Comparison of Single-Ion Conducting Polymer Gel Electrolytes for Sodium, Potassium, and Calcium Batteries: Influence of Polymer Chemistry, Cation Identity, Charge Density, and Solvent on Conductivity


Abstract
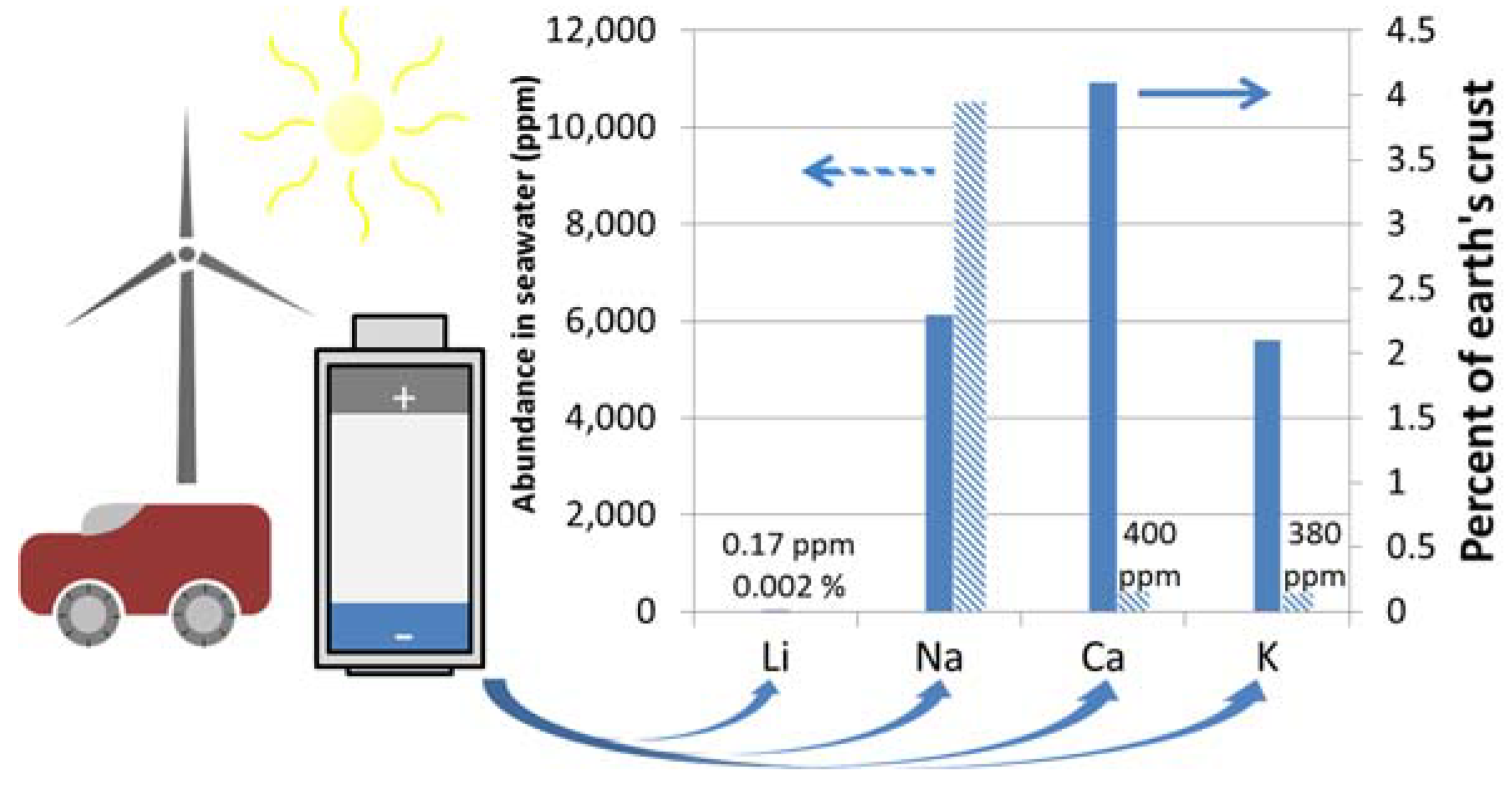
