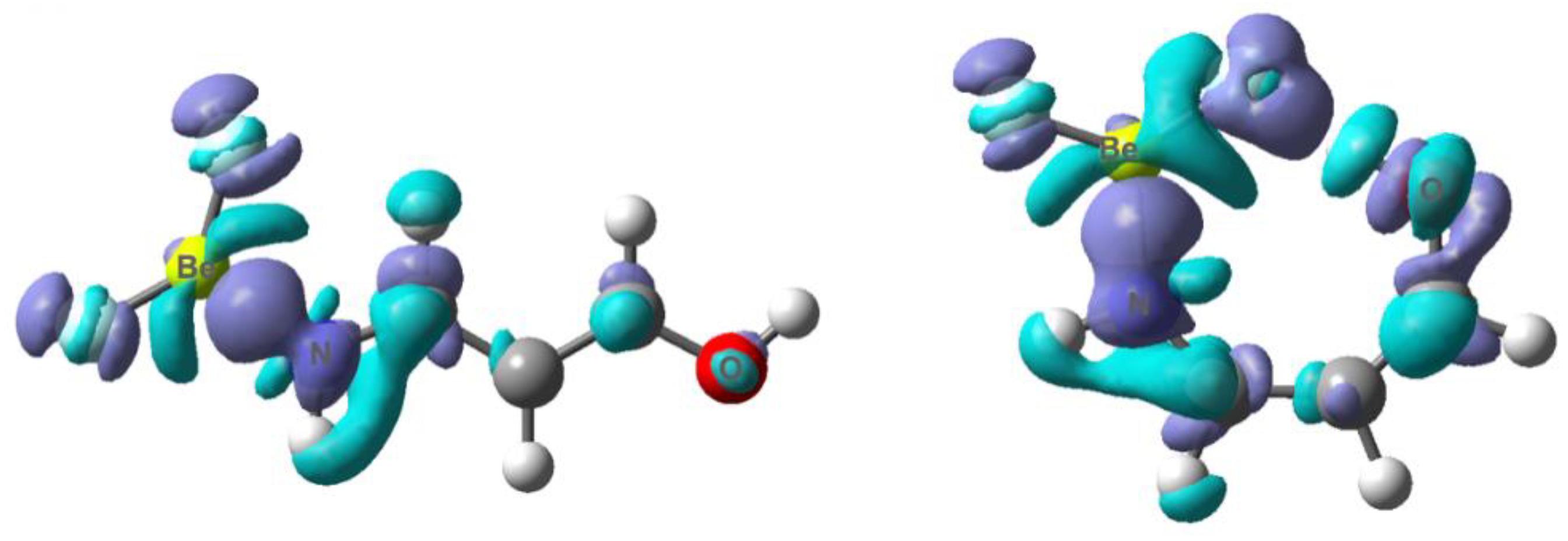
3.3. X = O, Y = NH
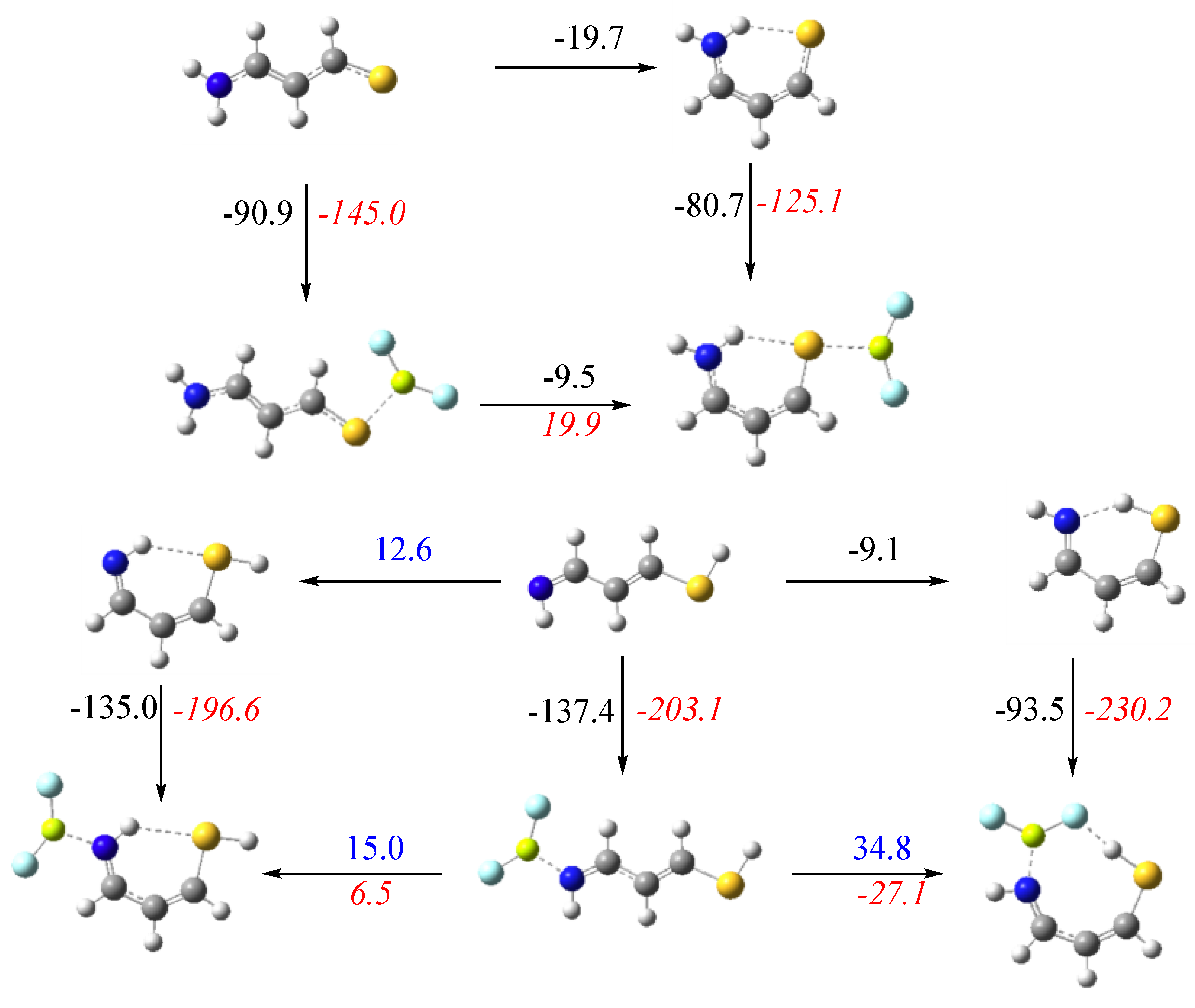
3.4. X = S, Y = NH
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Der Waals, J.D. On the Continuity of the Gaseous and Liquid State; University of Leiden: Leiden, The Netherlands, 1873. [Google Scholar]
- Pauling, L. The Structure and Entropy of Ice and of Other Crystals with Some Randomness of Atomic Arrangement. J. Am. Chem. Soc. 1935, 57, 2680–2684. [Google Scholar] [CrossRef]
- Latimer, W.M.; Rodebush, W.H. Polarity and ionization from the standpoint of the Lewis theory of valence. J. Am. Chem. Soc. 1920, 42, 1419–1433. [Google Scholar] [CrossRef] [Green Version]
- Pauling, L.; Corey, R.B.; Branson, H.R. The structure of proteins—2 Hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 1951, 37, 205–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller-Dethlefs, K.; Hobza, P. Noncovalent interactions: A challenge for experiment and theory. Chem. Rev. 2000, 100, 143–167. [Google Scholar] [CrossRef] [PubMed]
- Hobza, P.; Zahradník, R.; Müller-Dethlefs, K. The world of non-covalent interactions: 2006. Collect. Czech. Chem. Commun. 2006, 71, 443–531. [Google Scholar] [CrossRef] [Green Version]
- Hobza, P.; Müller-Dethlefs, K. Non-Covalent Interactions. Theory and Experiment; RSC Theoretical and Computational Chemistry Series 2; The Royal Society of Chemistry: Cambridge, UK, 2010. [Google Scholar]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Roza, A.O.d.l.; Dilabio, G.A. Non-Covalent Interactions in Quantum Chemistry and Physics. Theory and Applications; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Neel, A.J.; Hilton, M.J.; Sigman, M.S.; Toste, F.D. Exploiting non-covalent pi interactions for catalyst design. Nature 2017, 543, 637–646. [Google Scholar] [CrossRef]
- Davis, H.J.; Phipps, R.J. Harnessing non-covalent interactions to exert control over regioselectivity and site-selectivity in catalytic reactions. Chem. Sci. 2017, 8, 864–877. [Google Scholar] [CrossRef] [Green Version]
- Storch, G.; Trapp, O. By-design enantioselective self-amplification based on non-covalent product-catalyst interactions. Nat. Chem. 2017, 9, 179–187. [Google Scholar] [CrossRef]
- Barnberger, J.; Ostler, F.; Mancheno, O.G. Frontiers in Halogen and Chalcogen-Bond Donor Organocatalysis. ChemCatChem 2019, 11, 5198–5211. [Google Scholar] [CrossRef]
- Saper, N.I.; Ohgi, A.; Small, D.W.; Semba, K.; Nakao, Y.; Hartwig, J.F. Nickel-catalysed anti-Markovnikov hydroarylation of unactivated alkenes with unactivated arenes facilitated by non-covalent interactions. Nat. Chem. 2020, 12, 276–283. [Google Scholar] [CrossRef]
