
Multi-Omics Investigation of Fatty Acid Content Variations in Common Carp (Cyprinus carpio) Muscle: Integrating Genome, Transcriptome, and Lipid Profiling Data

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. FA Profiles Analysis of Common Carp Muscle
2.3. Genome Re-Sequencing, Genotyping, and Diversity Analysis
2.4. GWAS
2.5. Candidate Gene Identification and Enrichment Analysis
2.6. Conserved FA-Associated Genes Between Common Carp and Rainbow Trout
2.7. Identifying DEGs Related to TPUFA Content
2.8. Quantitative Real-Time PCR Validation of the Core Genes Related to the TPUFA Content
2.9. Genomic Selection for Content of Different FAs
3. Results
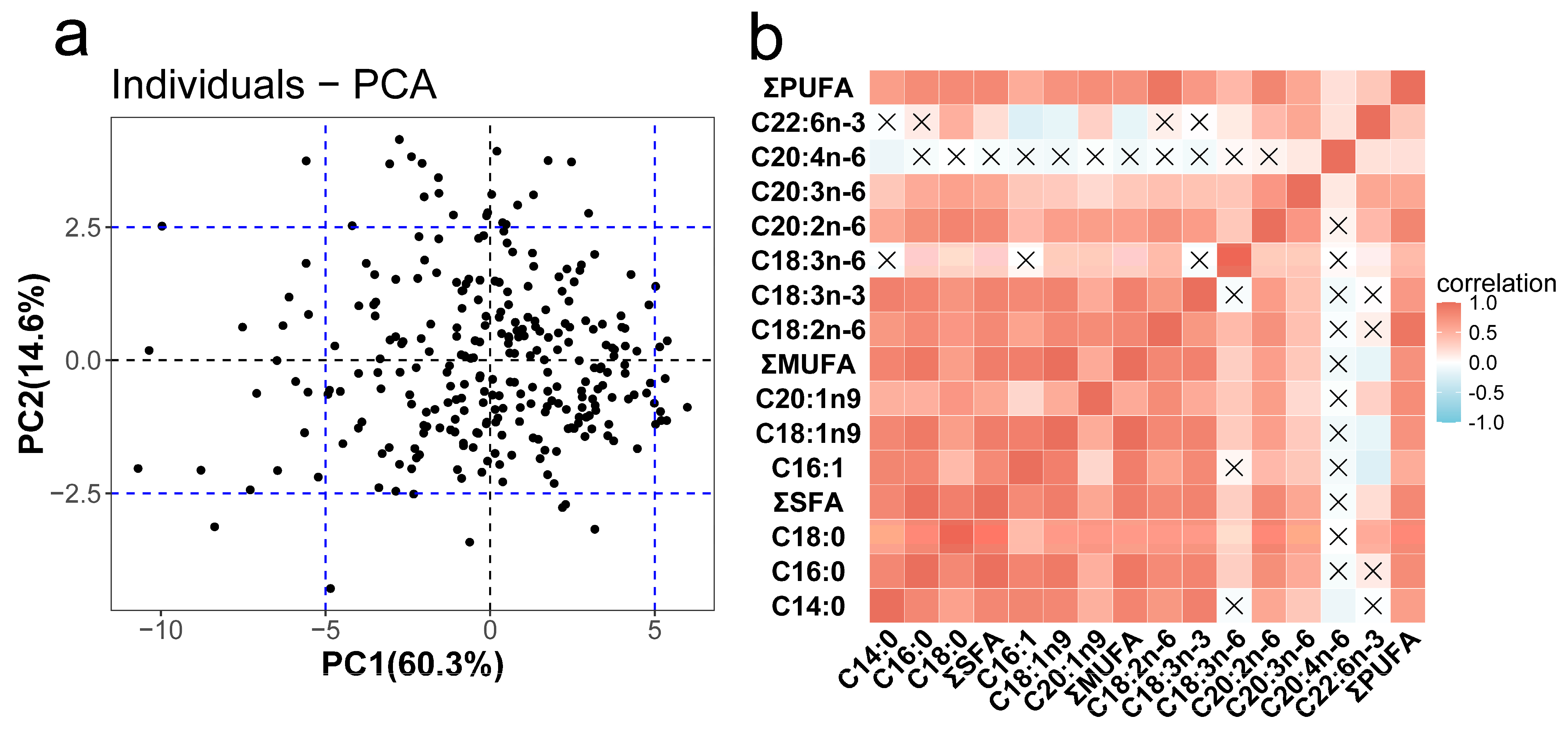
3.1. Diverse Muscular FA Contents
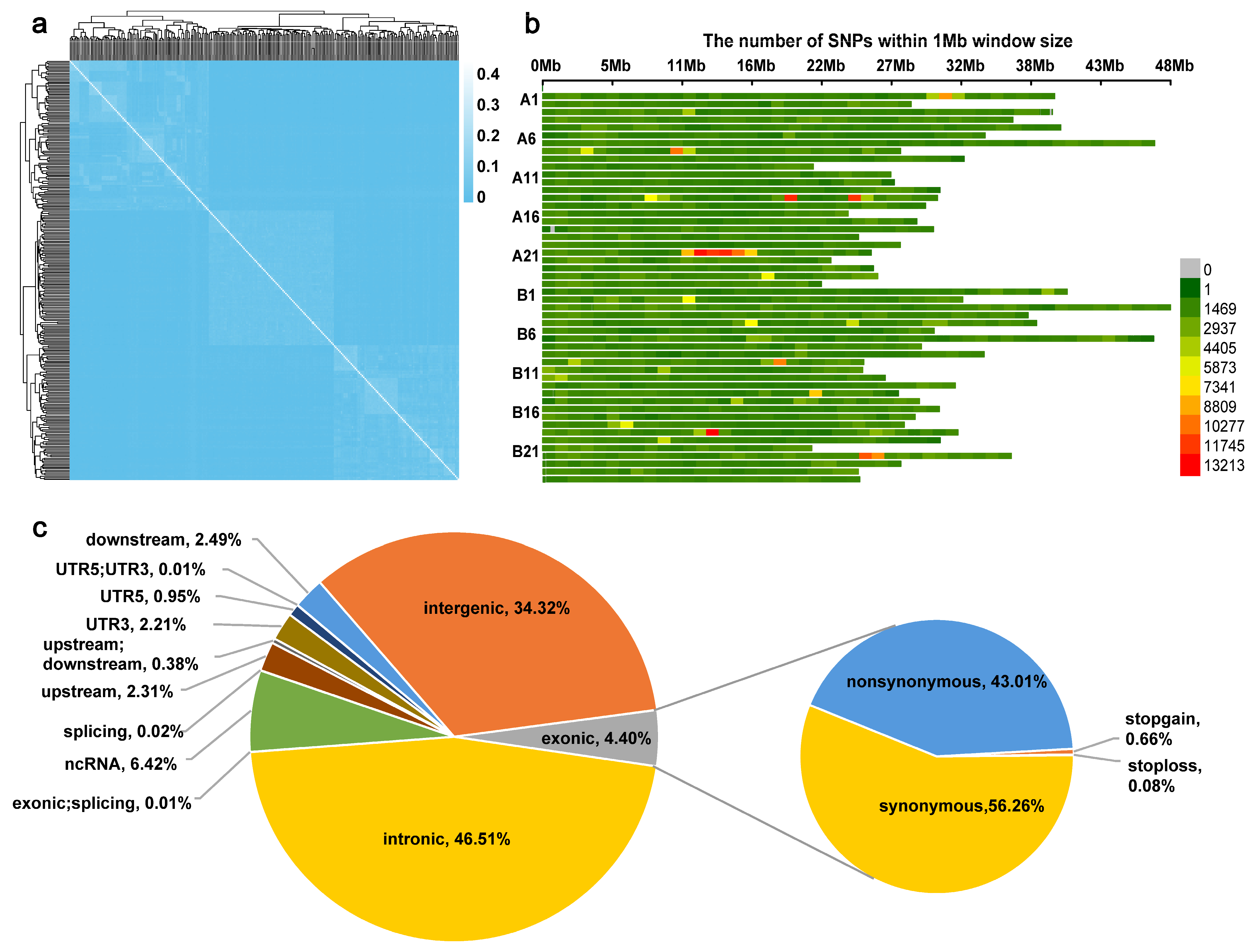
3.2. SNP Distribution Biases and Genetic Diversities
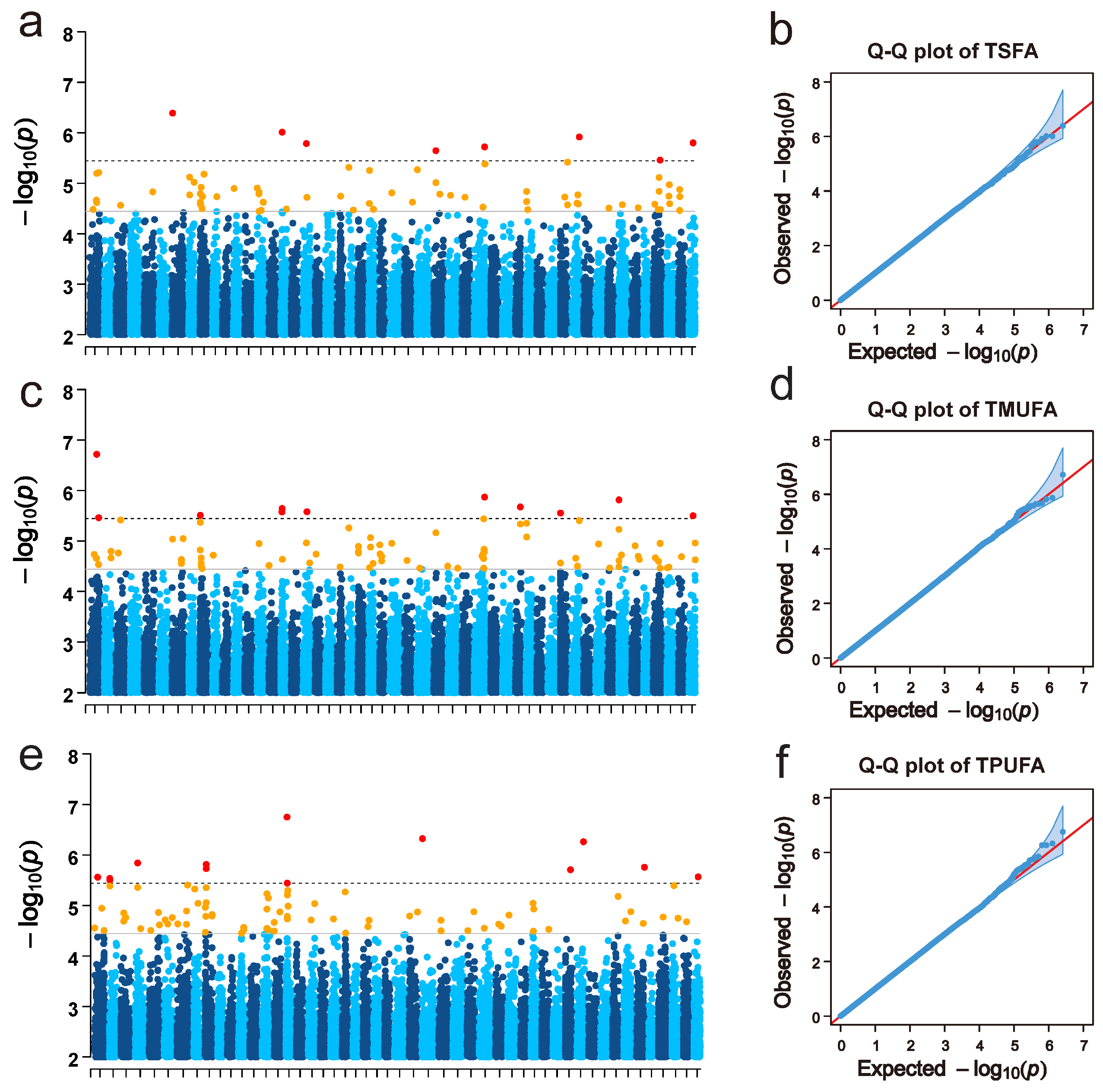
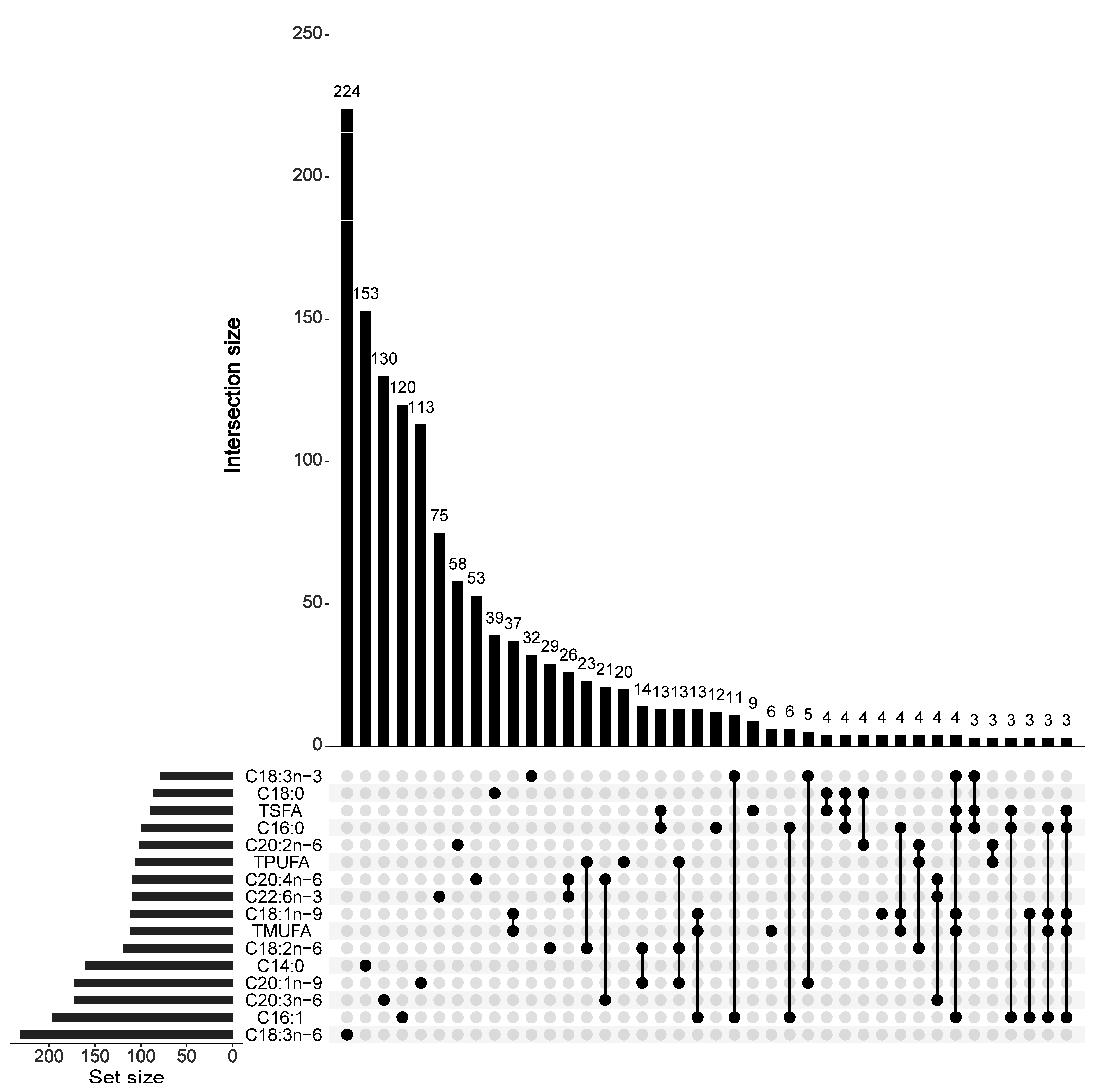
3.3. FA Content-Associated SNPs, Candidate Genes, and Inferred Functions
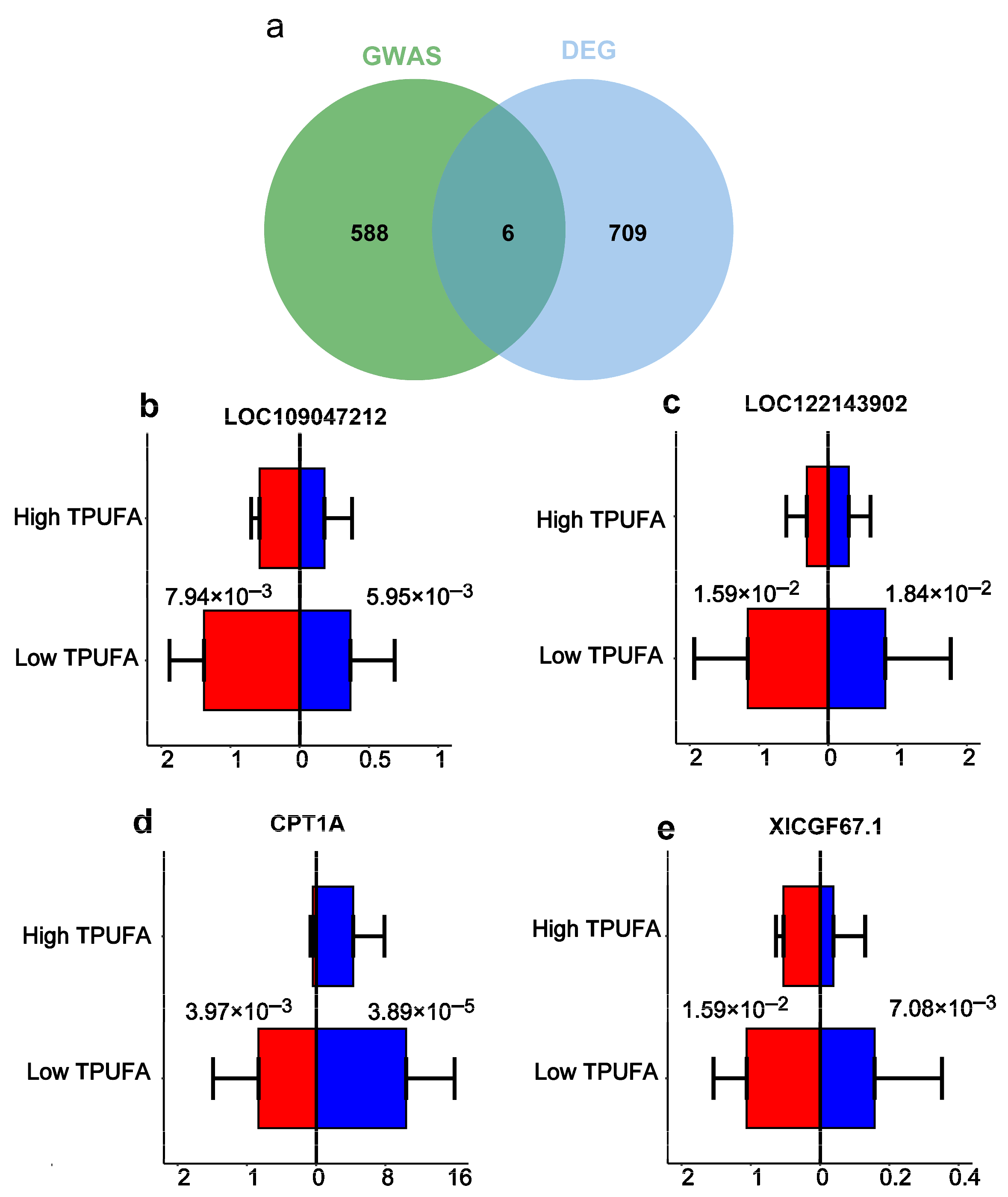
3.4. Conserved FA-Associated Genes Between Common Carp and Rainbow Trout
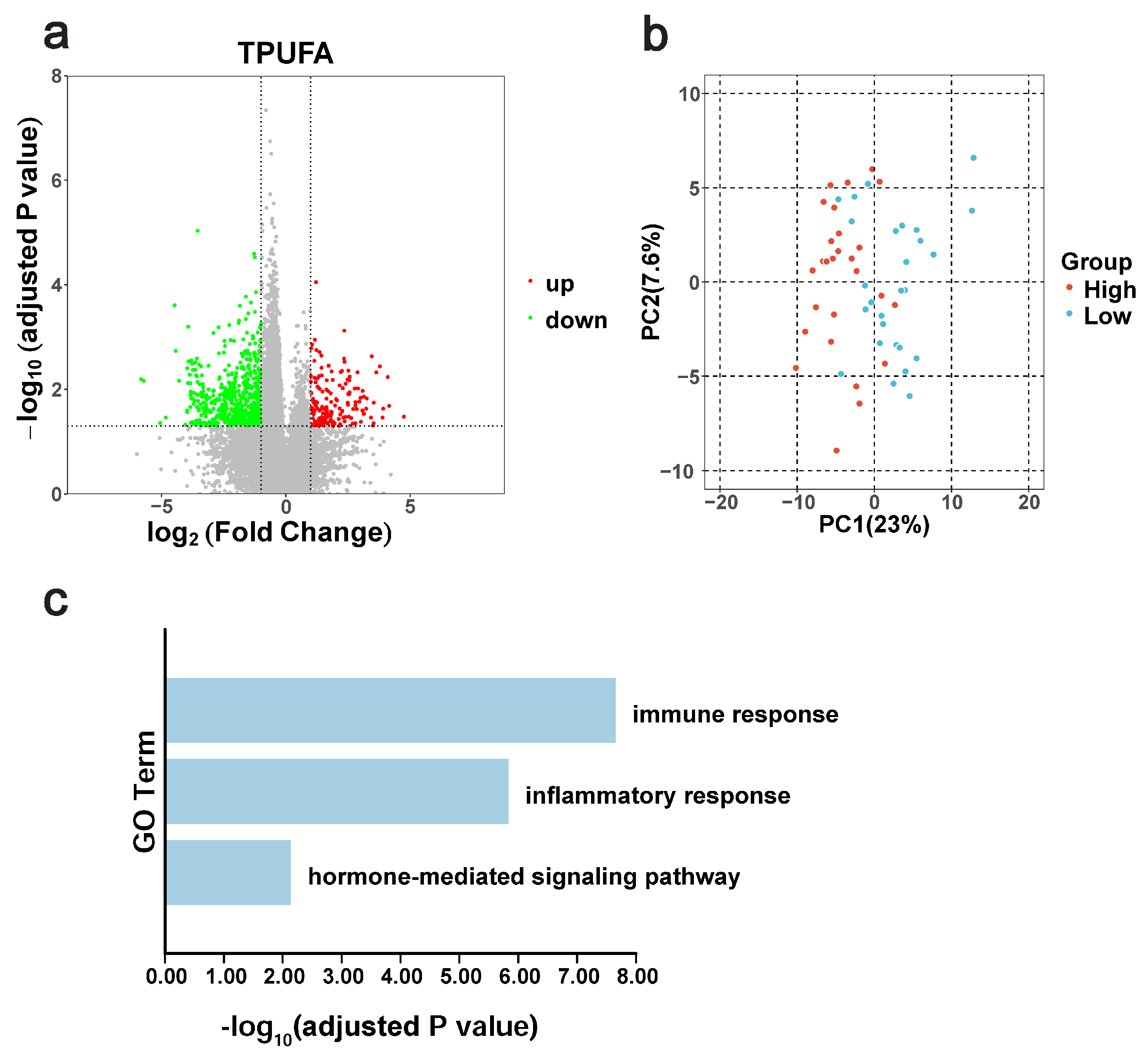
3.5. Differential Gene Expression Between the High-TPUFA and Low-TPUFA Groups
3.6. qRT-PCR Validated the Core Genes Related to TPUFA Content
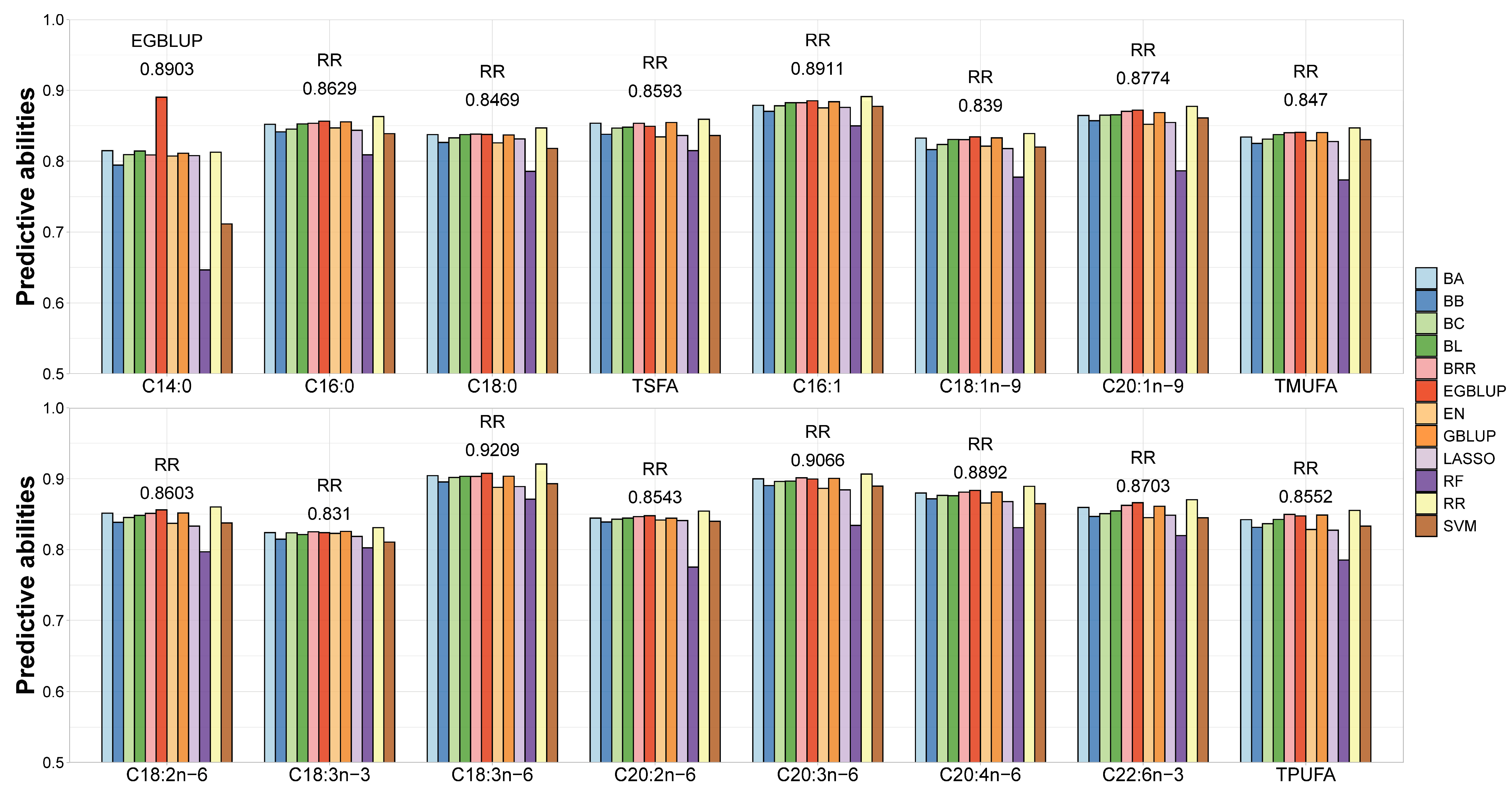
3.7. Using the Associated SNPs to Estimate FA Content
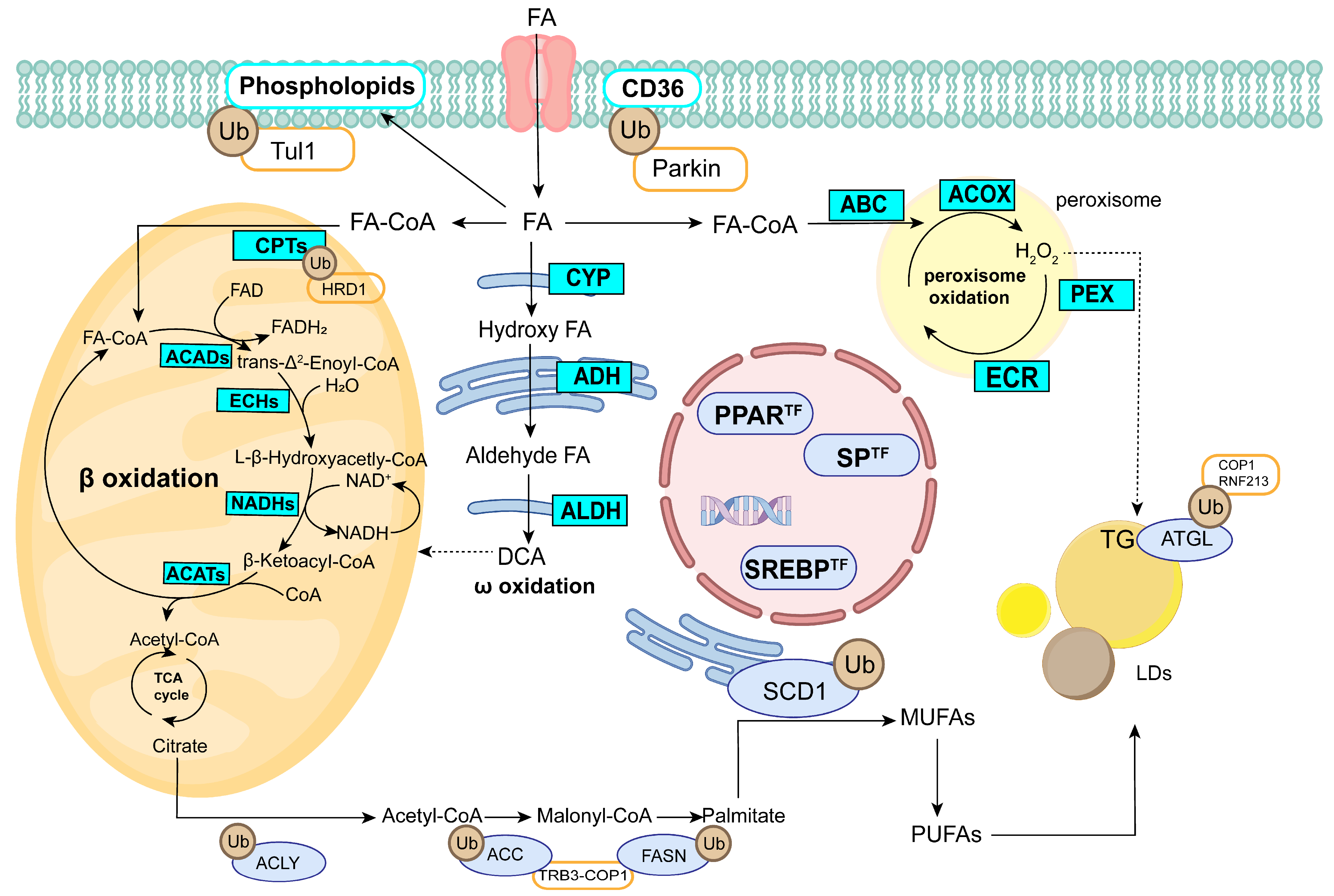
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| FAs | Fatty acids |
| PUFAs | polyunsaturated fatty acids |
| GWAS | genome-wide association studies |
| TPUFA | total PUFA |
| DEGs | differentially expressed genes |
| EPA | eicosapentaenoic acid |
| DHA | docosahexaenoic acid |
| CD36 | cluster of differentiation 36 |
| FABPs | fatty acid-binding proteins |
| FATPs | fatty acid transporter proteins |
| FFARs | free fatty acid receptors |
| SCDs | stearoyl-CoA desaturases |
| ACCs | acetyl-CoA carboxylases |
| FASNs | fatty acid synthases |
| ELOVLs | elongases of very long chain fatty acids |