:1. Introduction
2. Results and Discussion
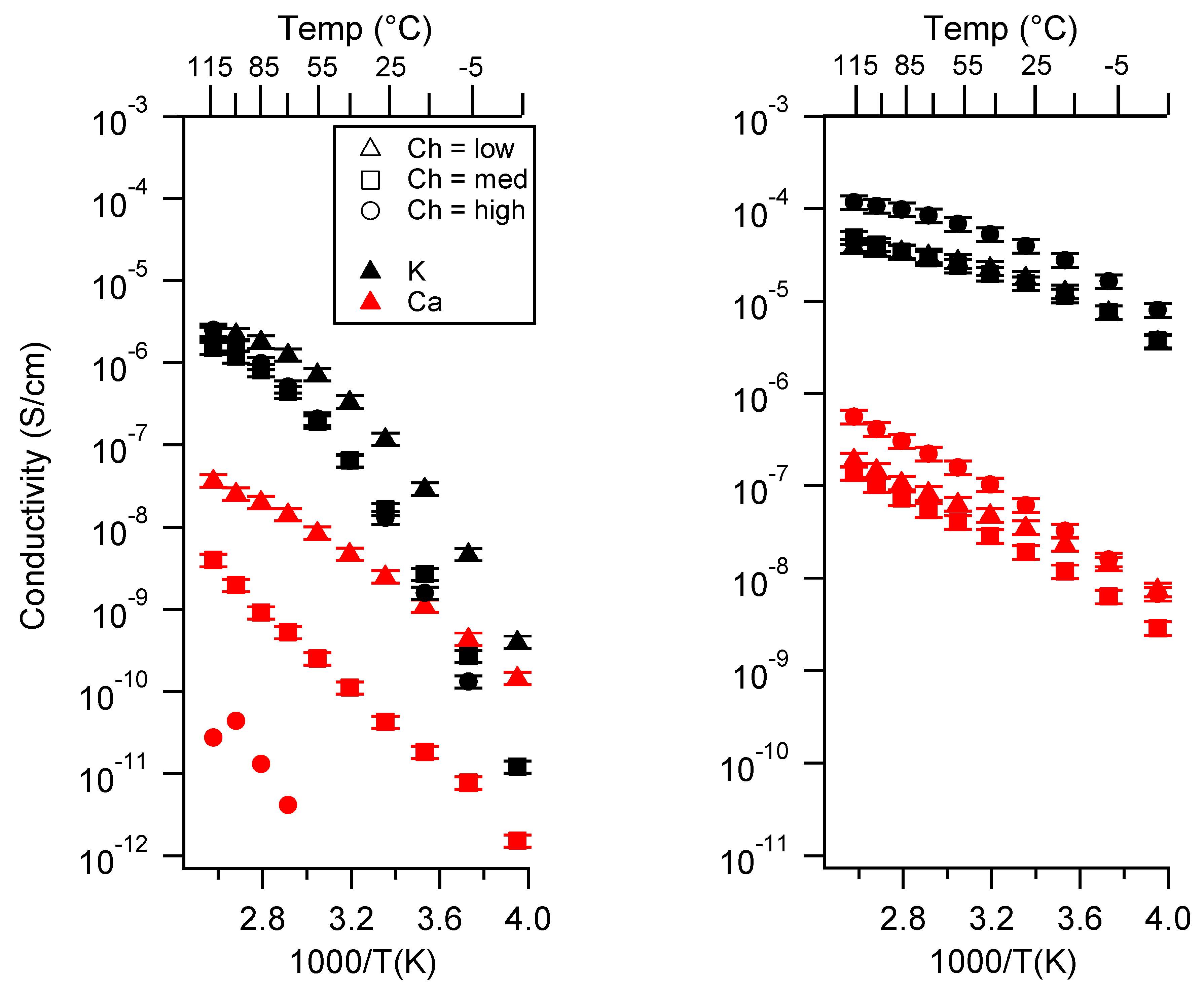
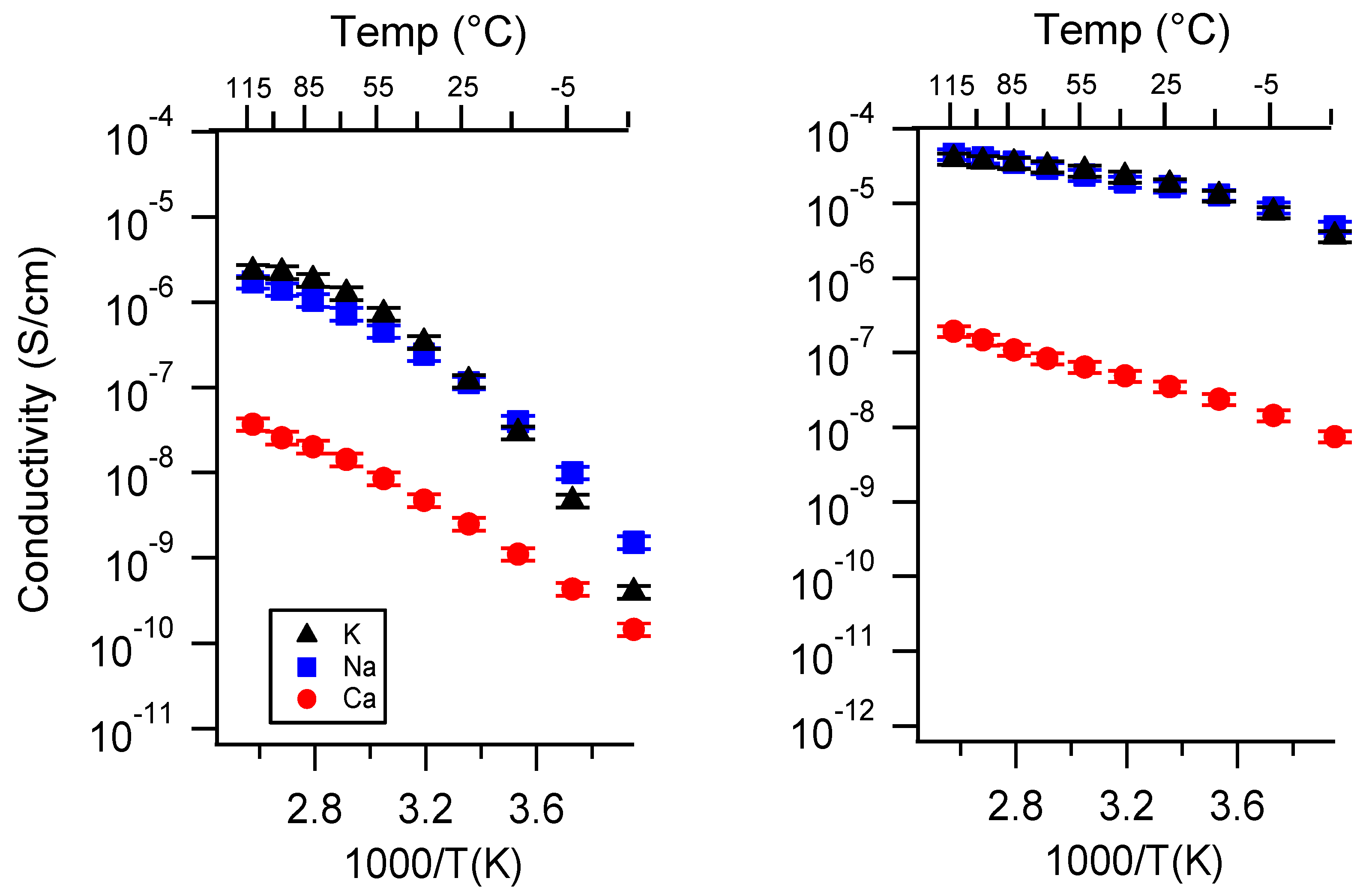
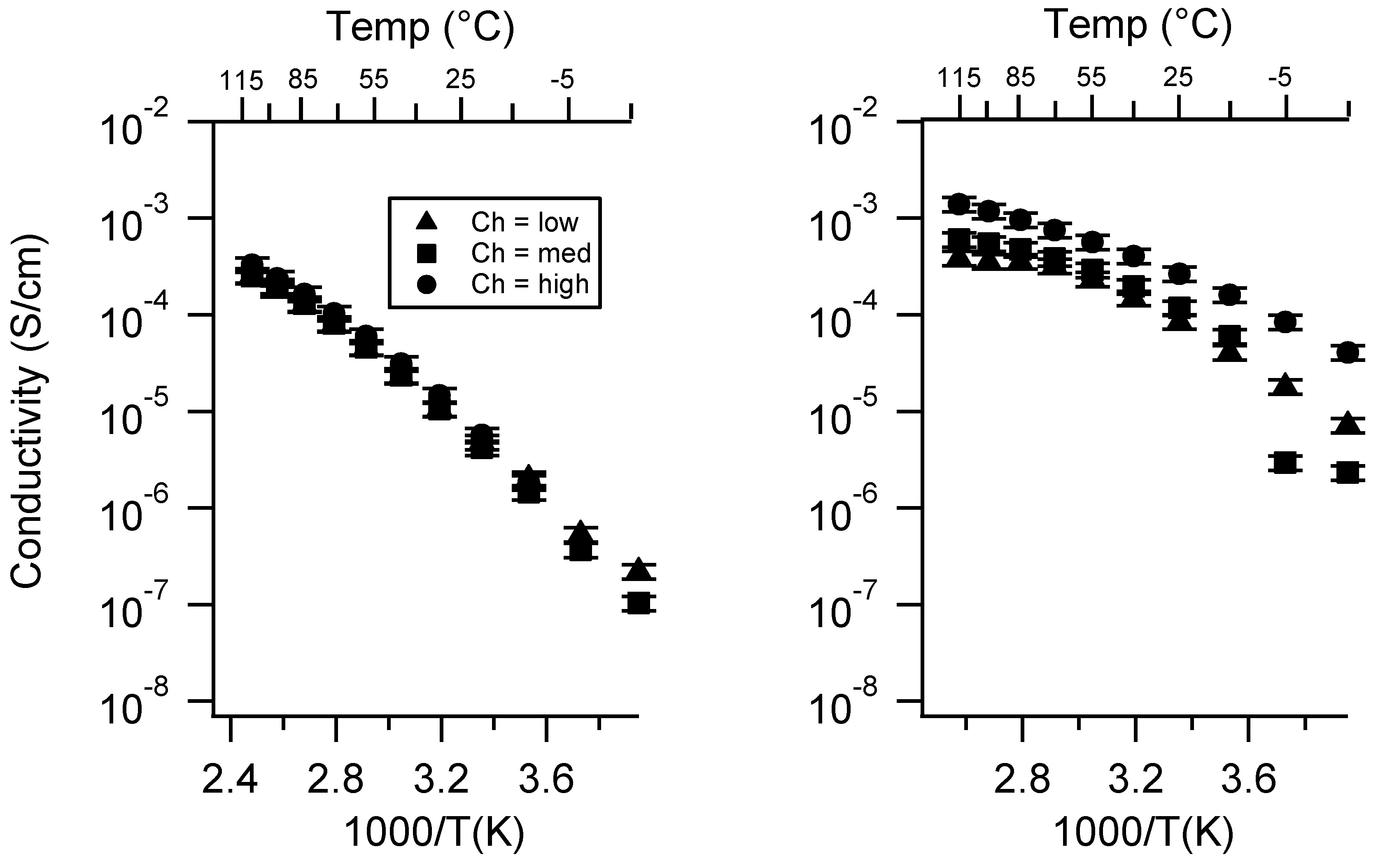
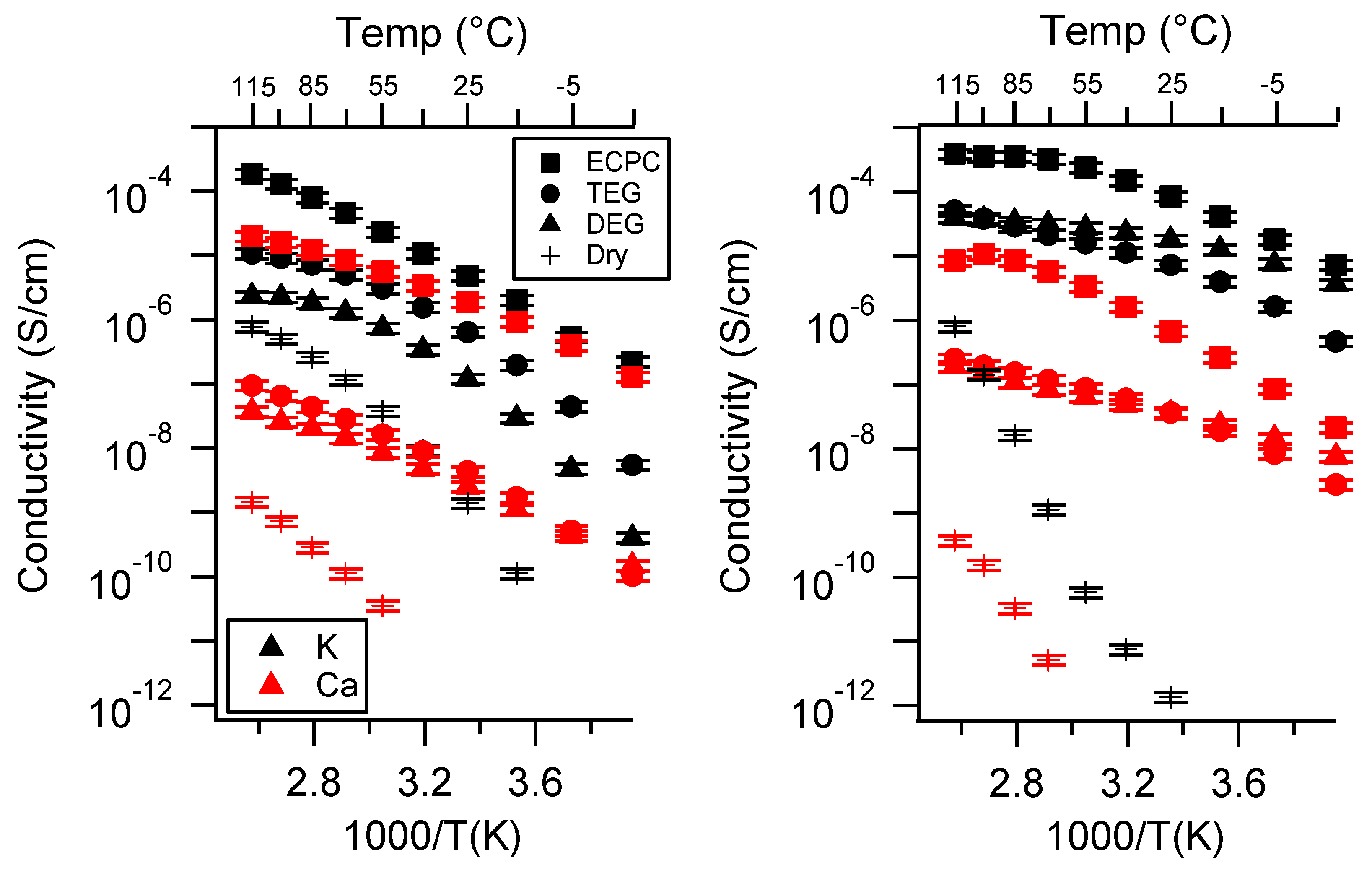
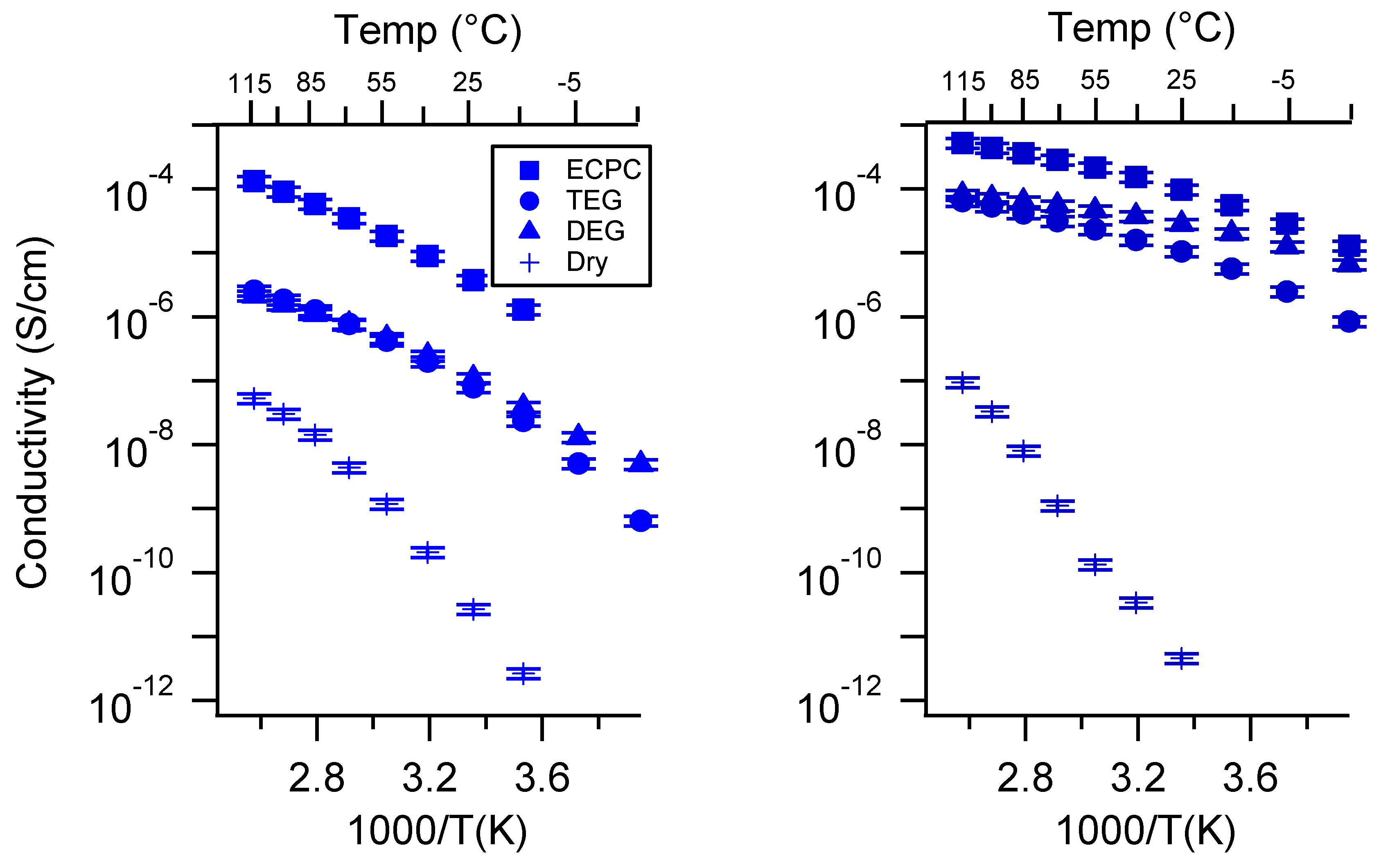