- Guerra, C.F.; Bickelhaupt, F.M.; Snijders, J.G.; Baerends, E.J. The nature of the hydrogen bond in DNA base pairs: The role of charge transfer and resonance assistance. Chem. Eur. J. 1999, 5, 3581–3594. [Google Scholar] [CrossRef]
- Guerra, C.F.; Bickelhaupt, F.M.; Snijders, J.G.; Baerends, E.J. Hydrogen bonding in DNA base pairs: Reconciliation of theory and experiment. J. Am. Chem. Soc. 2000, 122, 4117–4128. [Google Scholar] [CrossRef] [Green Version]
- Cerny, J.; Hobza, P. Non-covalent interactions in biomacromolecules. Phys. Chem. Chem. Phys. 2007, 9, 5291–5303. [Google Scholar] [CrossRef] [PubMed]
- Cockroft, S.L.; Hunter, C.A. Chemical double-mutant cycles: Dissecting non-covalent interactions. Chem. Soc. Rev. 2007, 36, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Hobza, P. Noncovalent interactions in biochemistry. WIREs Comput. Mol. Sci. 2011, 1, 3–17. [Google Scholar] [CrossRef]
- Maharramov, A.M.; Mahmudov, K.T.; Kopylovich, M.N.; Pombeiro, A.J.L. Non-covalent Interactions in the Synthesis and Design of New Compounds; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Biedermann, F.; Schneider, H.J. Experimental Binding Energies in Supramolecular Complexes. Chem. Rev. 2016, 116, 5216–5300. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.; Frangville, P.; Meyer, F. Halogen bonding for molecular recognition: New developments in materials and biological sciences. Chem. Comm. 2020, 56, 4970–4981. [Google Scholar] [CrossRef]
- Sarkar, A.; Behera, T.; Sasmal, R.; Capelli, R.; Empereur-mot, C.; Mahato, J.; Agasti, S.S.; Pavan, G.M.; Chowdhury, A.; George, S.J. Cooperative Supramolecular Block Copolymerization for the Synthesis of Functional Axial Organic Heterostructures. J. Am. Chem. Soc. 2020, 142, 11528–11539. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Aerogen Bonding Interaction: A New Supramolecular Force? Angew. Chem. Int. Ed. 2015, 54, 7340–7343. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen Bond: A Sister Noncovalent Bond to Halogen Bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef]
- Sanz, P.; Yáñez, M.; Mó, O. Competition between X···H···Y Intramolecular Hydrogen Bonds and X····Y (X = O, S, and Y = Se, Te) Chalcogen-Chalcogen Interactions. J. Phys. Chem. A 2002, 106, 4661–4668. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. Molecular Complexes between Silicon Derivatives and Electron-Rich Groups. J. Phys. Chem. A 2001, 105, 743–749. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889. [Google Scholar] [CrossRef]
- Grabowski, S.J. Tetrel bond-sigma-hole bond as a preliminary stage of the S(N)2 reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef]
- Bauzà, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef]