| FADs | fatty acid desaturases |
| FACLs | fatty-acid-CoA ligases |
| ACSs | acyl-CoA synthetases |
| CPTs | arnitine palmitoyltransferases |
| ACADs | acyl-CoA dehydrogenases |
| ECHs | enoyl-CoA hydratases |
| HADHs | hydroxy acyl-CoA dehydrogenases |
| ACATs | acetyl-CoA acetyltransferases |
| NDEs | NAD(P)H dehydrogenases |
| ACOXs | acyl-CoA oxidases |
| ECRs | enoyl-CoA reductases |
| PEXs | peroxisomal biogenesis factors |
| ADHs | alcohol dehydrogenases |
| ALDHs | aldehyde dehydrogenases |
| PPARs | peroxisome proliferator-activated receptors |
| SREBPs | sterol regulatory element binding proteins |
| lncRNAs | long non-coding RNAs |
| MUFA | monounsaturated fatty acid |
| GS | Genomic Selection |
| TSFA | total saturated FA |
| TMUFA | total monounsaturated fatty acids |
| ncRNA | non-coding RNA |
| Ho | observed heterozygosity |
| PCs | Principal components |
| MLM | mixed linear model |
| Q-Q | quantile-quantile |
| PVE | proportion of variance explained |
| GO | Gene Ontology |
| BH | Benjamini-Hochberg |
| FPKMs | fragments per kilobase per million mapped reads |
| FDR | false discovery rate |
| qRT-PCR | quantitative real-time PCR |
| BVs | breeding values |
| GBLUP | genomic best linear unbiased prediction |
| EGBLUP | empirical best linear unbiased prediction |
| RR | ridge regression |
| EN | LASSOelastic net |
| BRR | Bayesian ridge regression |
| BL | Bayesian LASSO |
| BA | Bayes A |
| BB | Bayes B |
| RF | random forest regression |
| SVM | support vector machine |
| BC | Bayes C |
| SD | standard deviation |
| MSEP | Mean squared error of prediction |
| PCA | Principal component analysis |
| GPCR | G protein-coupled receptor |
| ROS | response to reactive oxygen species |
| ARP2 | microfilament-associated proteins 2 |
| ARP3 | microfilament-associated proteins 3 |
| ALOX5 | polyunsaturated fatty acid 5-lipoxygenase |
| NLR | NOD-like Receptor |
| CARD | Caspase recruitment domain |
| CPT1A | carnitine O-palmitoyltransferase 1, liver isoform-like |