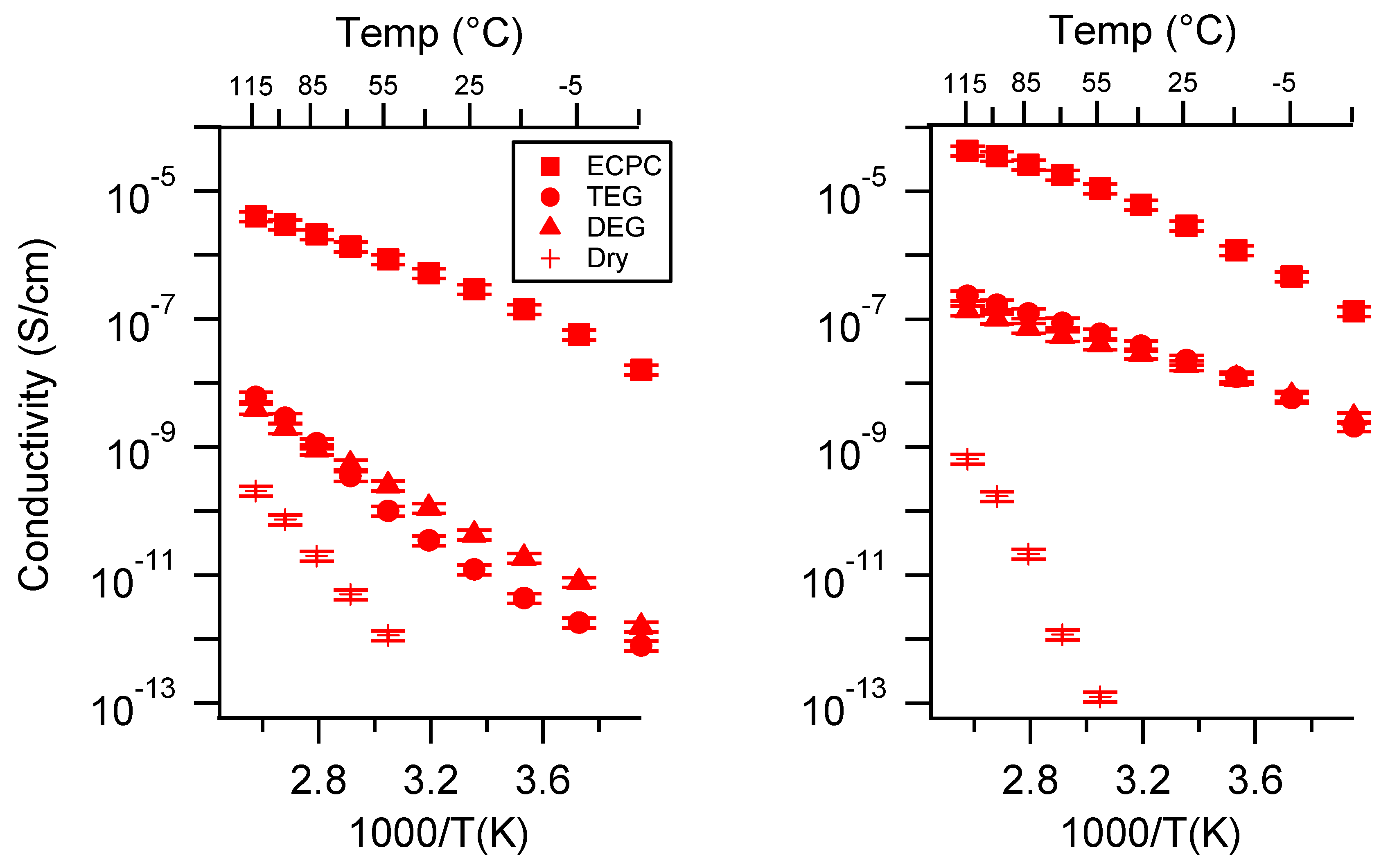
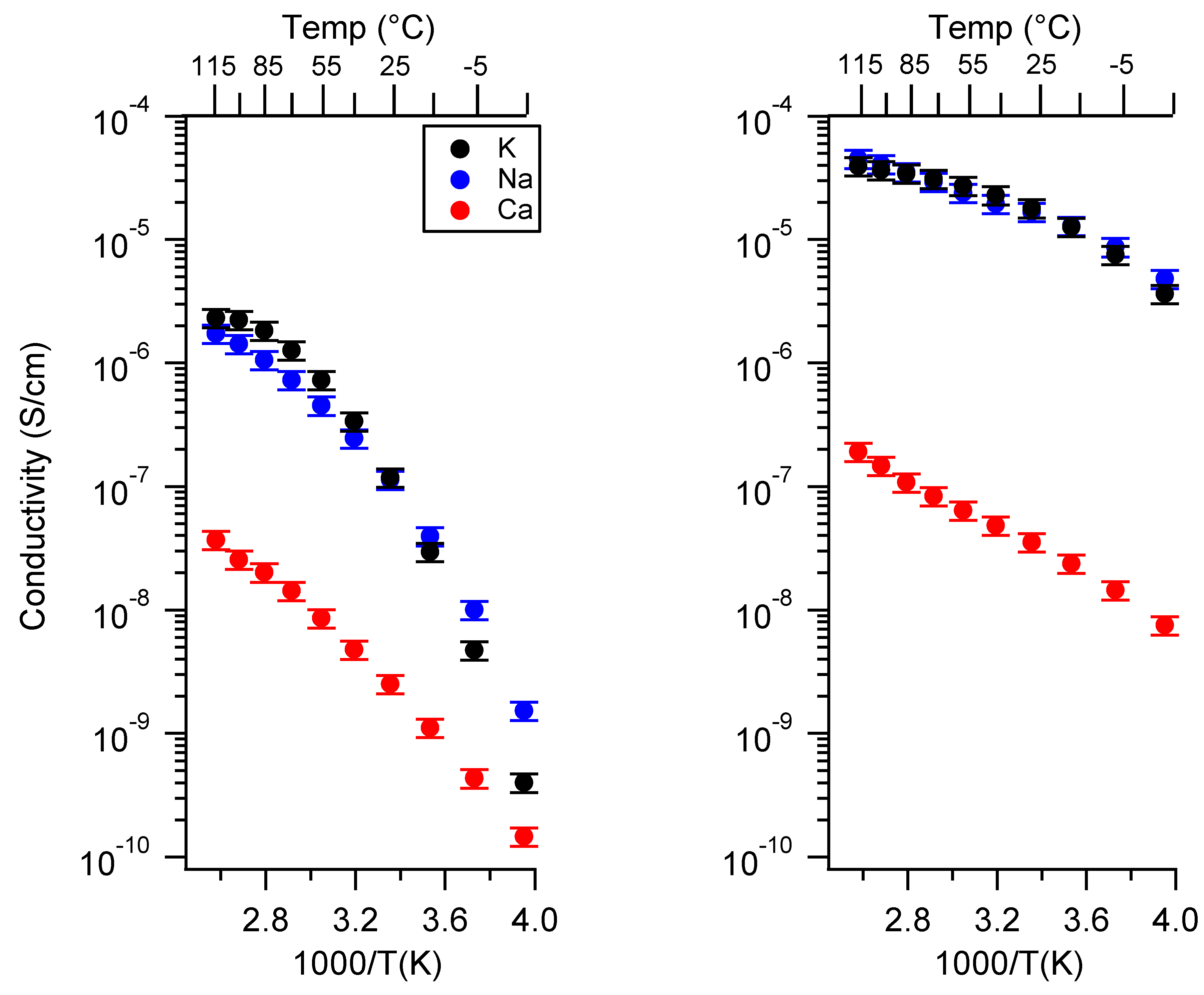
2.1. Effect of the Cation
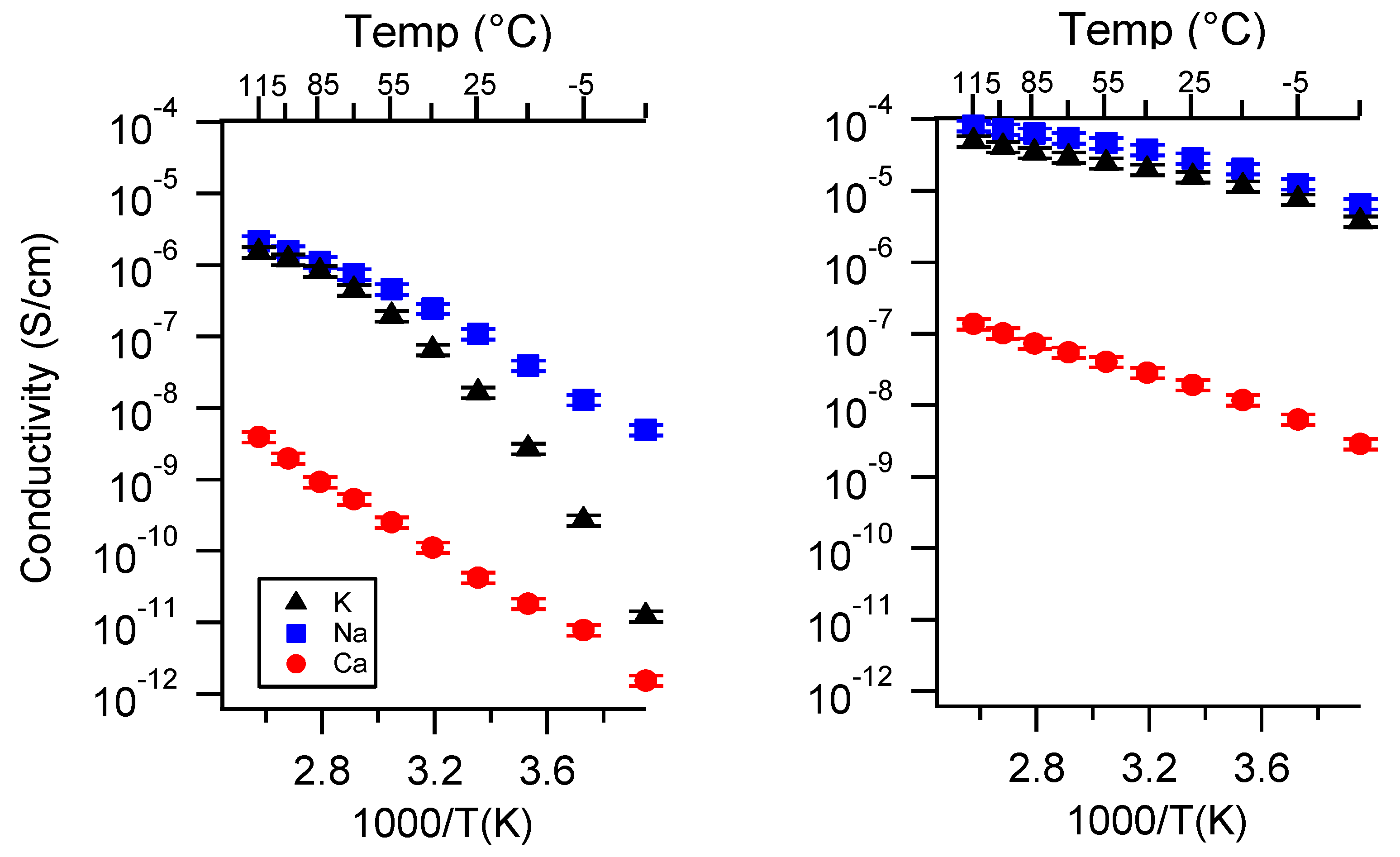
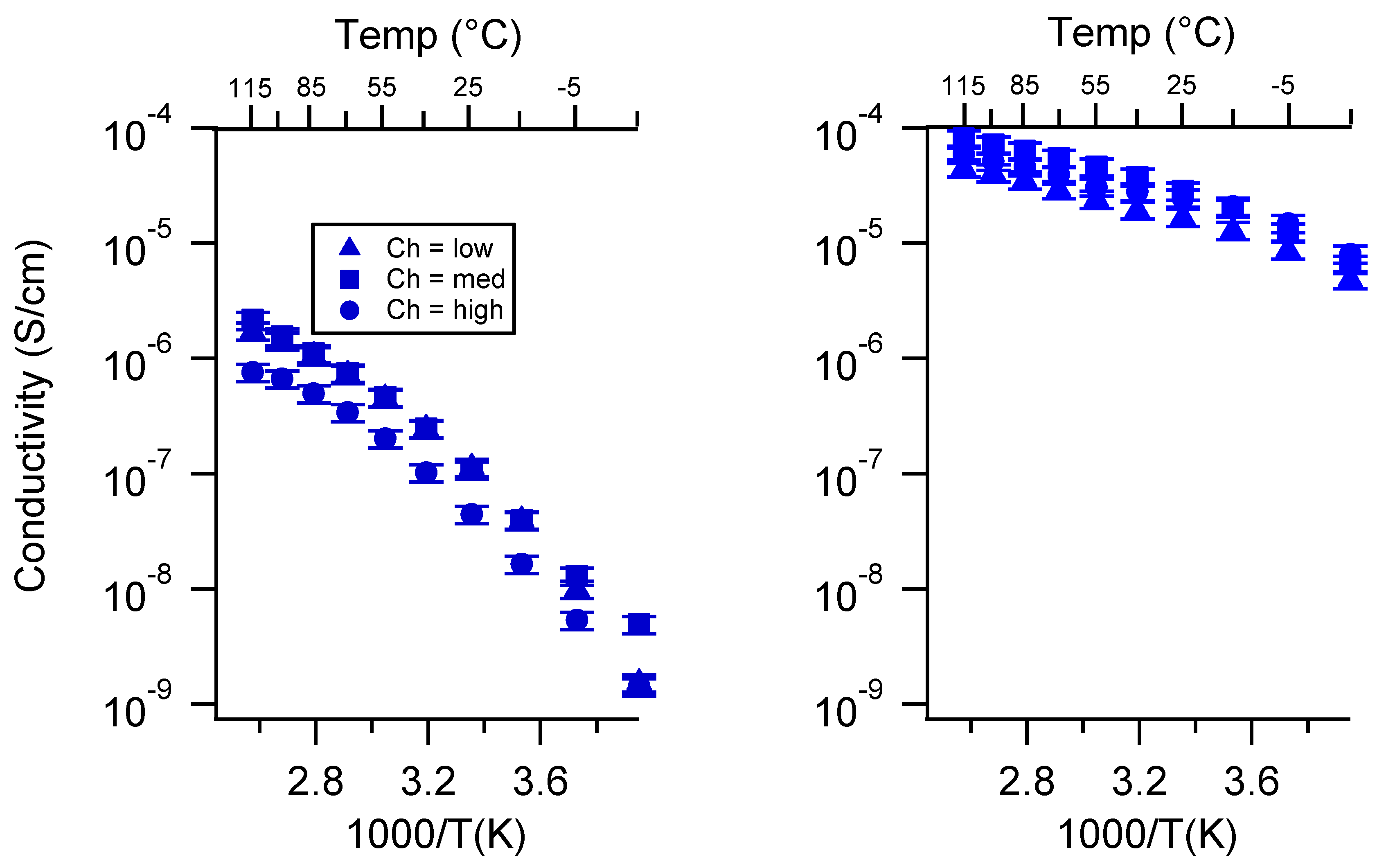
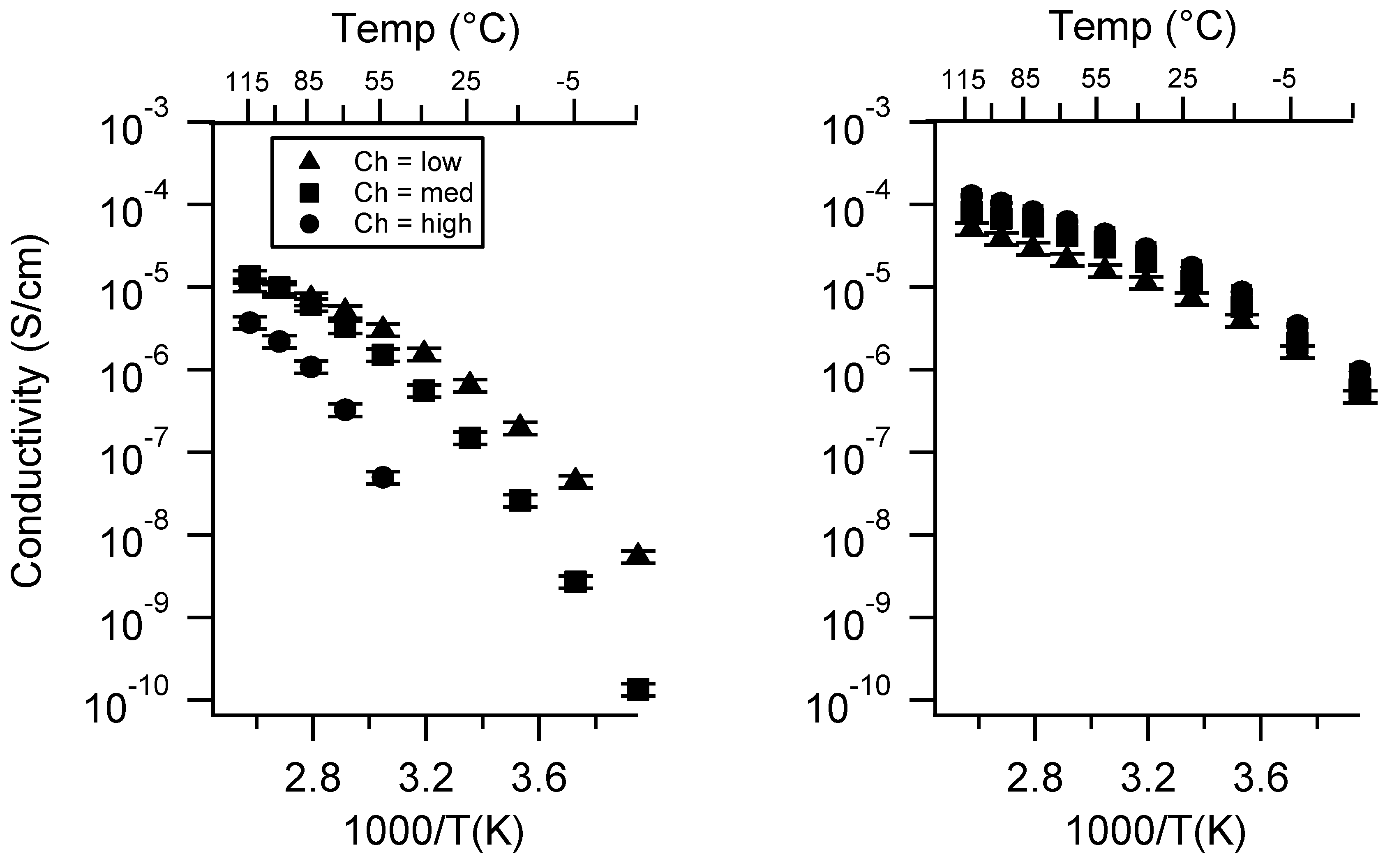