- Grabowski, S.J. Boron and other Triel Lewis Acid Centers: From Hypovalency to Hypervalency. ChemPhysChem 2014, 15, 2985–2993. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Del Bene, J.E.; Mó, O.; Yáñez, M. New Insights into Factors Influencing B-N Bonding in X:BH3-nFn and X:BH3-nCln for X = N-2, HCN, LiCN, H2CNH, NF3, NH3 and n = 0-3: The Importance of Deformation. Chem. Eur. J. 2010, 16, 11897–11905. [Google Scholar] [CrossRef]
- Bauzà, A.; Alkorta, I.; Elguero, J.; Mooibroek, T.J.; Frontera, A. Spodium Bonds: Noncovalent Interactions Involving Group 12 Elements. Angew. Chem. Int. Ed. 2020, 59, 17482–17487. [Google Scholar] [CrossRef] [PubMed]
- Legon, A.C.; Walker, N.R. What’s in a name? ‘Coinage-metal’ non-covalent bonds and their definition. Phys. Chem. Chem. Phys. 2018, 20, 19332–19338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Understanding Regium Bonds and their Competition with Hydrogen Bonds in Au2:HX Complexes. ChemPhysChem 2019, 20, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Regium bonds between Mn clusters (M = Cu, Ag, Au and n = 2–6) and nucleophiles NH3 and HCN. Phys. Chem. Chem. Phys. 2018, 20, 22498–22509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenlid, J.H.; Brinck, T. Extending the σ-Hole Concept to Metals: An Electrostatic Interpretation of the Effects of Nanostructure in Gold and Platinum Catalysis. J. Am. Chem. Soc. 2017, 139, 11012–11015. [Google Scholar] [CrossRef]
- Alkorta, I.; Legon, A.C. Non-Covalent Interactions Involving Alkaline-Earth Atoms and Lewis Bases B: An ab Initio Investigation of Beryllium and Magnesium Bonds, B···MR2 (M = Be or Mg, and R = H, F or CH3). Inorganics 2019, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Mó, O.; Montero-Campillo, M.M.; Alkorta, I.; Elguero, J.; Yáñez, M. Ternary Complexes Stabilized by Chalcogen and Alkaline-Earth Bonds: Crucial Role of Cooperativity and Secondary Noncovalent Interactions. Chem. Eur. J. 2019, 25, 11688–11695. [Google Scholar] [CrossRef]
- Alkorta, I.; Hill, J.G.; Legon, A.C. An ab initio investigation of alkali–metal non-covalent bonds B⋯LiR and B⋯NaR (R = F, H or CH3) formed with simple Lewis bases B: The relative inductive effects of F, H and CH3. Phys. Chem. Chem. Phys. 2020, 22, 16421–16430. [Google Scholar] [CrossRef]
- Yáñez, M.; Sanz, P.; Mó, O.; Alkorta, I.; Elguero, J. Beryllium Bonds, do they exist? J. Chem. Theor. Comput. 2009, 5, 2763–2771. [Google Scholar] [CrossRef]
- Montero-Campillo, M.M.; Mó, O.; Yáñez, M.; Alkorta, I.; Elguero, J. The Beryllium Bond. In Advances in Inorganic Chemistry; VanEldik, R., Puchta, R., Eds.; Elsevier: San Diego, CA, USA; Academic Press, Inc.: Cambridge, MA, USA, 2019; Volume 73, pp. 73–121. [Google Scholar]