| MYLKA | myosin, light chain kinase a |
| UPS | ubiquitin proteasome system |
| ATGLs | adipose triglyceride lipases |
| ACLY | ATP-citrate lyase |
References
- Panov, A.V.; Mayorov, V.I.; Dikalov, S.I. Role of Fatty Acids Β-Oxidation in the Metabolic Interactions between Organs. Int. J. Mol. Sci. 2024, 25, 12740. [Google Scholar] [CrossRef] [PubMed]
- Casares, D.; Escribá, P.V.; Rosselló, C.A. Membrane Lipid Composition: Effect on Membrane and Organelle Structure, Function and Compartmentalization and Therapeutic Avenues. Int. J. Mol. Sci. 2019, 20, 2167. [Google Scholar] [CrossRef] [PubMed]
- Graber, R.; Sumida, C.; Nunez, E.A. Fatty Acids and Cell Signal Transduction. J. Lipid Mediat. Cell Signal. 1994, 9, 91–116. [Google Scholar] [PubMed]
- Kapoor, B.; Kapoor, D.; Gautam, S.; Singh, R.; Bhardwaj, S. Dietary Polyunsaturated Fatty Acids (Pufas): Uses and Potential Health Benefits. Curr. Nutr. Rep. 2021, 10, 232–242. [Google Scholar] [CrossRef]
- De Smet, S. Meat, Poultry, and Fish Composition: Strategies for Optimizing Human Intake of Essential Nutrients. Anim. Front. 2012, 2, 10–16. [Google Scholar] [CrossRef]
- Tocher, D.R. Omega-3 Long-Chain Polyunsaturated Fatty Acids and Aquaculture in Perspective. Aquaculture 2015, 449, 94–107. [Google Scholar] [CrossRef]
- Carr, I.; Glencross, B.; Santigosa, E. The Importance of Essential Fatty Acids and Their Ratios in Aquafeeds to Enhance Salmonid Production, Welfare, and Human Health. Front. Anim. Sci. 2023, 4, 1147081. [Google Scholar] [CrossRef]
- Li, X.; Liu, C.; Zhang, R.; Li, Y.; Ye, D.; Wang, H.; He, M.; Sun, Y. Biosynthetic Deficiency of Docosahexaenoic Acid Causes Nonalcoholic Fatty Liver Disease and Ferroptosis-Mediated Hepatocyte Injury. J. Biol. Chem. 2024, 300, 107405. [Google Scholar] [CrossRef]
- Samovski, D.; Jacome-Sosa, M.; Abumrad, N.A. Fatty Acid Transport and Signaling: Mechanisms and Physiological Implications. Annu. Rev. Physiol. 2023, 85, 317–337. [Google Scholar] [CrossRef]