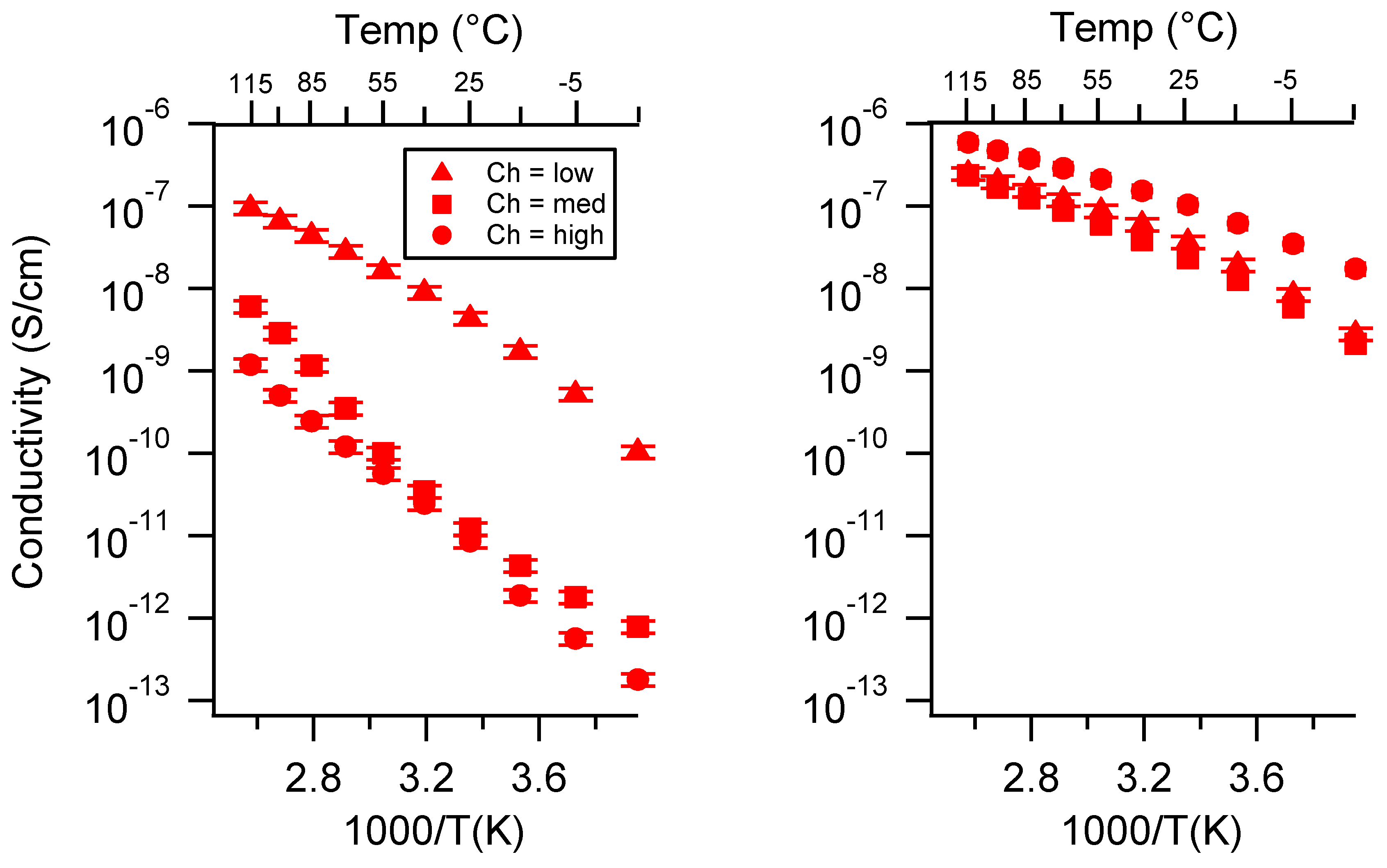
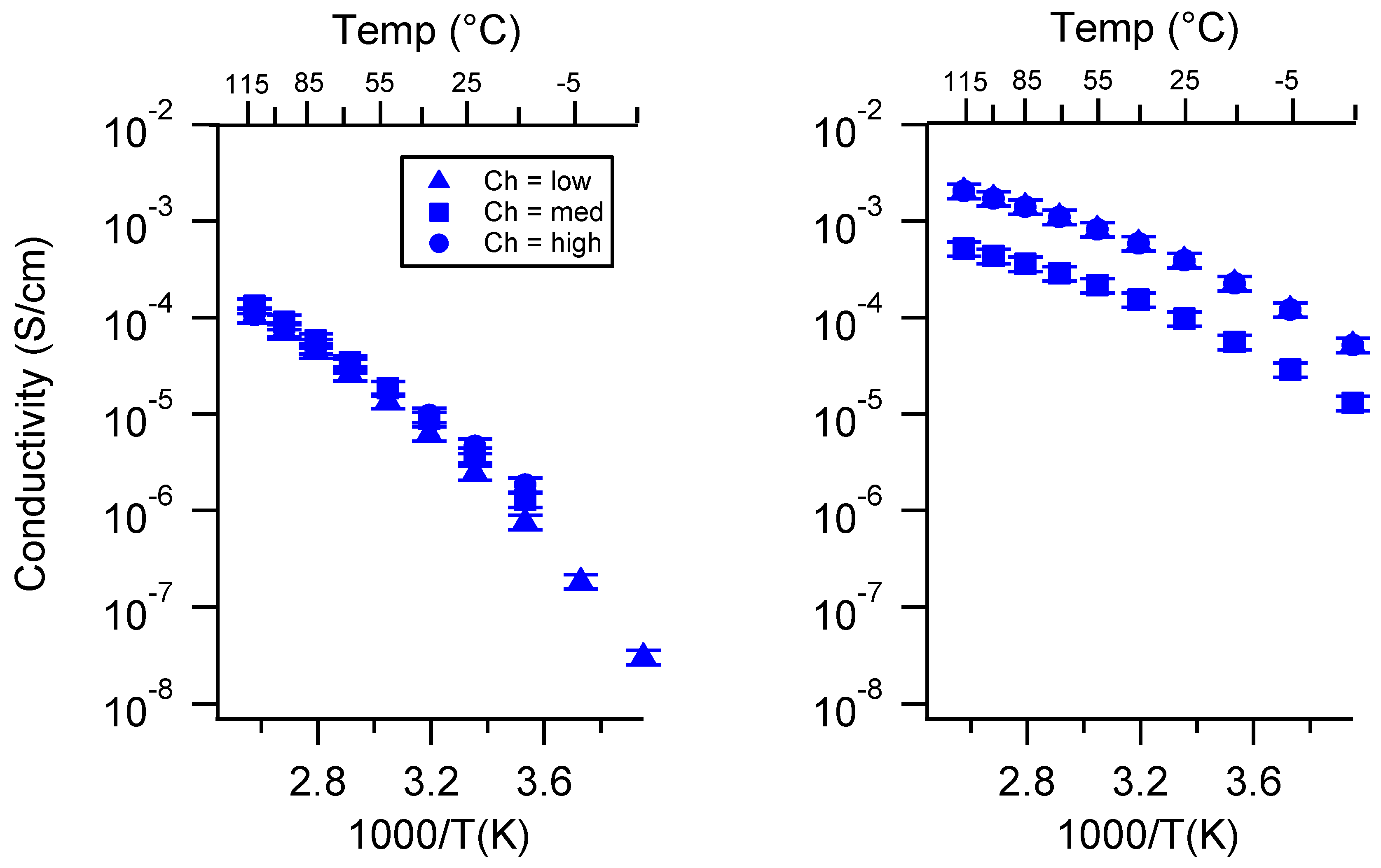
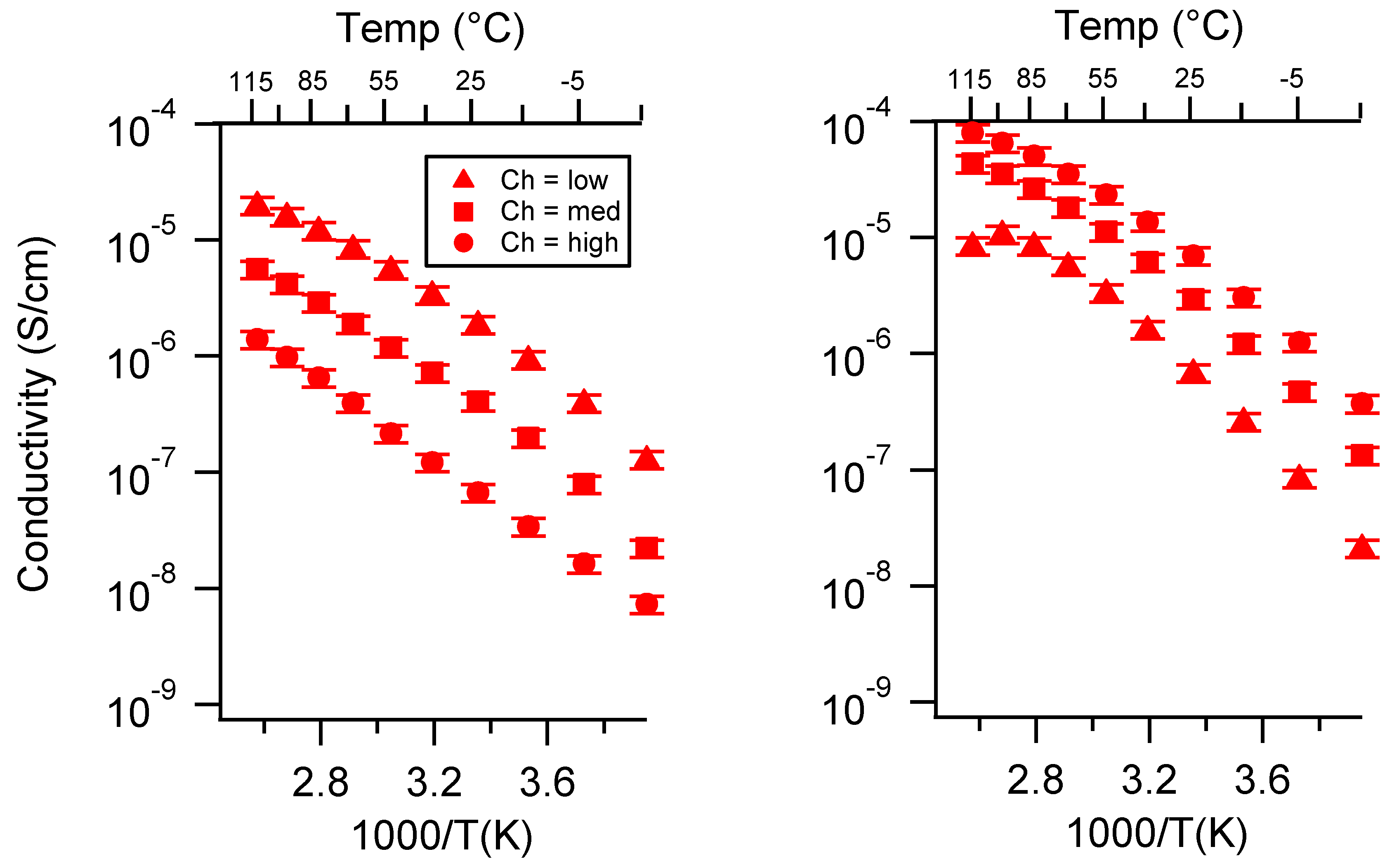
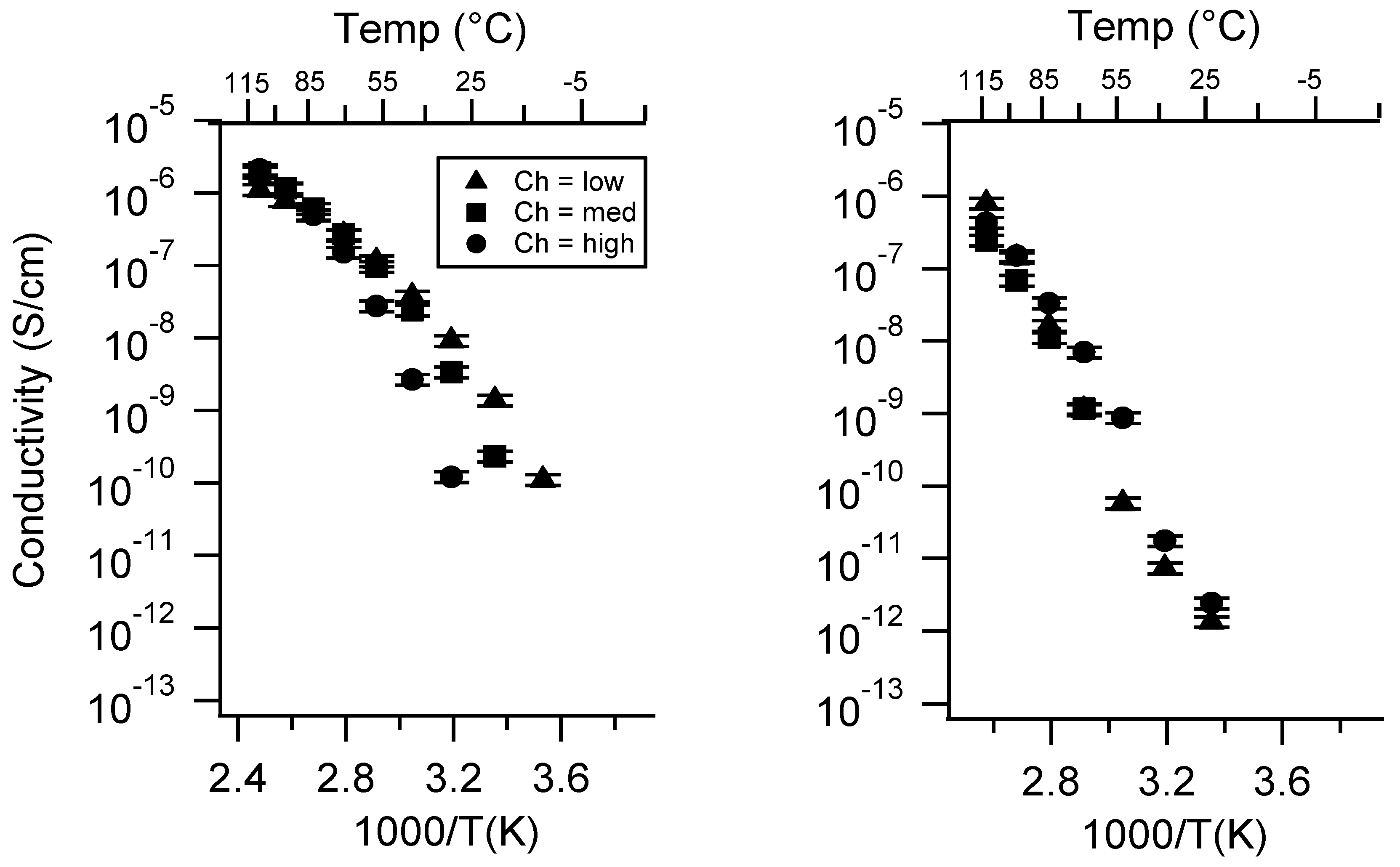
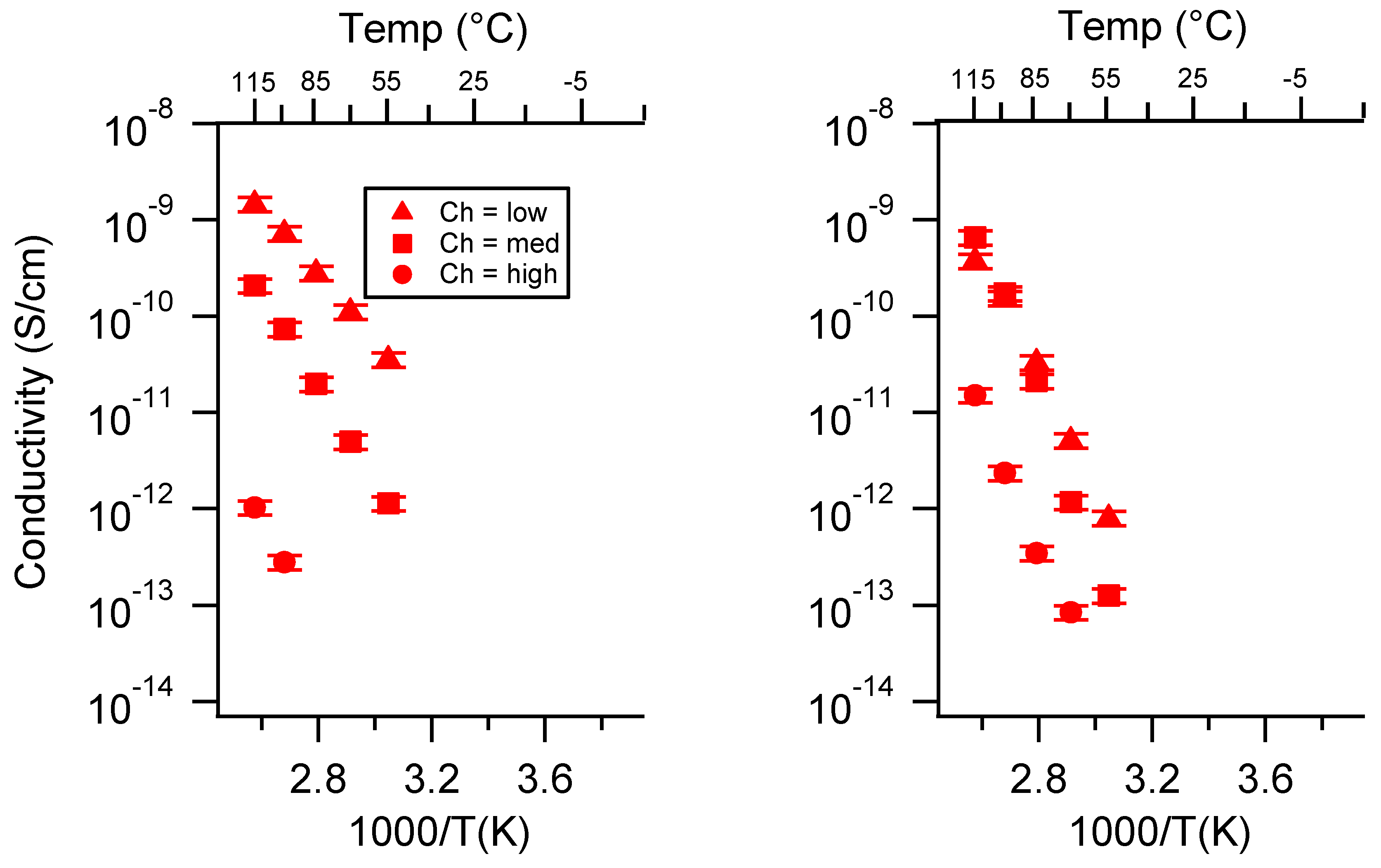
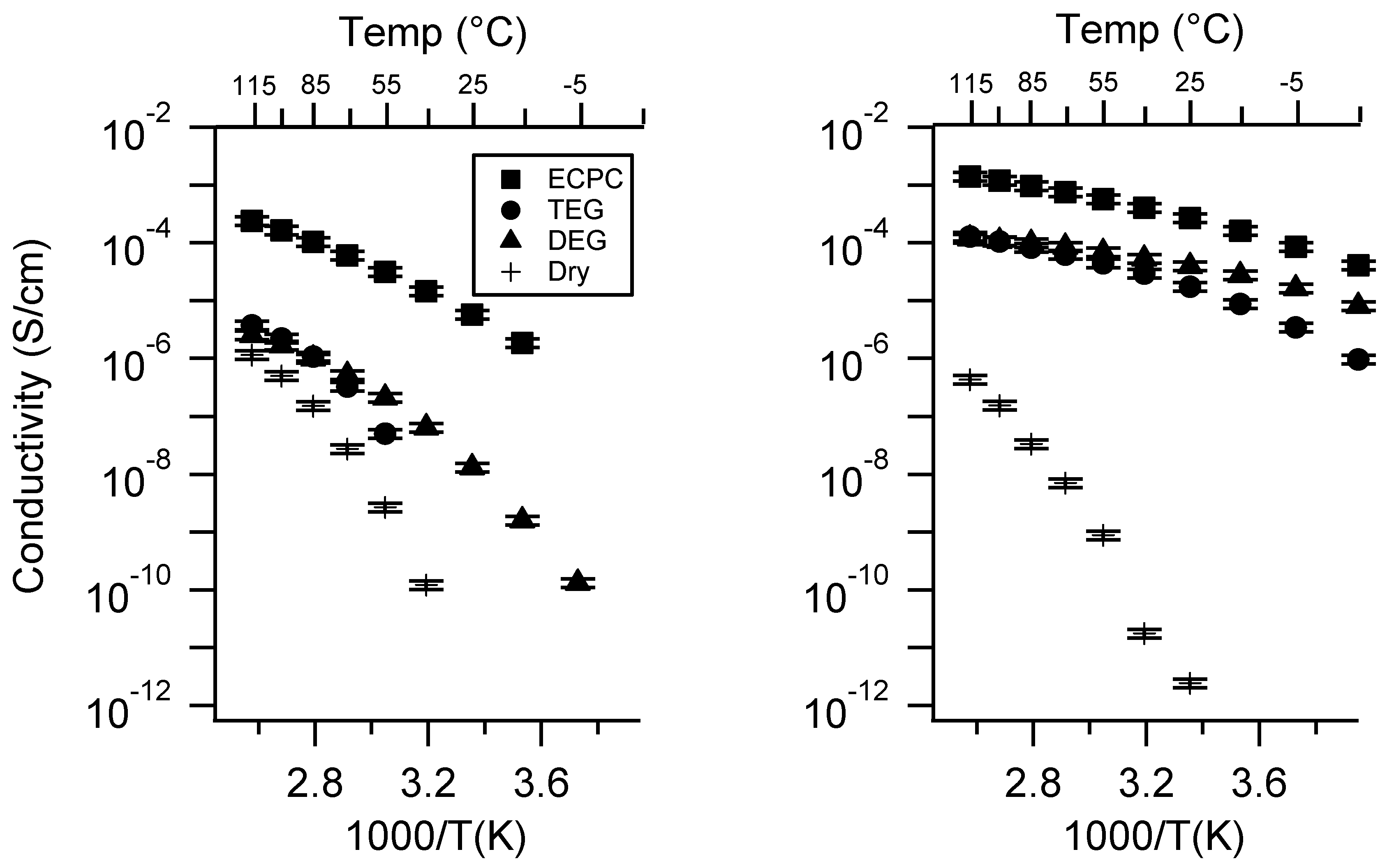