- Albrecht, L.; Boyd, R.J.; Mó, O.; Yáñez, M. Changing Weak Halogen Bonds into Strong Ones through Cooperativity with Beryllium Bonds. J. Phys. Chem. A 2014, 118, 4205–4213. [Google Scholar] [CrossRef]
- McDowell, S.A.C.; Hamilton, D.S. Cooperative effects of hydrogen, halogen and beryllium bonds on model halogen-bonded FCl... YZ (YZ = BF, CO, N-2) complexes in FX‘... FCl... YZ trimers (FX‘ = FH, FCl, F2Be). Mol. Phys. 2015, 113, 1991–1997. [Google Scholar] [CrossRef]
- Yu, D.; Wu, D.; Li, Y.; Li, S.Y. On the formation of beryllium bonds where radicals act as electron donors. Theor. Chem. Acc. 2016, 135. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. On the Importance of π-Hole Beryllium Bonds: Theoretical Study and Biological Implications. Chem. Eur. J. 2017, 23, 5375–5380. [Google Scholar] [CrossRef] [PubMed]
- Casals-Sainz, J.L.; Jiménez-Grávalos, F.; Costales, A.; Francisco, E.; Pendás, A.M. Beryllium Bonding in the Light of Modern Quantum Chemical Topology Tools. J. Phys. Chem. A 2018, 122, 849–858. [Google Scholar] [CrossRef] [Green Version]
- Alikhani, M.E. Beryllium bonding: Insights from the sigma- and pi-hole analysis. J. Mol. Model. 2020, 26, 94. [Google Scholar] [CrossRef]
- Alkorta, I.; Blanco, F.; Deyà, P.M.; Elguero, J.; Estarellas, C.; Frontera, A.; Quiñonero, D. Cooperativity in multiple unusual weak bonds. Theor. Chem. Acc. 2010, 126, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Mahadevi, A.S.; Sastry, G.N. Cooperativity in Noncovalent Interactions. Chem. Rev. 2016, 116, 2775–2825. [Google Scholar] [CrossRef]
- Mó, O.; Yáñez, M.; Elguero, J. Cooperative (nonpairwise) effects in water trimers: An ab initio molecular orbital study. J. Chem. Phys. 1992, 97, 6628–6638. [Google Scholar] [CrossRef]
- Albrecht, L.; Boyd, R.J. Atomic energy analysis of cooperativity, anti-cooperativity, and non-cooperativity in small clusters of methanol, water, and formaldehyde. Comp. Theor. Chem. 2015, 1053, 328–336. [Google Scholar] [CrossRef]
- Adasme-Carreno, F.; Alzate-Morales, J.; Ireta, J. Modeling cooperative effects in halogen-bonded infinite linear chains. Phys. Chem. Chem. Phys. 2017, 19, 18529–18538. [Google Scholar] [CrossRef]
- Roman, M.; Cannizzo, C.; Pinault, T.; Isare, B.; Andrioletti, B.; van der Schoot, P.; Bouteiller, L. Supramolecular Balance: Using Cooperativity to Amplify Weak Interactions. J. Am. Chem. Soc. 2010, 132, 16818–16824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabowski, S.J. Cooperativity of hydrogen and halogen bond interactions. Theor. Chem. Acc. 2013, 132, 10. [Google Scholar] [CrossRef]
- Domagala, M.; Lutynska, A.; Palusiak, M. Halogen bond versus hydrogen bond: The many-body interactions approach. Int. J. Quant. Chem. 2017, 117, e25348. [Google Scholar] [CrossRef]