- Storch, J.; Thumser, A.E. The Fatty Acid Transport Function of Fatty Acid-Binding Proteins. Biochim. Biophys. Acta 2000, 1486, 28–44. [Google Scholar] [CrossRef]
- Gimeno, R.E. Fatty Acid Transport Proteins. Curr. Opin. Lipidol. 2007, 18, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Kashihara, D.; Ichimura, A.; Kimura, I.; Tsujimoto, G.; Hirasawa, A. Role of Free Fatty Acid Receptors in the Regulation of Energy Metabolism. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2014, 1841, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Loix, M.; Zelcer, N.; Bogie, J.F.J.; Hendriks, J.J.A. The Ubiquitous Role of Ubiquitination in Lipid Metabolism. Trends Cell Biol. 2024, 34, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yu, H.; Gao, R.; Liu, M.; Xie, W. A Comprehensive Review of the Family of Very-Long-Chain Fatty Acid Elongases: Structure, Function, and Implications in Physiology and Pathology. Eur. J. Med. Res. 2023, 28, 532. [Google Scholar] [CrossRef]
- Los, D.A.; Murata, N. Structure and Expression of Fatty Acid Desaturases. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1998, 1394, 3–15. [Google Scholar] [CrossRef]
- Houten, S.M.; Wanders, R.J. A General Introduction to the Biochemistry of Mitochondrial Fatty Acid Β-Oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef]
- Kerner, J.; Hoppel, C. Fatty Acid Import into Mitochondria. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2000, 1486, 1–17. [Google Scholar] [CrossRef]
- Kashyap, I.; Deb, R.; Battineni, A.; Nagotu, S. Acyl Coa Oxidase: From Its Expression, Structure, Folding, and Import to Its Role in Human Health and Disease. Mol. Genet. Genom. 2023, 298, 1247–1260. [Google Scholar] [CrossRef]
- Poirier, Y.; Antonenkov, V.D.; Glumoff, T.; Hiltunen, J.K. Peroxisomal Β-Oxidation—A Metabolic Pathway with Multiple Functions. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2006, 1763, 1413–1426. [Google Scholar] [CrossRef]