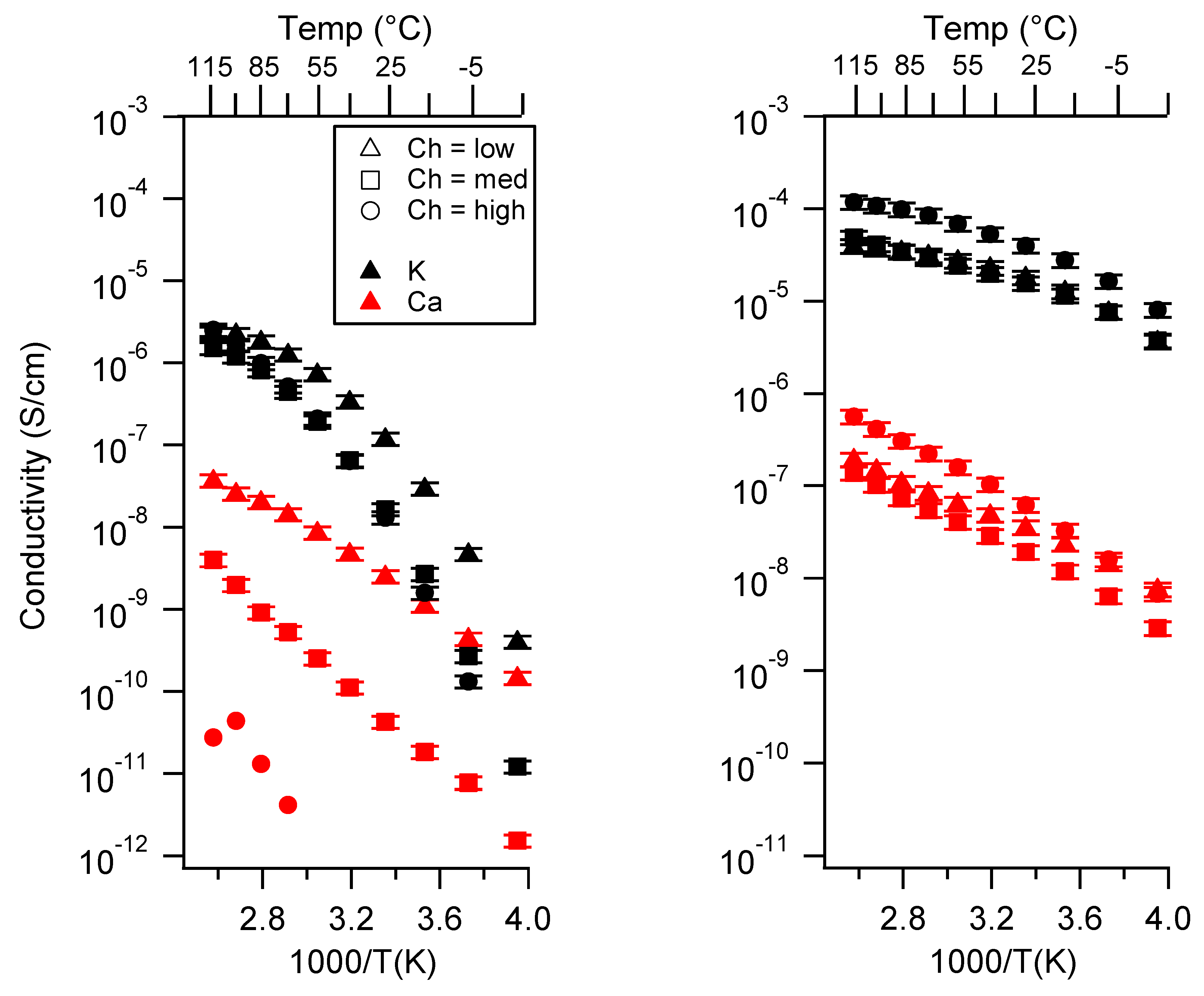
2.2. Effect of Charge Density
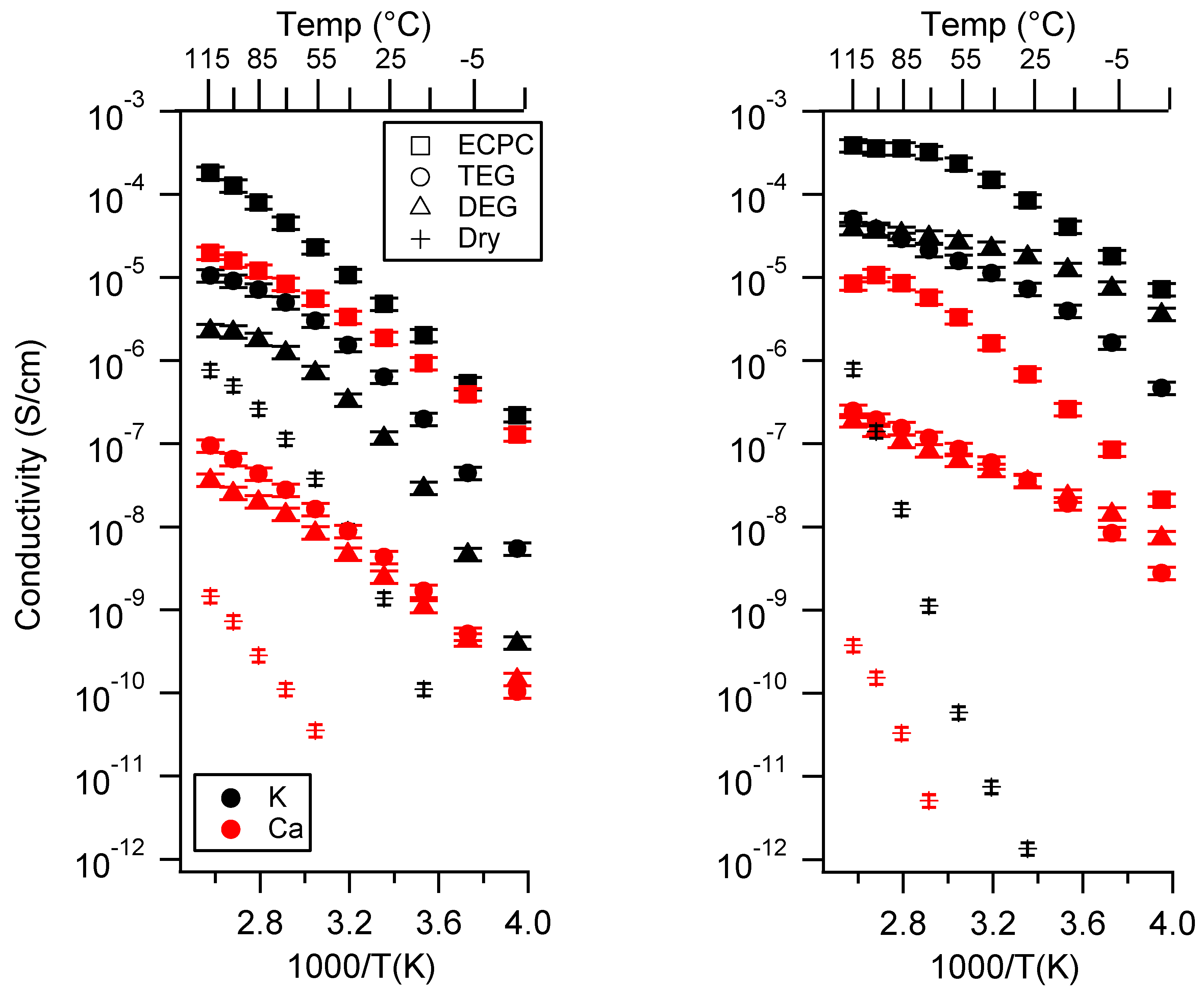
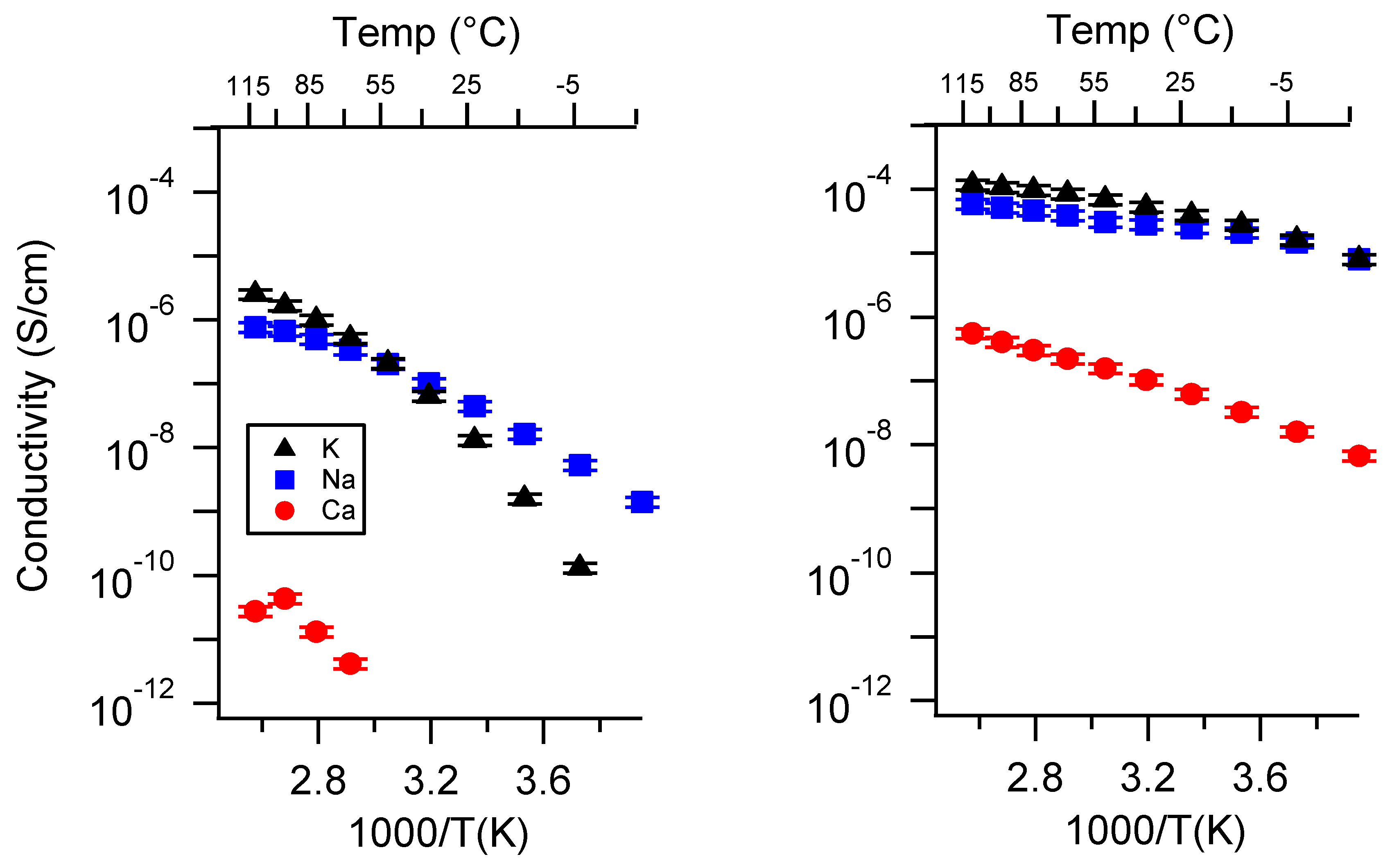
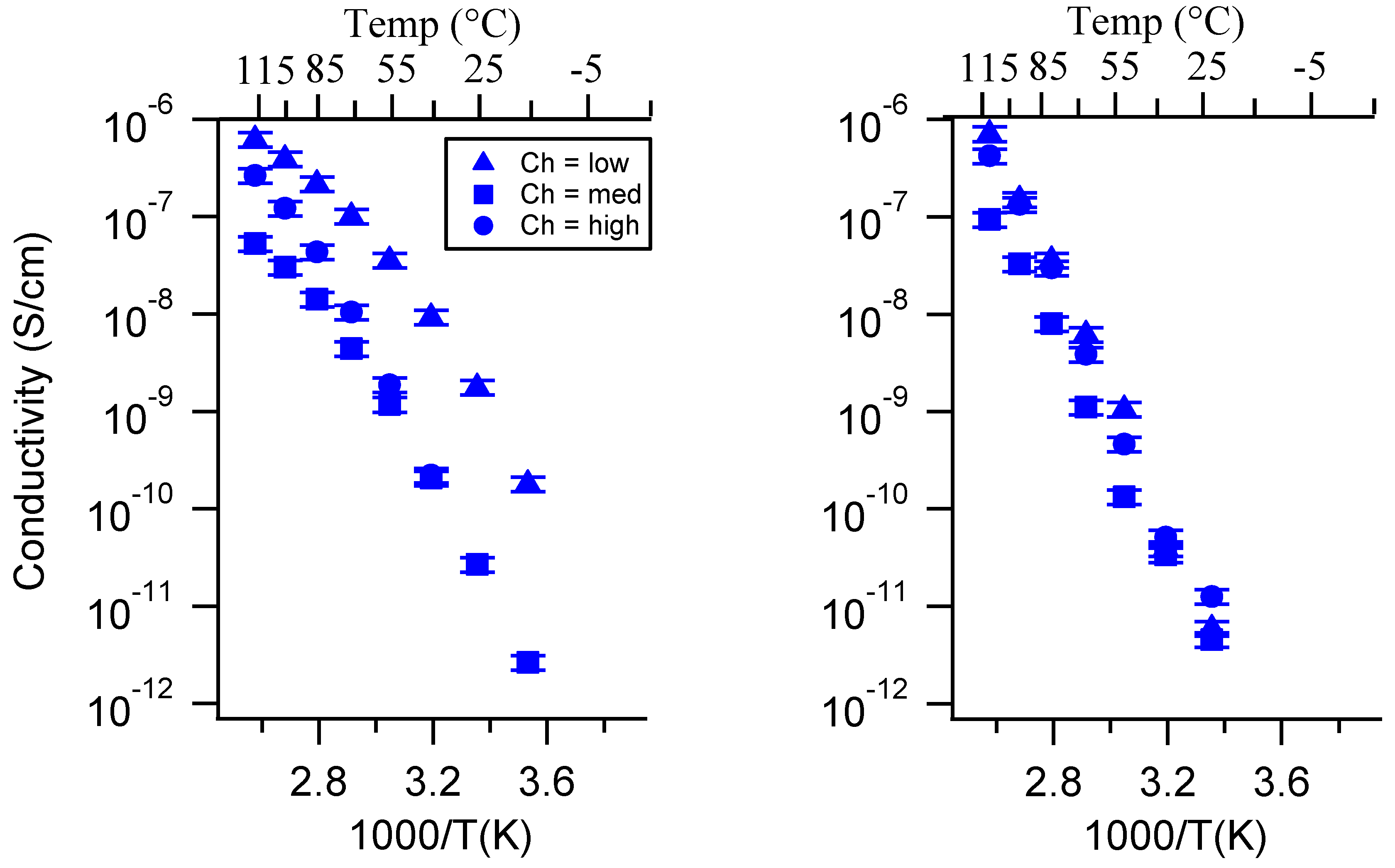
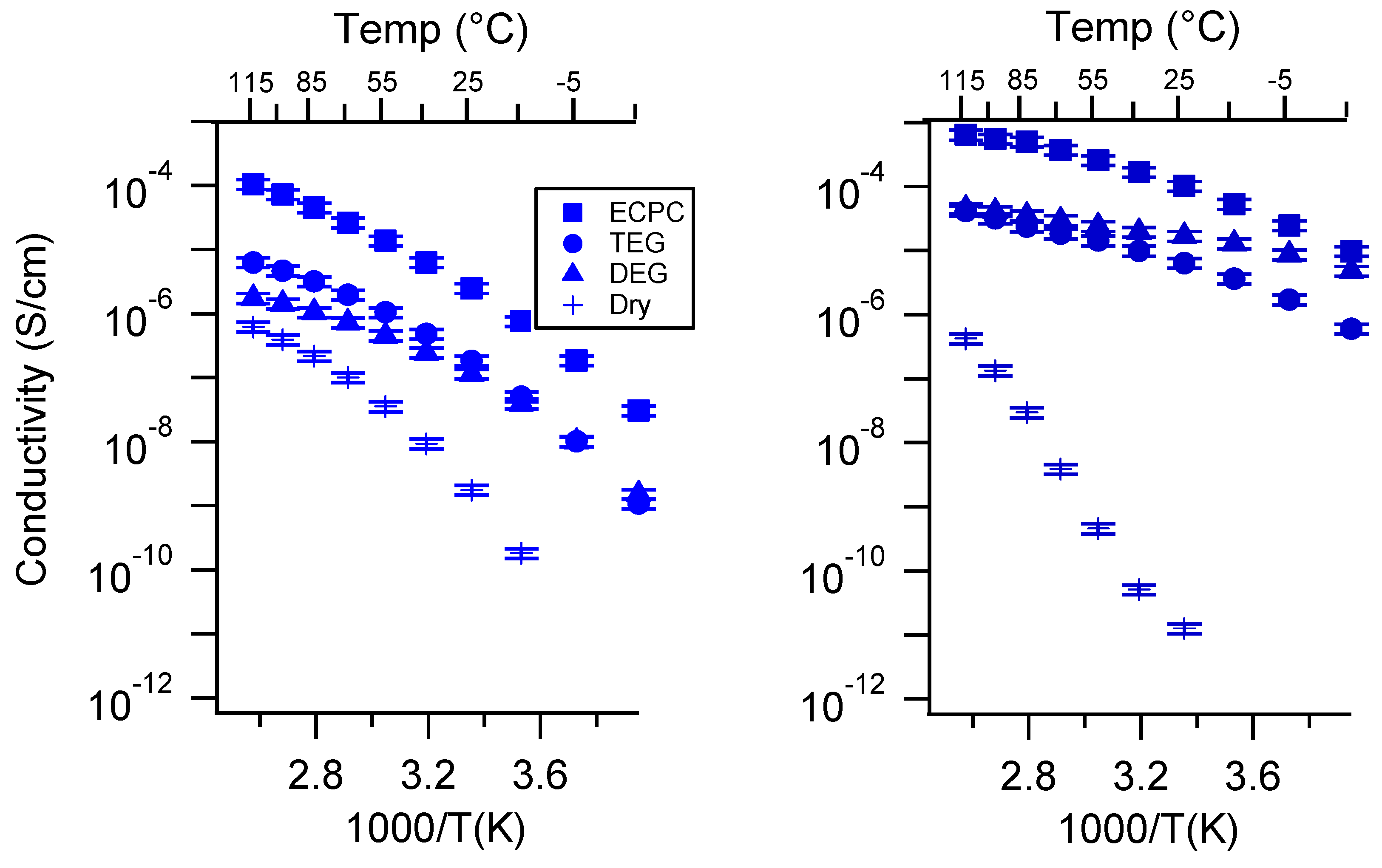