- Ciancaleoni, G. Cooperativity between hydrogen- and halogen bonds: The case of selenourea. Phys. Chem. Chem. Phys. 2018, 20, 8506–8514. [Google Scholar] [CrossRef]
- Yang, J.; Yu, Q.; Yang, F.-L.; Lu, K.; Yan, C.-X.; Dou, W.; Yang, L.; Zhou, P.-P. Competition and cooperativity of hydrogen-bonding and tetrel-bonding interactions involving triethylene diamine (DABCO), H2O and CO2 in air. New J. Chem. 2020, 44, 2328–2338. [Google Scholar] [CrossRef]
- Hou, M.C.; Zhu, Y.F.; Li, Q.Z.; Scheiner, S. Tuning the Competition between Hydrogen and Tetrel Bonds by a Magnesium Bond. Chemphyschem 2020, 21, 212–219. [Google Scholar] [CrossRef]
- Liu, M.; Yang, L.; Li, Q.; Li, W.; Cheng, J.; Xiao, B.; Yu, X. Modulating the strength of tetrel bonding through beryllium bonding. J. Mol. Model. 2016, 22, 192. [Google Scholar] [CrossRef]
- George, J.; Deringer, V.L.; Dronskowski, R. Cooperativity of Halogen, Chalcogen, and Pnictogen Bonds in Infinite Molecular Chains by Electronic Structure Theory. J. Phys. Chem. A 2014, 118, 3193–3200. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Del Bene, J.E.; Mó, O.; Montero-Campillo, M.M.; Yáñez, M. Mutual Influence of Pnicogen Bonds and Beryllium Bonds: Energies and Structures in the Spotlight. J. Phys. Chem. A 2020, 124, 5871–5878. [Google Scholar] [CrossRef]
- Albrecht, L.; Boyd, R.J.; Mó, O.; Yáñez, M. Cooperativity between hydrogen bonds and beryllium bonds in (H2O)(n)BeX2 (n=1-3, X = H, F) complexes. A new perspective. Phys. Chem. Chem. Phys. 2012, 14, 14540–14547. [Google Scholar] [CrossRef]
- Gilli, G.; Bellucci, F.; Ferretti, V.; Bertolasi, V. Evidence for resonance-assisted hydrogen-bonding from crystal-structure correlations on the enol form of the beta-diketone fragment. J. Am. Chem. Soc. 1989, 111, 1023–1028. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 theory. J. Chem. Phys. 2007, 126, 84108. [Google Scholar] [CrossRef] [PubMed]
- Moller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 0618–0622. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, R.; Pople, J.A. Approximate 4th-order perturbation-theory of electron correlation energy. Int. J. Quant. Chem. 1978, 14, 91–100. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Headgordon, M. A 5th-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Electron density shift description of non-bonding intramolecular interactions. Comput. Theor. Chem. 2012, 991, 124–133. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of pople-santry-segal cndo method to cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1088. [Google Scholar] [CrossRef]