- Pratama, A.M.; Sharma, M.; Naidu, S.; Bömmel, H.; Prabhuswamimath, S.C.; Madhusudhan, T.; Wihadmadyatami, H.; Bachhuka, A.; Karnati, S. Peroxisomes and Ppars: Emerging Role as Master Regulators of Cancer Metabolism. Mol. Metab. 2024, 90, 102044. [Google Scholar] [CrossRef]
- Miura, Y. The Biological Significance of Ω-Oxidation of Fatty Acids. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2013, 89, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-H.; Wen, R.; Yang, N.; Zhang, T.-N.; Liu, C.-F. Roles of Protein Post-Translational Modifications in Glucose and Lipid Metabolism: Mechanisms and Perspectives. Mol. Med. 2023, 29, 93. [Google Scholar] [CrossRef]
- Zhu, Y.; Lin, X.; Zhou, X.; Prochownik, E.V.; Wang, F.; Li, Y. Posttranslational Control of Lipogenesis in the Tumor Microenvironment. J. Hematol. Oncol. 2022, 15, 120. [Google Scholar] [CrossRef]
- Zhu, J.; Sun, Y.; Luo, J.; Wu, M.; Li, J.; Cao, Y. Specificity Protein 1 Regulates Gene Expression Related to Fatty Acid Metabolism in Goat Mammary Epithelial Cells. Int. J. Mol. Sci. 2015, 16, 1806–1820. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, L.l.C.; Siersbæk, M.; Mandrup, S. Ppars: Fatty Acid Sensors Controlling Metabolism. Semin. Cell Dev. Biol. 2012, 23, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Jeon, T.-I.; Osborne, T.F. Srebps: Metabolic Integrators in Physiology and Metabolism. Trends Endocrinol. Metab. 2012, 23, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Ahonen, M.A.; Taskinen, J.H.; Latorre, J.; Ortega, F.J.; Pa, N.H.; Olkkonen, V. Microrna-221-3p Alters Human Adipocyte Lipid Storage by Regulating Glucose and Lipid Metabolism. Atherosclerosis 2020, 315, e99. [Google Scholar] [CrossRef]
- Zhou, Q.; Kong, D.; Li, W.; Shi, Z.; Liu, Y.; Sun, R.; Ma, X.; Qiu, C.; Liu, Z.; Hou, Y.; et al. Lncrna Hoxb-As3 Binding to Ptbp1 Protein Regulates Lipid Metabolism by Targeting Srebp1 in Endometrioid Carcinoma. Life Sci. 2023, 320, 121512. [Google Scholar] [CrossRef]
- Xu, P.; Zhang, X.; Wang, X.; Li, J.; Liu, G.; Kuang, Y.; Xu, J.; Zheng, X.; Ren, L.; Wang, G.; et al. Genome Sequence and Genetic Diversity of the Common Carp, Cyprinus carpio. Nat. Genet. 2014, 46, 1212–1219. [Google Scholar] [CrossRef]