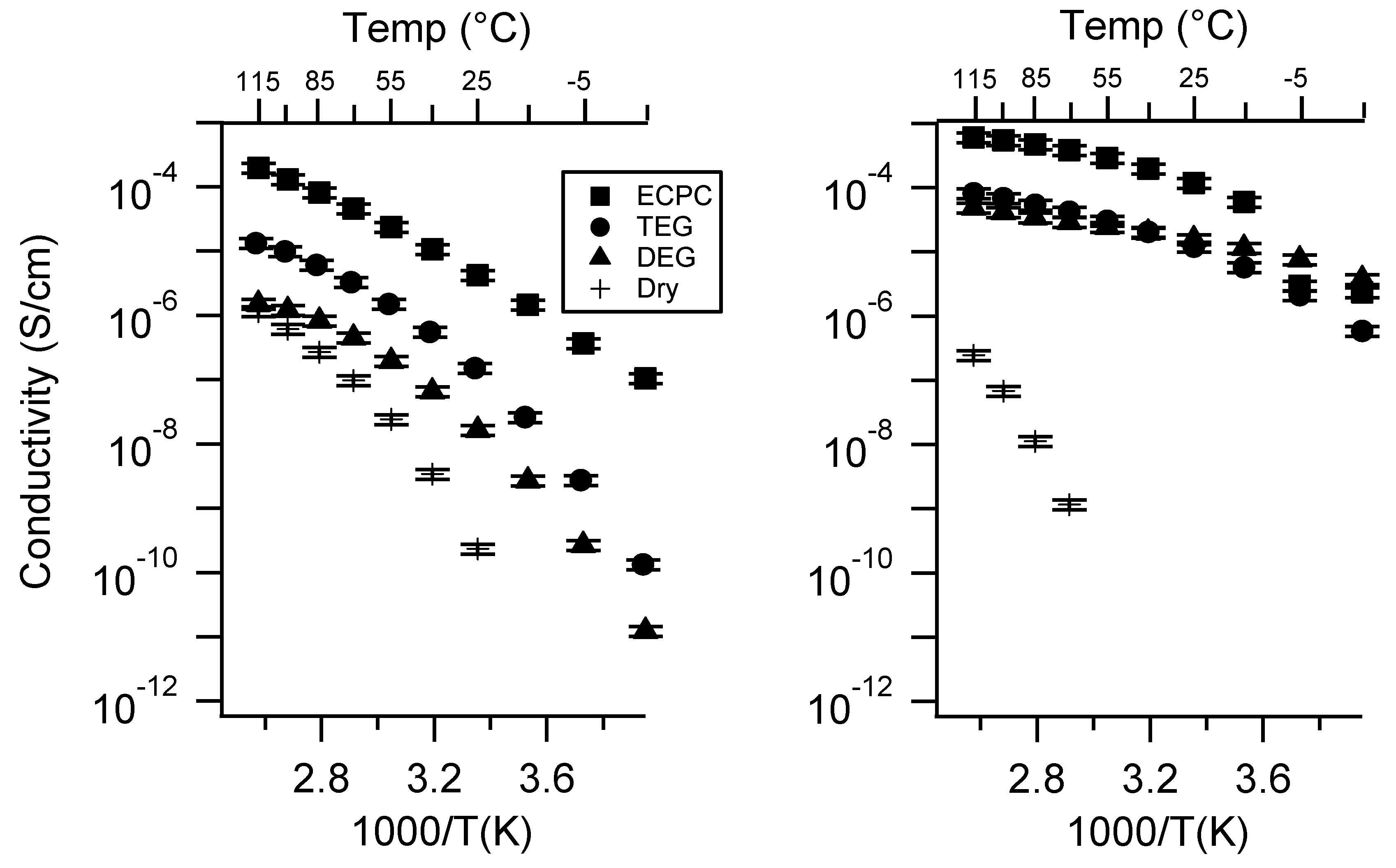
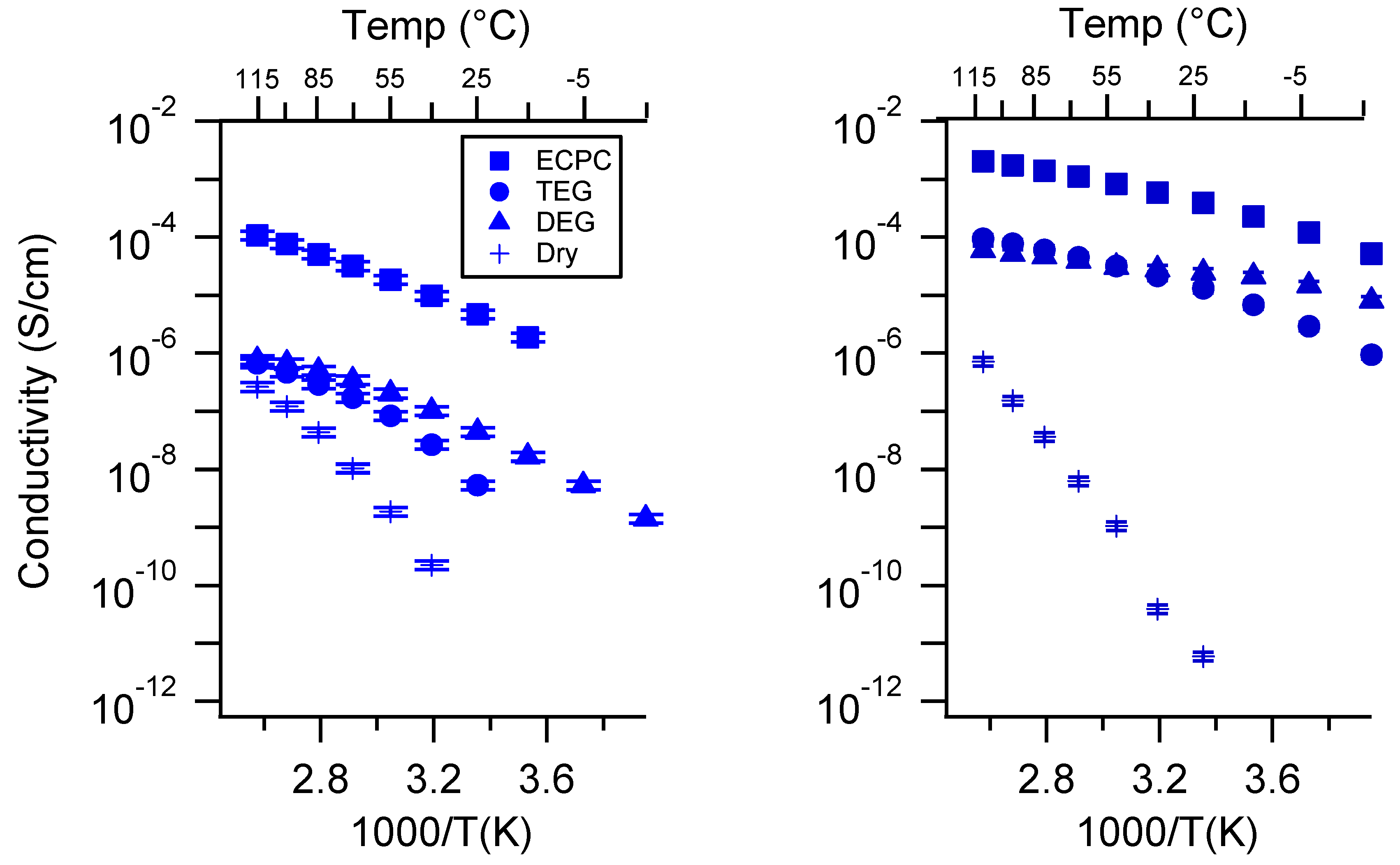
2.3. Effect of Swelling Condition
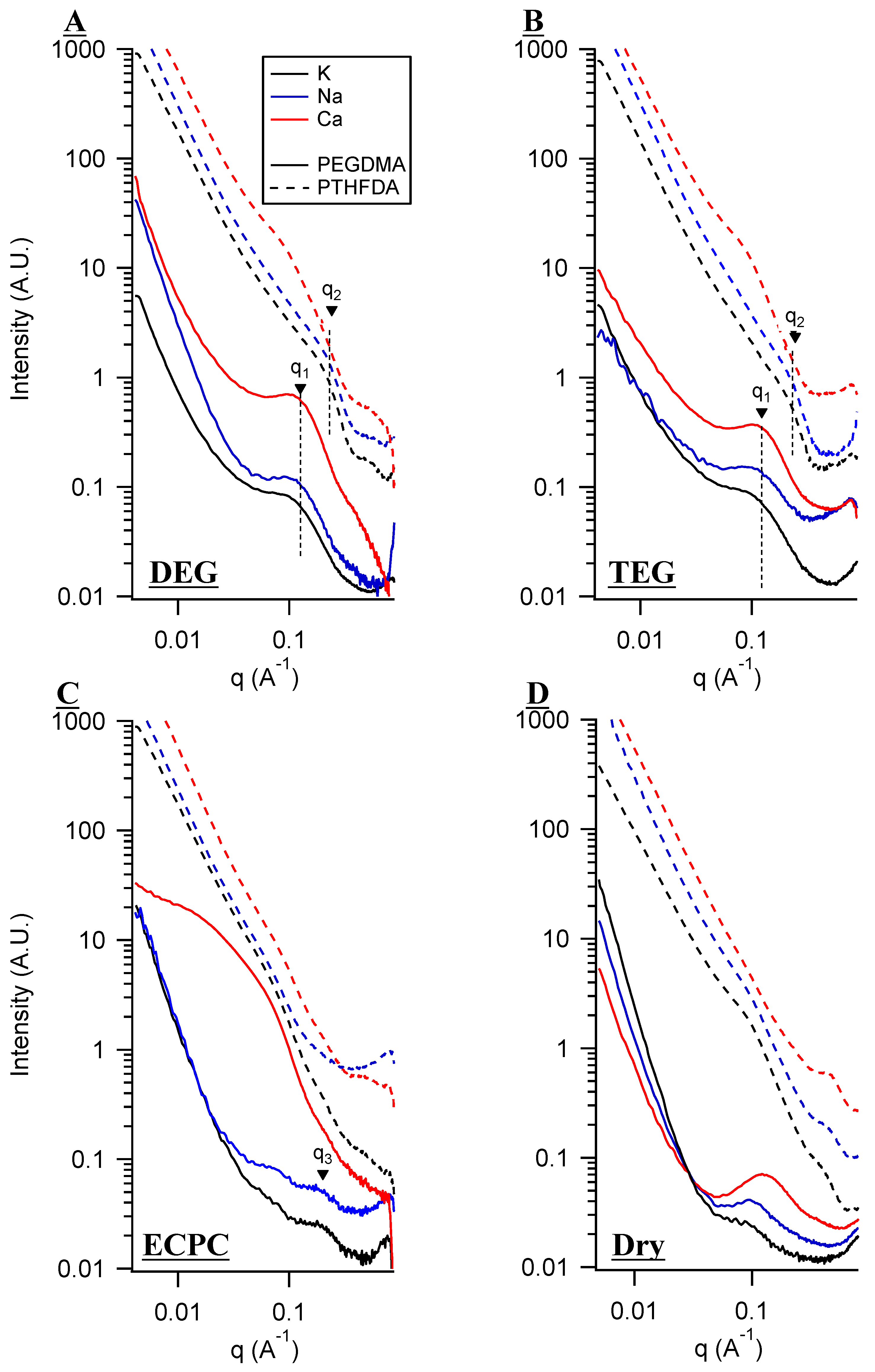
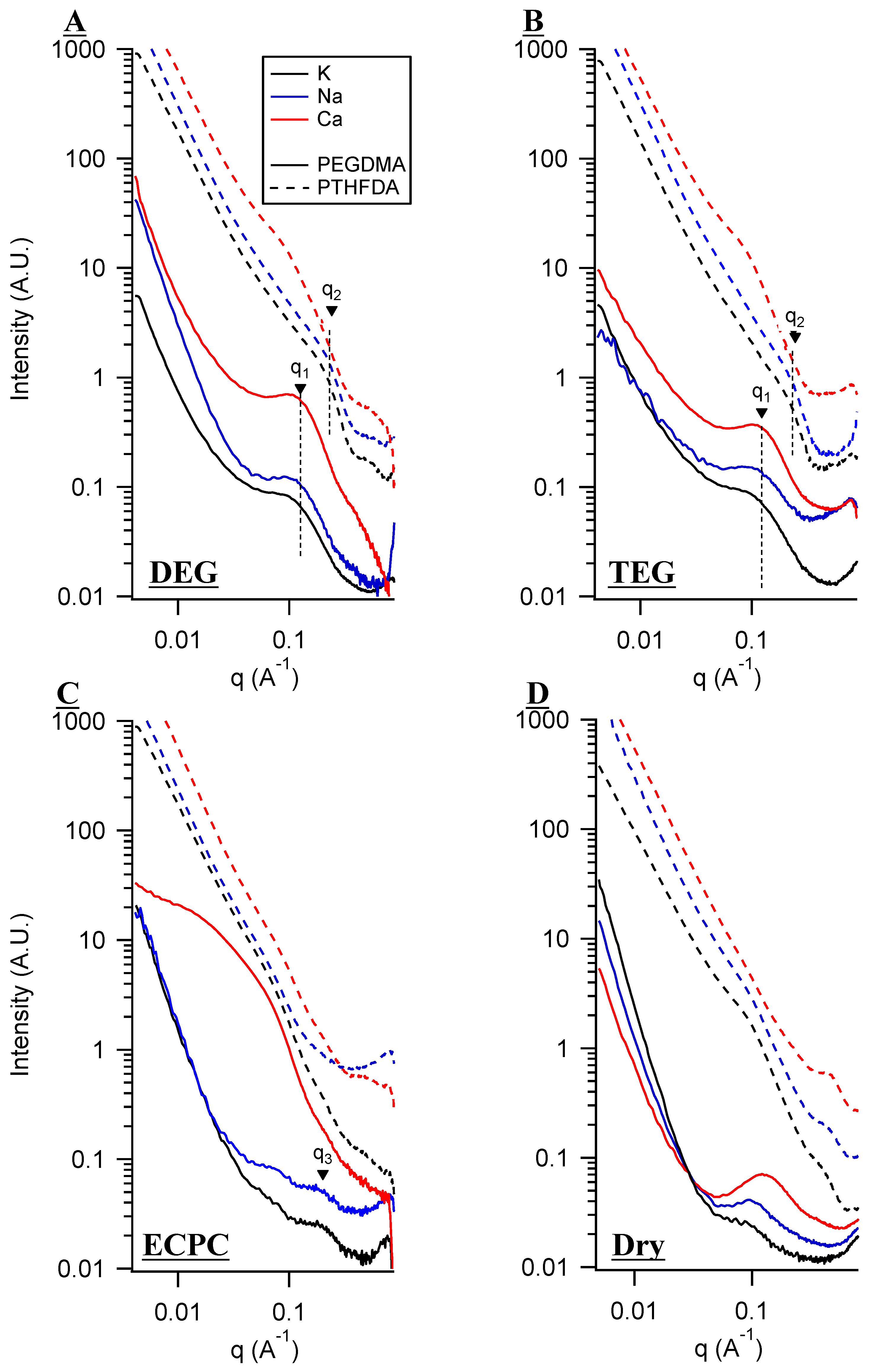
3. Small Angle X-Ray Scattering (SAXS) and Solvent Uptake