- Iribarren, I.; Sánchez-Sanz, G.; Alkorta, I.; Elguero, J.; Trujillo, C. Evaluation of Electron Density Shifts in Noncovalent Interactions. J. Phys. Chem. A 2021, 125, 4741–4749. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Mó, O.; Yáñez, M.; Del Bene, J.E. Do coupling constants and chemical shifts provide evidence for the existence of resonance-assisted hydrogen bonds? Mol. Phys. 2004, 102, 2563–2574. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Mó, O.; Yáñez, M.; Del Bene, J.E. Are resonance-assisted hydrogen bonds ‘resonance assisted’? A theoretical NMR study. Chem. Phys. Lett. 2005, 411, 411–415. [Google Scholar] [CrossRef]
- Kurczab, R.; Mitoraj, M.P.; Michalak, A.; Ziegler, T. Theoretical Analysis of the Resonance Assisted Hydrogen Bond Based on the Combined Extended Transition State Method and Natural Orbitals for Chemical Valence Scheme. J. Phys. Chem. A 2010, 114, 8581–8590. [Google Scholar] [CrossRef] [PubMed]
- Gora, R.W.; Maj, M.; Grabowski, S.J. Resonance-assisted hydrogen bonds revisited. Resonance stabilization vs. charge delocalization. Phys. Chem. Chem. Phys. 2013, 15, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Vela, J.M.; Romero-Montalvo, E.; del Rio-Lima, A.; Martin Pendas, A.; Hernandez-Rodriguez, M.; Rinza, T.R. Hydrogen-Bond Weakening through pi Systems: Resonance-Impaired Hydrogen Bonds (RIHB). Chem. Eur. J. 2017, 23, 16605–16611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosch, A.A.; van der Lubbe, S.C.C.; Guerra, C.F. Nature of Intramolecular Resonance Assisted Hydrogen Bonding in Malonaldehyde and Its Saturated Analogue. J. Phys. Chem. A 2018, 122, 1813–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guevara-Vela, J.M.; Gallegos, M.; Valentin-Rodriguez, M.A.; Costales, A.; Rocha-Rinza, T.; Pendas, A.M. On the Relationship between Hydrogen Bond Strength and the Formation Energy in Resonance-Assisted Hydrogen Bonds. Molecules 2021, 26, 4196. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montero-Campillo, M.M.; Mó, O.; Yáñez, M. Malonaldehyde-like Systems: BeF2 Clusters—A Subtle Balance between Hydrogen Bonds, Beryllium Bonds, and Resonance. Sci 2022, 4, 7. https://doi.org/10.3390/sci4010007
Montero-Campillo MM, Mó O, Yáñez M. Malonaldehyde-like Systems: BeF2 Clusters—A Subtle Balance between Hydrogen Bonds, Beryllium Bonds, and Resonance. Sci. 2022; 4(1):7. https://doi.org/10.3390/sci4010007
Chicago/Turabian StyleMontero-Campillo, M. Merced, Otilia Mó, and Manuel Yáñez. 2022. "Malonaldehyde-like Systems: BeF2 Clusters—A Subtle Balance between Hydrogen Bonds, Beryllium Bonds, and Resonance" Sci 4, no. 1: 7. https://doi.org/10.3390/sci4010007
APA StyleMontero-Campillo, M. M., Mó, O., & Yáñez, M. (2022). Malonaldehyde-like Systems: BeF2 Clusters—A Subtle Balance between Hydrogen Bonds, Beryllium Bonds, and Resonance. Sci, 4(1), 7. https://doi.org/10.3390/sci4010007