- Jiang, X.; Li, Y.; Tian, J.; Li, C.; Ge, Y.; Hu, X.; Cheng, L.; Shi, X.; Shi, L.; Jia, Z. Nutritional Components, Biochemical Characteristics, Enzyme Activities, and Growth Differences of Five Freshwater Fish Species? Fishes 2022, 7, 285. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Kim, W.K.; Bae, K.-H.; Lee, S.C.; Lee, E.-W. Lipid Metabolism and Ferroptosis. Biology 2021, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Yang, C.-R.; Wang, Y.-X.; Xu, Z.-M.; Li, S.-Q.; Li, J.-C.; Sun, X.-Q.; Wang, H.-W.; Wang, Q.; Zhang, Y.; et al. Fads2b Plays a Dominant Role in ∆6/∆5 Desaturation Activities Compared with Fads2a in Common Carp (Cyprinus carpio). Int. J. Mol. Sci. 2023, 24, 10638. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Wang, Y.X.; Yang, C.R.; Li, S.Q.; Li, J.C.; Sun, X.Q.; Wang, H.W.; Wang, Q.; Zhang, Y.; Li, J.T. Dominant Elongase Activity of Elovl5a but Higher Expression of Elovl5b in Common Carp (Cyprinus carpio). Int. J. Mol. Sci. 2022, 23, 14666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, X.Q.; Ye, Y.Q.; Wang, Q.; Li, Q.S.; Zhao, R.; Wang, H.W.; Li, J.T. Association between the Polymorphisms of Fads2a and Fads2b and Poly-Unsaturated Fatty Acids in Common Carp (Cyprinus carpio). Animals 2021, 11, 1780. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Z.M.; Wang, Q.; Li, Q.S.; Sun, X.Q.; Li, J.T. The Promoter Snps Were Associated with Both the Contents of Poly-Unsaturated Fatty Acids (Pufas) and the Expressions of Pufa-Related Genes in Common Carp. Biology 2023, 12, 524. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Q.S.; Ye, Y.Q.; Wang, Q.; Sun, X.Q.; Zhao, R.; Li, J.T. Association Analysis between Genetic Variants of Elovl5a and Elovl5b and Poly-Unsaturated Fatty Acids in Common Carp (Cyprinus carpio). Biology 2022, 11, 466. [Google Scholar] [CrossRef]
- R Core Team, R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014; Volume 1.