4. Conclusions
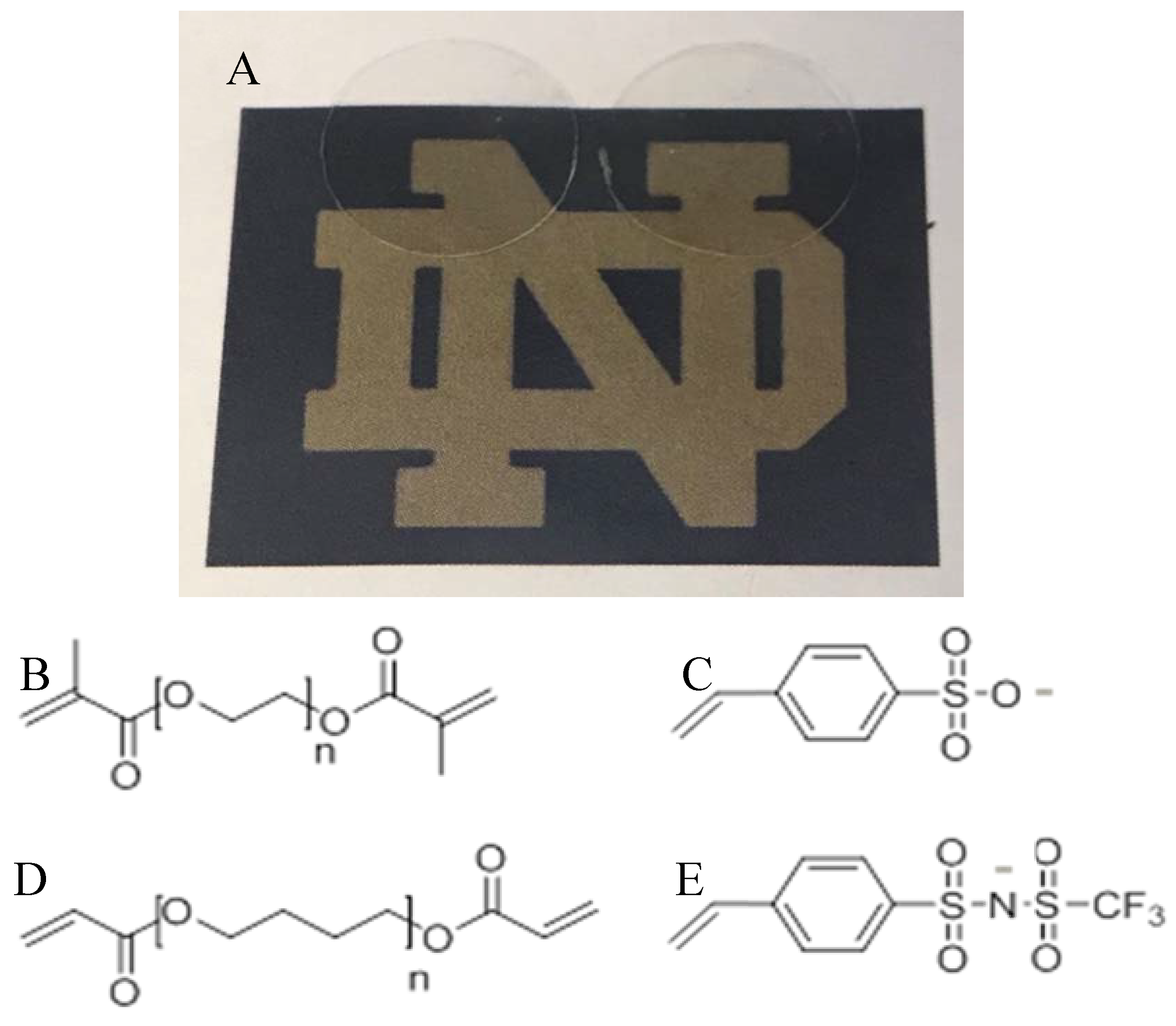
5. Experimental
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
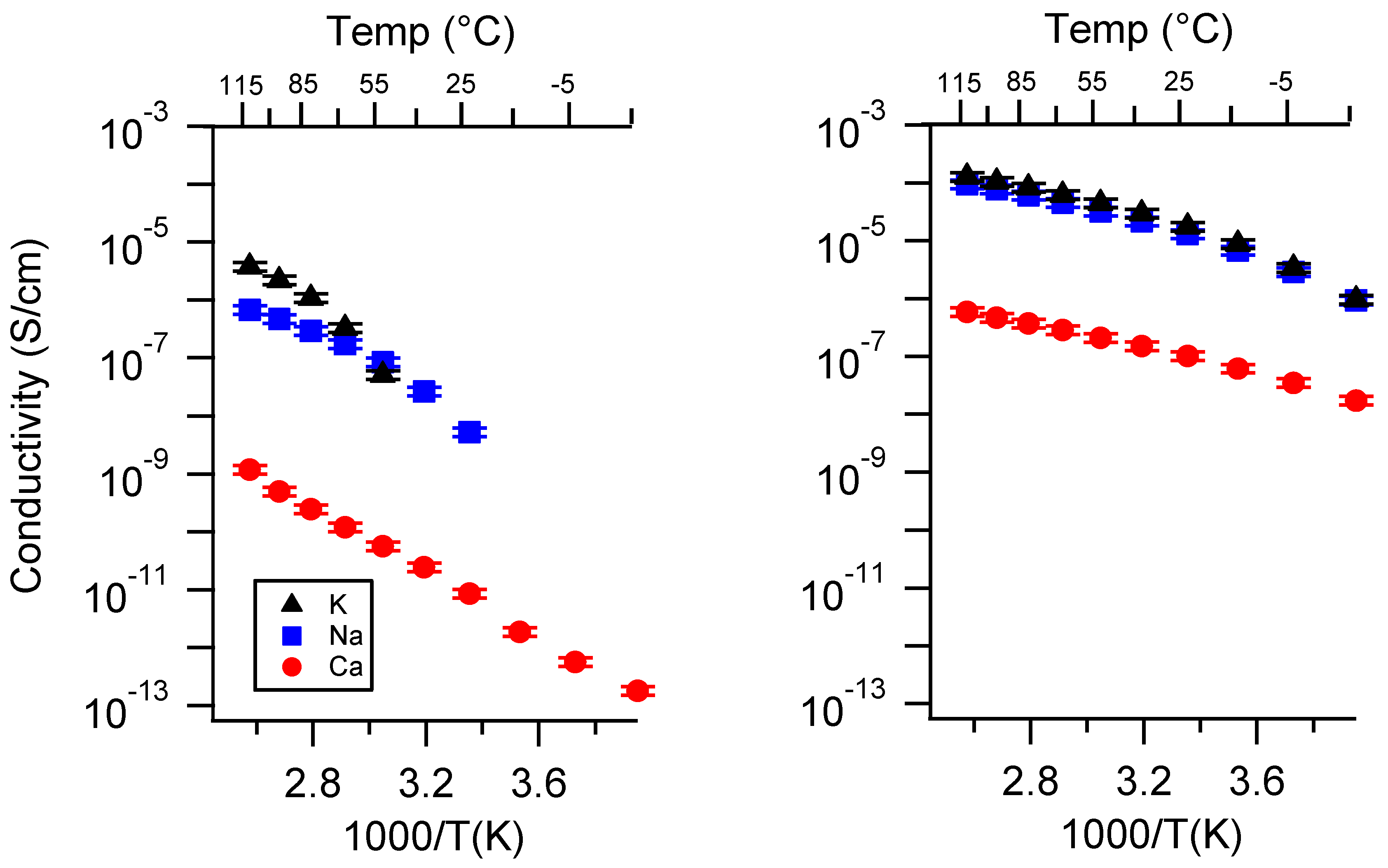
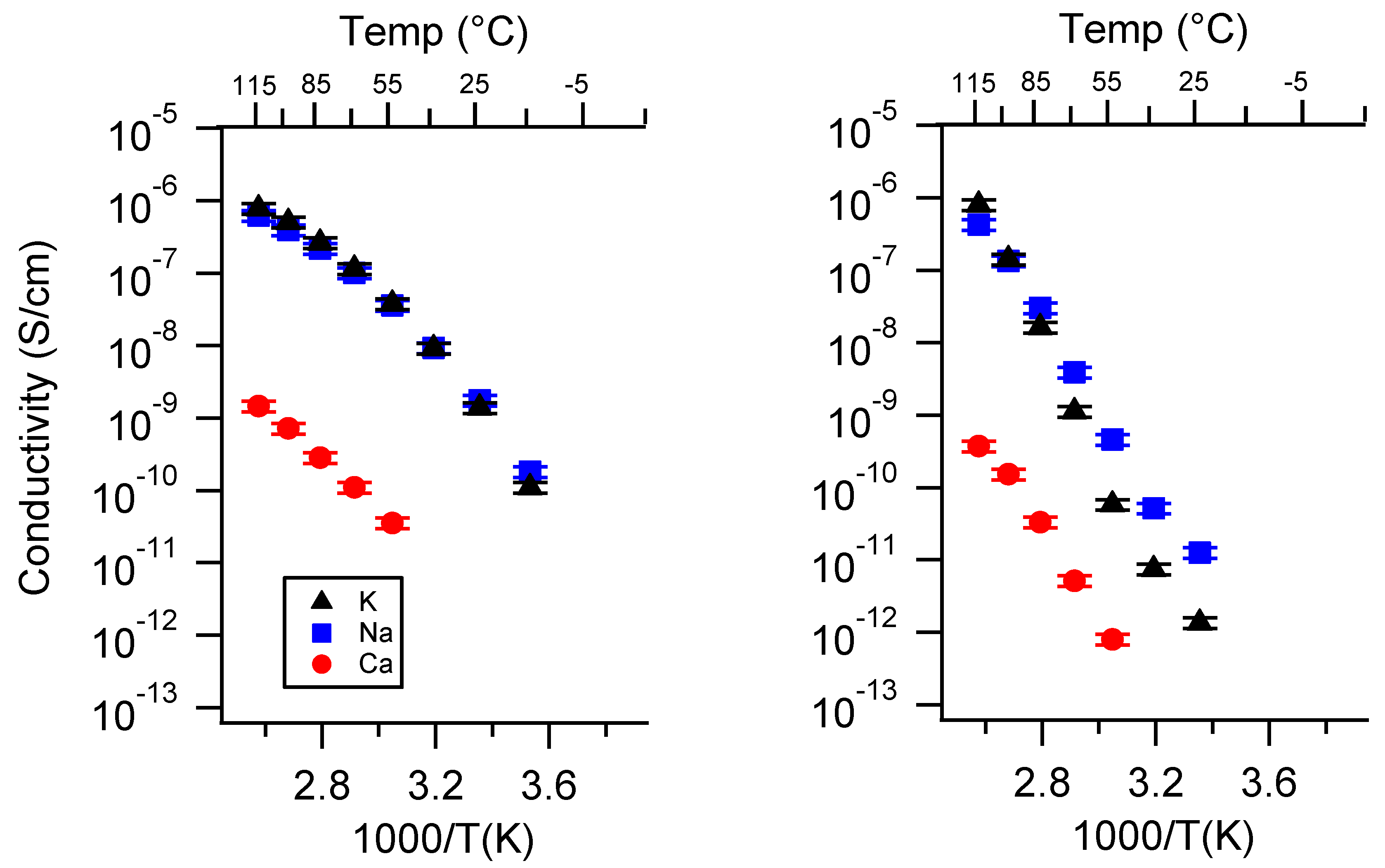
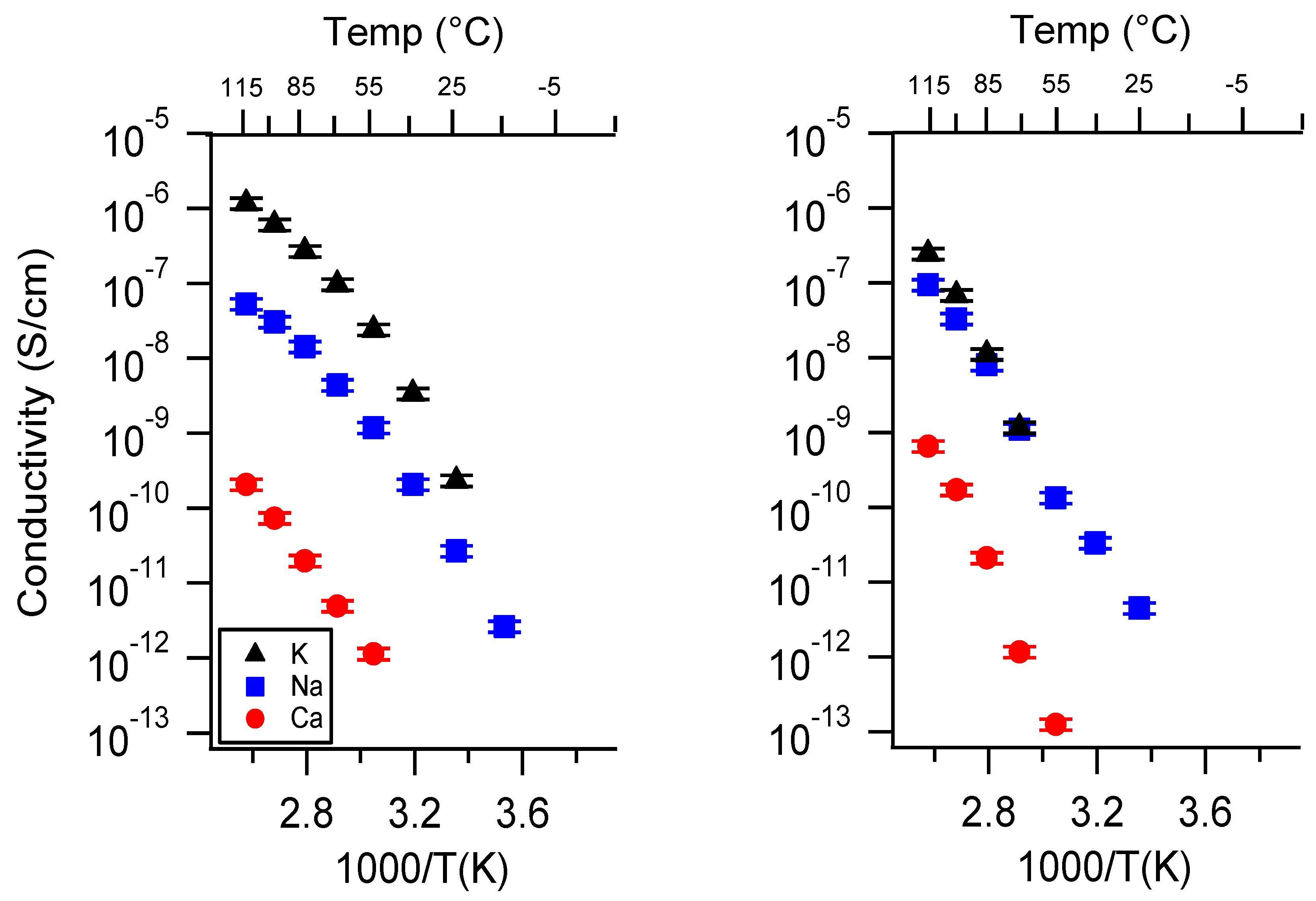