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, J.-T.; Wang, Q.; Huang Yang, M.-D.; Li, Q.-S.; Cui, M.-S.; Dong, Z.-J.; Wang, H.-W.; Yu, J.-H.; Zhao, Y.-J.; Yang, C.-R.; et al. Parallel Subgenome Structure and Divergent Expression Evolution of Allo-Tetraploid Common Carp and Goldfish. Nat. Genet. 2021, 53, 1493–1503. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of Samtools and Bcftools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A Mapreduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. Plink: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. Annovar: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and Vcftools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-Wide Efficient Mixed-Model Analysis for Association Studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef]
- Zhang, W.; Bin, Y.; Zhang, J.; Cui, L.; Ma, J.; Chen, C.; Ai, H.; Xiao, S.; Ren, J.; Huang, L. Genome-Wide Association Studies for Fatty Acid Metabolic Traits in Five Divergent Pig Populations. Sci. Rep. 2016, 6, 24718. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. Rmvp: A Memory-Efficient, Visualization-Enhanced, and Parallel-Accelerated Tool for Genome-Wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef]
- Shim, H.; Chasman, D.I.; Smith, J.D.; Mora, S.; Ridker, P.M.; Nickerson, D.A.; Krauss, R.M.; Stephens, M. A Multivariate Genome-Wide Association Analysis of 10 Ldl Subfractions, and Their Response to Statin Treatment, in 1868 Caucasians. PLoS ONE 2015, 10, e0120758. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. Bedtools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. Tbtools-Ii: A “One for All, All for One” Bioinformatics Platform for Biological Big-Data Mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 2018, 57, 289–300. [Google Scholar] [CrossRef]
- Blay, C.; Haffray, P.; D’Ambrosio, J.; Prado, E.; Dechamp, N.; Nazabal, V.; Bugeon, J.; Enez, F.; Causeur, D.; Eklouh-Molinier, C.; et al. Genetic Architecture and Genomic Selection of Fatty Acid Composition Predicted by Raman Spectroscopy in Rainbow Trout. BMC Genom. 2021, 22, 788. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One Fastq Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with Hisat2 and Hisat-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. Stringtie Enables Improved Reconstruction of a Transcriptome from Rna-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for Rna-Seq Data with Deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ginestet, C. Ggplot2: Elegant Graphics for Data Analysis. J. R. Stat. Soc. Ser. A Stat. Soc. 2011, 174, 245–246. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative Pcr and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Mann, H.B.; Whitney, D.R. On a Test of Whether One of Two Random Variables Is Stochastically Larger Than the Other. Ann. Math. Stat. 1947, 18, 50–60. [Google Scholar] [CrossRef]
- Charmet, G.; Tran, L.-G.; Auzanneau, J.; Rincent, R.; Bouchet, S. Bwgs: A R Package for Genomic Selection and Its Application to a Wheat Breeding Programme. PLoS ONE 2020, 15, e0222733. [Google Scholar] [CrossRef] [PubMed]
- Endelman, J.B. Ridge Regression and Other Kernels for Genomic Selection with R Package Rrblup. Plant Genome 2011, 4. [Google Scholar] [CrossRef]
- Zhuang, Z.; Wu, J.; Qiu, Y.; Ruan, D.; Ding, R.; Xu, C.; Zhou, S.; Zhang, Y.; Liu, Y.; Ma, F.; et al. Improving the Accuracy of Genomic Prediction for Meat Quality Traits Using Whole Genome Sequence Data in Pigs. J. Anim. Sci. Biotechnol. 2023, 14, 67. [Google Scholar] [CrossRef]
- Talukdar, S.; Olefsky, J.M.; Osborn, O. Targeting Gpr120 and Other Fatty Acid-Sensing Gpcrs Ameliorates Insulin Resistance and Inflammatory Diseases. Trends Pharmacol. Sci. 2011, 32, 543–550. [Google Scholar] [CrossRef]
- Oteng, A.-B.; Liu, L. Gpcr-Mediated Effects of Fatty Acids and Bile Acids on Glucose Homeostasis. Front. Endocrinol. 2023, 14, 1206063. [Google Scholar] [CrossRef]
- Alvarez-Curto, E.; Milligan, G. Metabolism Meets Immunity: The Role of Free Fatty Acid Receptors in the Immune System. Biochem. Pharmacol 2016, 114, 3–13. [Google Scholar] [CrossRef]
- Paintlia, A.S.; Paintlia, M.K.; Singh, A.K.; Singh, I. Modulation of Rho-Rock Signaling Pathway Protects Oligodendrocytes against Cytokine Toxicity Via Ppar-A-Dependent Mechanism. Glia 2013, 61, 1500–1517. [Google Scholar] [CrossRef]
- Jahani, V.; Kavousi, A.; Mehri, S.; Karimi, G. Rho Kinase, a Potential Target in the Treatment of Metabolic Syndrome. Biomed. Pharmacother. 2018, 106, 1024–1030. [Google Scholar] [CrossRef]
- Hasegawa, K.; Takenaka, N.; Yamamoto, M.; Sakoda, Y.; Aiba, A.; Satoh, T. Regulation of De Novo Lipid Synthesis by the Small Gtpase Rac1 in the Adipogenic Differentiation of Progenitor Cells from Mouse White Adipose Tissue. Int. J. Mol. Sci. 2023, 24, 4608. [Google Scholar] [CrossRef]
- Chan, M.P.; Takenaka, N.; Abe, Y.; Satoh, T. Insulin-Stimulated Translocation of the Fatty Acid Transporter Cd36 to the Plasma Membrane Is Mediated by the Small Gtpase Rac1 in Adipocytes. Cell. Signal. 2024, 117, 111102. [Google Scholar] [CrossRef] [PubMed]
- Enkler, L.; Szentgyörgyi, V.; Pennauer, M.; Prescianotto-Baschong, C.; Riezman, I.; Wiesyk, A.; Avraham, R.E.; Spiess, M.; Zalckvar, E.; Kucharczyk, R.; et al. Arf1 Coordinates Fatty Acid Metabolism and Mitochondrial Homeostasis. Nat. Cell Biol. 2023, 25, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Han, H.; Wu, X.; Wu, J.; Ren, Z. Arp2/3 Regulates Fatty Acid Synthesis by Modulating Lipid Droplets’ Motility. Int. J. Mol. Sci. 2022, 23, 8730. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shao, L.; Pan, C.; Ye, J.; Ding, Z.; Wu, J.; Du, Q.; Ren, Y.; Zhu, C. Elevated Level of Mitochondrial Reactive Oxygen Species Via Fatty Acid Β-Oxidation in Cancer Stem Cells Promotes Cancer Metastasis by Inducing Epithelial-Mesenchymal Transition. Stem Cell Res. Ther. 2019, 10, 175. [Google Scholar] [CrossRef]
- Antollini, S.S.; Barrantes, F.J. Fatty Acid Regulation of Voltage- and Ligand-Gated Ion Channel Function. Front. Physiol. 2016, 7, 573. [Google Scholar] [CrossRef]
- Klotz, L.O.; Steinbrenner, H. Cellular Adaptation to Xenobiotics: Interplay between Xenosensors, Reactive Oxygen Species and Foxo Transcription Factors. Redox Biol. 2017, 13, 646–654. [Google Scholar] [CrossRef]
- Weng, L.; Tang, W.-S.; Wang, X.; Gong, Y.; Liu, C.; Hong, N.-N.; Tao, Y.; Li, K.-Z.; Liu, S.-N.; Jiang, W.; et al. Surplus Fatty Acid Synthesis Increases Oxidative Stress in Adipocytes and Induces Lipodystrophy. Nat. Commun. 2024, 15, 133. [Google Scholar] [CrossRef]
- Ferreira, A.V.; Alarcon-Barrera, J.C.; Domínguez-Andrés, J.; Bulut, Ö.; Kilic, G.; Debisarun, P.A.; Röring, R.J.; Özhan, H.N.; Terschlüsen, E.; Ziogas, A.; et al. Fatty Acid Desaturation and Lipoxygenase Pathways Support Trained Immunity. Nat. Commun. 2023, 14, 7385. [Google Scholar] [CrossRef]
- Phelan, N.; O’Connor, A.; Kyaw Tun, T.; Correia, N.; Boran, G.; Roche, H.M.; Gibney, J. Hormonal and Metabolic Effects of Polyunsaturated Fatty Acids in Young Women with Polycystic Ovary Syndrome: Results from a Cross-Sectional Analysis and a Randomized, Placebo-Controlled, Crossover Trial. Am. J. Clin. Nutr. 2011, 93, 652–662. [Google Scholar] [CrossRef]
- Paul, R.; Luo, M.; Mo, X.; Lu, J.; Yeo, S.K.; Guan, J.-L. Fak Activates Akt-Mtor Signaling to Promote the Growth and Progression of Mmtv-Wnt1-Driven Basal-Like Mammary Tumors. Breast Cancer Res. 2020, 22, 59. [Google Scholar] [CrossRef]
- Su, W.Y.; Tian, L.Y.; Guo, L.P.; Huang, L.Q.; Gao, W.Y. Pi3k Signaling-Regulated Metabolic Reprogramming: From Mechanism to Application. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188952. [Google Scholar] [CrossRef] [PubMed]
- Mock, T.S.; Francis, D.S.; Jago, M.K.; Glencross, B.D.; Smullen, R.P.; Turchini, G.M. Endogenous Biosynthesis of N-3 Long-Chain Pufa in Atlantic Salmon. Br. J. Nutr. 2019, 121, 1108–1123. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Shimizu, Y.; Tanaka, A.; Nogi, T.; Tabuchi, I.; Oyama, K.; Taniguchi, M.; Mannen, H.; Sasazaki, S. The Snp in the Promoter Region of the Bovine Elovl5 Gene Influences Economic Traits Including Subcutaneous Fat Thickness. Mol. Biol. Rep. 2013, 40, 3231–3237. [Google Scholar] [CrossRef]
- Zhao, L.; Li, F.; Liu, T.; Yuan, L.; Zhang, X.; Zhang, D.; Li, X.; Zhang, Y.; Zhao, Y.; Song, Q.; et al. Ovine Elovl5 and Fasn Genes Polymorphisms and Their Correlations with Sheep Tail Fat Deposition. Gene 2022, 807, 145954. [Google Scholar] [CrossRef] [PubMed]
- Casteels, M.; Foulon, V.; Mannaerts, G.P.; Van Veldhoven, P.P. Alpha-Oxidation of 3-Methyl-Substituted Fatty Acids and Its Thiamine Dependence. Eur. J. Biochem. 2003, 270, 1619–1627. [Google Scholar] [CrossRef]
- Xu, M.; Tan, J.; Dong, W.; Zou, B.; Teng, X.; Zhu, L.; Ge, C.; Dai, X.; Kuang, Q.; Zhong, S.; et al. The E3 Ubiquitin-Protein Ligase Trim31 Alleviates Non-Alcoholic Fatty Liver Disease by Targeting Rhbdf2 in Mouse Hepatocytes. Nat. Commun. 2022, 13, 1052. [Google Scholar] [CrossRef]
- Zou, Y.; Zhang, Y.; Li, M.; Cao, K.; Song, C.; Zhang, Z.; Cai, K.; Geng, D.; Chen, S.; Wu, Y.; et al. Regulation of Lipid Metabolism by E3 Ubiquitin Ligases in Lipid-Associated Metabolic Diseases. Int. J. Biol. Macromol. 2024, 265, 130961. [Google Scholar] [CrossRef]
- Chen, S.; Hu, Z.; He, H.; Liu, X. Fatty Acid Elongase7 Is Regulated Via Sp1 and Is Involved in Lipid Accumulation in Bovine Mammary Epithelial Cells. J. Cell. Physiol. 2018, 233, 4715–4725. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, J.; Dong, Y.; Yin, Z.; Li, Y.; Liu, Y.; You, C.; Monroig, Ó.; Tocher, D.R.; Wang, S. Sp1 Is Involved in Vertebrate Lc-Pufa Biosynthesis by Upregulating the Expression of Liver Desaturase and Elongase Genes. Int. J. Mol. Sci. 2019, 20, 5066. [Google Scholar] [CrossRef]
- Zong, Y.; Wang, X.; Cui, B.; Xiong, X.; Wu, A.; Lin, C.; Zhang, Y. Decoding the Regulatory Roles of Non-Coding Rnas in Cellular Metabolism and Disease. Mol. Ther. 2023, 31, 1562–1576. [Google Scholar] [CrossRef]
- Datsomor, A.K.; Olsen, R.E.; Zic, N.; Madaro, A.; Bones, A.M.; Edvardsen, R.B.; Wargelius, A.; Winge, P. Crispr/Cas9-Mediated Editing of Δ5 and Δ6 Desaturases Impairs Δ8-Desaturation and Docosahexaenoic Acid Synthesis in Atlantic Salmon (Salmo Salar L.). Sci. Rep. 2019, 9, 16888. [Google Scholar] [CrossRef] [PubMed]
- Hallerman, E.; Bredlau, J.; Camargo, L.S.A.; Dagli, M.L.Z.; Karembu, M.; Kovich, D.; Muia, A.N.; Murrone, M.L.; Rocha-Salavarrieta, P.J.; Romero-Aldemita, R.; et al. Enabling Regulatory Policy Globally Will Promote Realization of the Potential of Animal Biotechnology. CABI Agric. Biosci. 2024, 5, 1–28. [Google Scholar] [CrossRef]
- Alemu, A.; Åstrand, J.; Montesinos-López, O.; Sánchez, J.; Fernández-Gónzalez, J.; Tadesse, W.; Vetukuri, R.; Carlsson, A.; Ceplitis, A.; Crossa, J.; et al. Genomic Selection in Plant Breeding: Key Factors Shaping Two Decades of Progress. Mol. Plant 2024, 17, 552–578. [Google Scholar] [CrossRef]
- Yáñez, J.M.; Barría, A.; López, M.E.; Moen, T.; Garcia, B.F.; Yoshida, G.M.; Xu, P. Genome-Wide Association and Genomic Selection in Aquaculture. Rev. Aquac. 2023, 15, 645–675. [Google Scholar] [CrossRef]
- He, R.; Xue, H.; Pan, W. Statistical Power of Transcriptome-Wide Association Studies. Genet. Epidemiol. 2022, 46, 572–588. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean | SD | Min | Max | CV | |
|---|---|---|---|---|---|
| C14:0 | 0.17 | 0.15 | 0.00 | 2.20 | 0.89 |
| C16:0 | 3.80 | 1.51 | 0.91 | 10.10 | 0.40 |
| C18:0 | 1.18 | 0.45 | 0.24 | 3.13 | 0.38 |
| TSFA | 5.19 | 1.99 | 1.21 | 12.88 | 0.38 |
| C16:1 | 0.53 | 0.35 | 0.01 | 2.01 | 0.66 |
| C18:1n-9 | 6.89 | 3.15 | 1.65 | 18.11 | 0.46 |
| C20:1n-9 | 0.27 | 0.15 | 0.01 | 0.93 | 0.55 |
| TMUFA | 7.82 | 3.52 | 2.02 | 20.28 | 0.45 |
| C18:2n-6 | 4.66 | 1.96 | 0.02 | 11.31 | 0.42 |
| C18:3n-3 | 0.30 | 0.15 | 0.00 | 0.71 | 0.50 |
| C18:3n-6 | 0.10 | 0.07 | 0.00 | 0.47 | 0.68 |
| C20:2n-6 | 0.13 | 0.05 | 0.00 | 0.37 | 0.42 |
| C20:3n-6 | 0.25 | 0.15 | 0.00 | 1.01 | 0.58 |
| C20:4n-6 | 0.57 | 0.40 | 0.00 | 2.25 | 0.69 |
| C22:6n-3 | 0.48 | 0.44 | 0.00 | 2.38 | 0.92 |
| TPUFA | 6.48 | 2.50 | 1.13 | 14.01 | 0.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, K.; Wang, Q.; Chen, Y.; Cao, Y.; Zhao, R.; Yu, S.; Sun, X.; Zhang, Y.; Li, J. Multi-Omics Investigation of Fatty Acid Content Variations in Common Carp (Cyprinus carpio) Muscle: Integrating Genome, Transcriptome, and Lipid Profiling Data. Fishes 2025, 10, 234. https://doi.org/10.3390/fishes10050234
Wang K, Wang Q, Chen Y, Cao Y, Zhao R, Yu S, Sun X, Zhang Y, Li J. Multi-Omics Investigation of Fatty Acid Content Variations in Common Carp (Cyprinus carpio) Muscle: Integrating Genome, Transcriptome, and Lipid Profiling Data. Fishes. 2025; 10(5):234. https://doi.org/10.3390/fishes10050234
Chicago/Turabian StyleWang, Kaikuo, Qi Wang, Yingjie Chen, Yiming Cao, Ran Zhao, Shuangting Yu, Xiaoqing Sun, Yan Zhang, and Jiongtang Li. 2025. "Multi-Omics Investigation of Fatty Acid Content Variations in Common Carp (Cyprinus carpio) Muscle: Integrating Genome, Transcriptome, and Lipid Profiling Data" Fishes 10, no. 5: 234. https://doi.org/10.3390/fishes10050234
APA StyleWang, K., Wang, Q., Chen, Y., Cao, Y., Zhao, R., Yu, S., Sun, X., Zhang, Y., & Li, J. (2025). Multi-Omics Investigation of Fatty Acid Content Variations in Common Carp (Cyprinus carpio) Muscle: Integrating Genome, Transcriptome, and Lipid Profiling Data. Fishes, 10(5), 234. https://doi.org/10.3390/fishes10050234