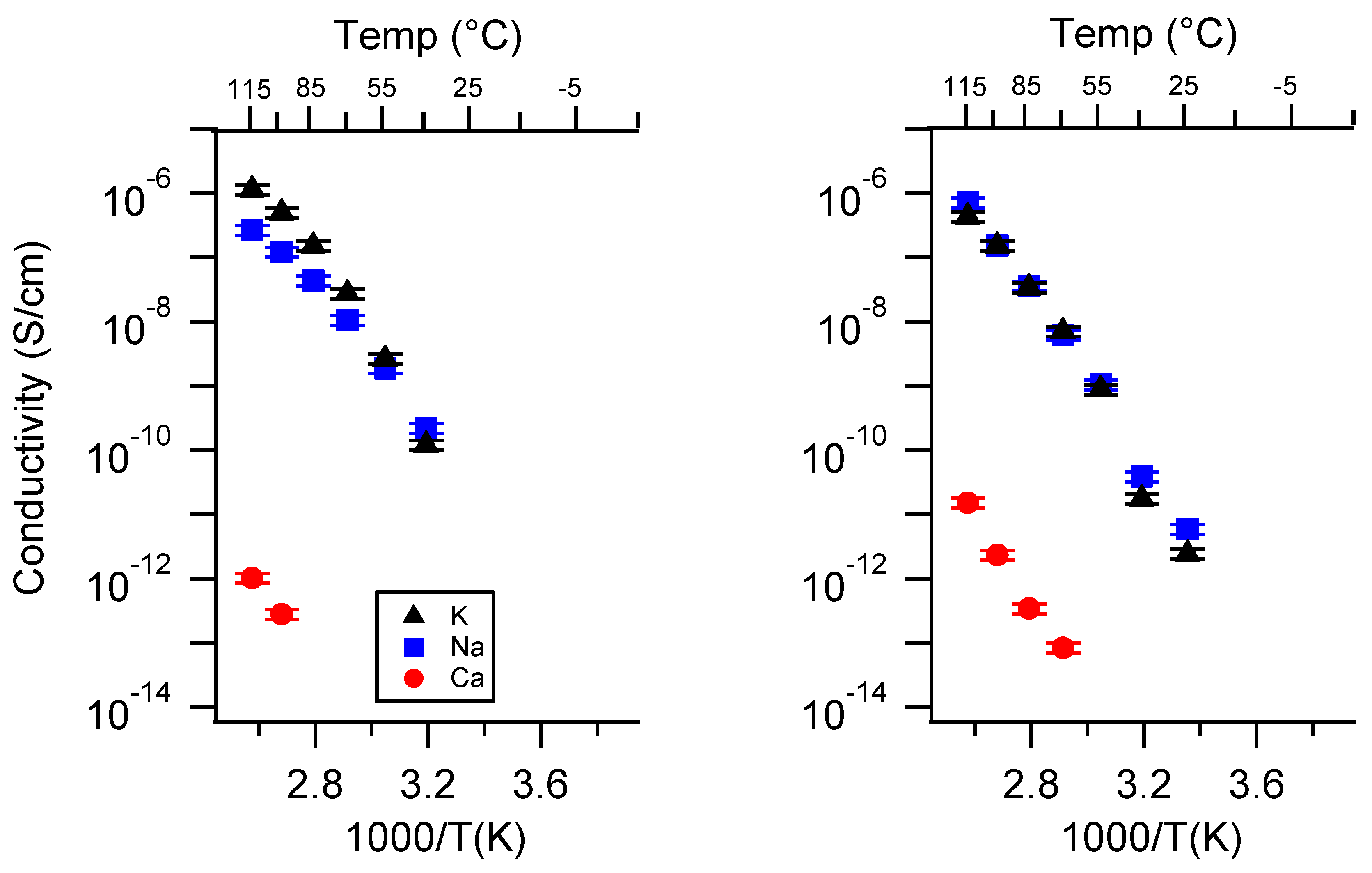
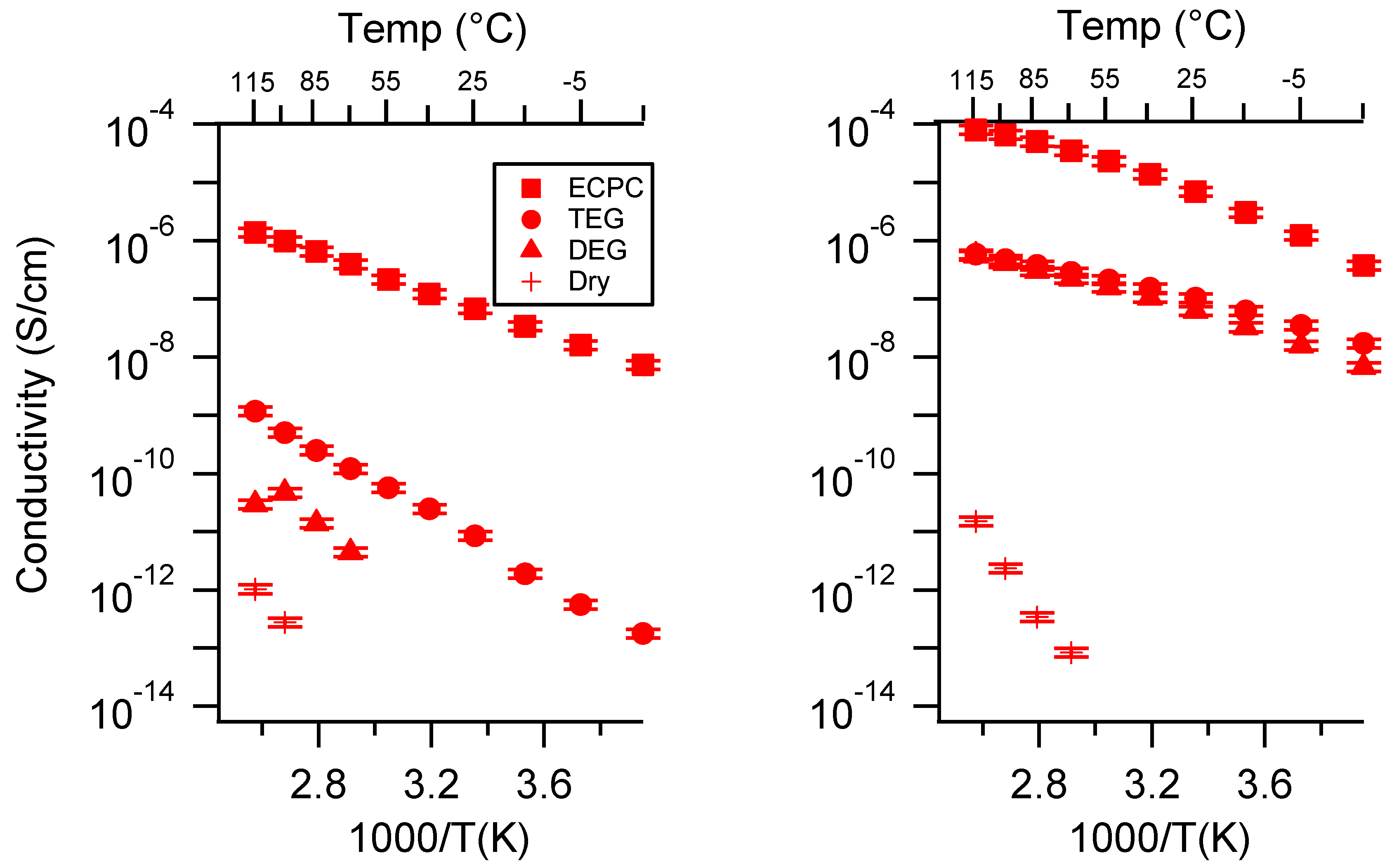
Appendix A. All SIPE Conductivity as a Function of Cation
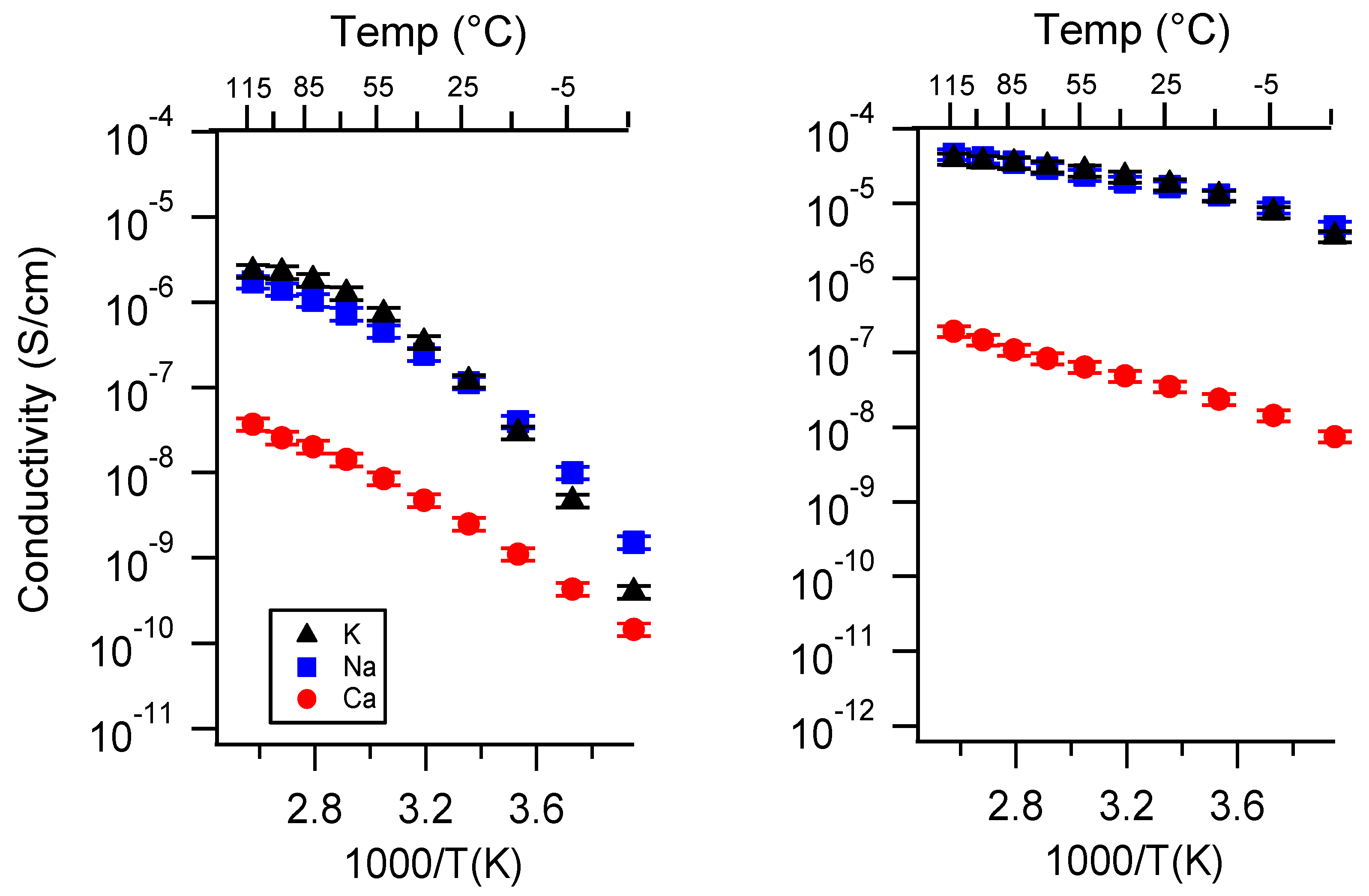
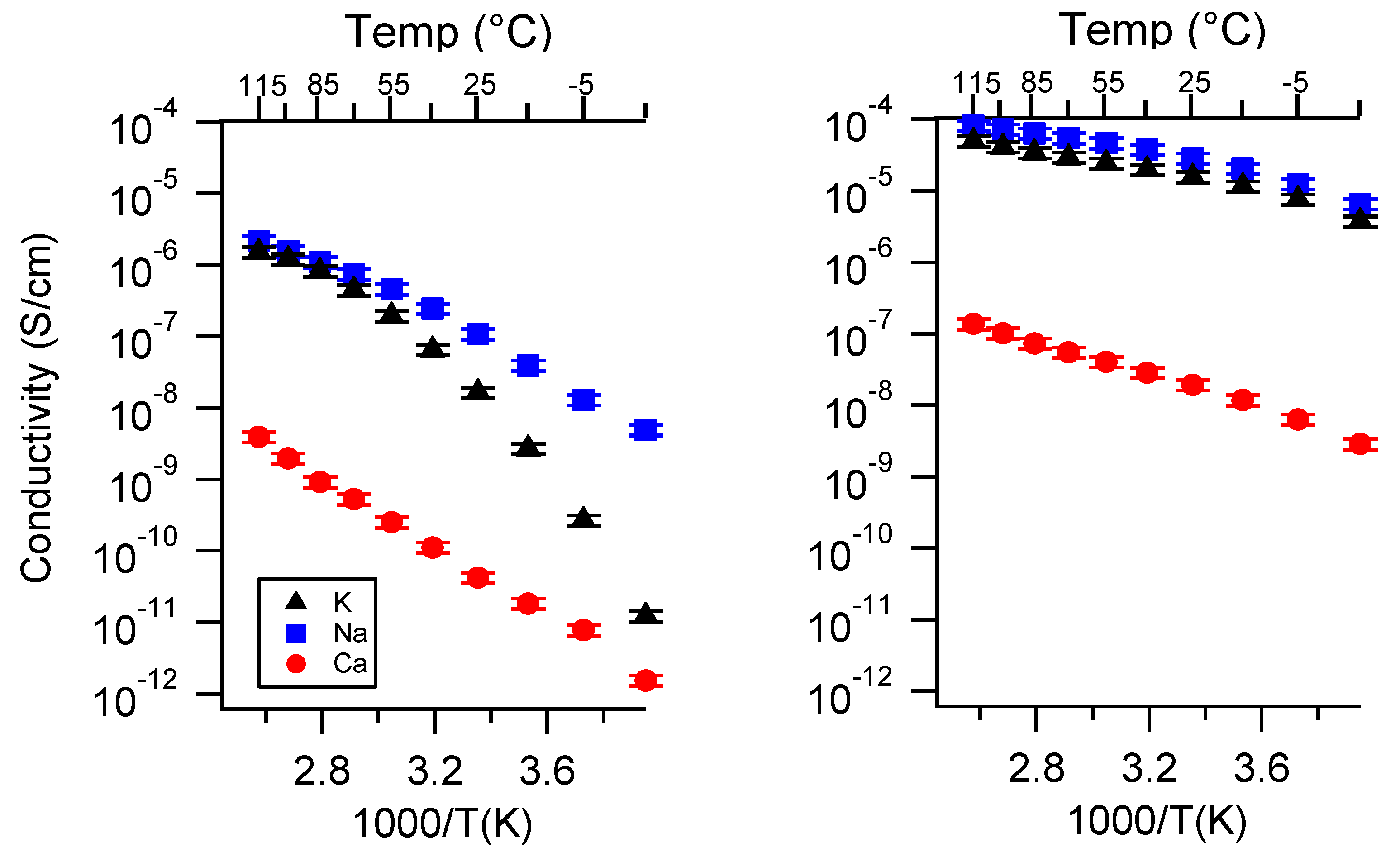
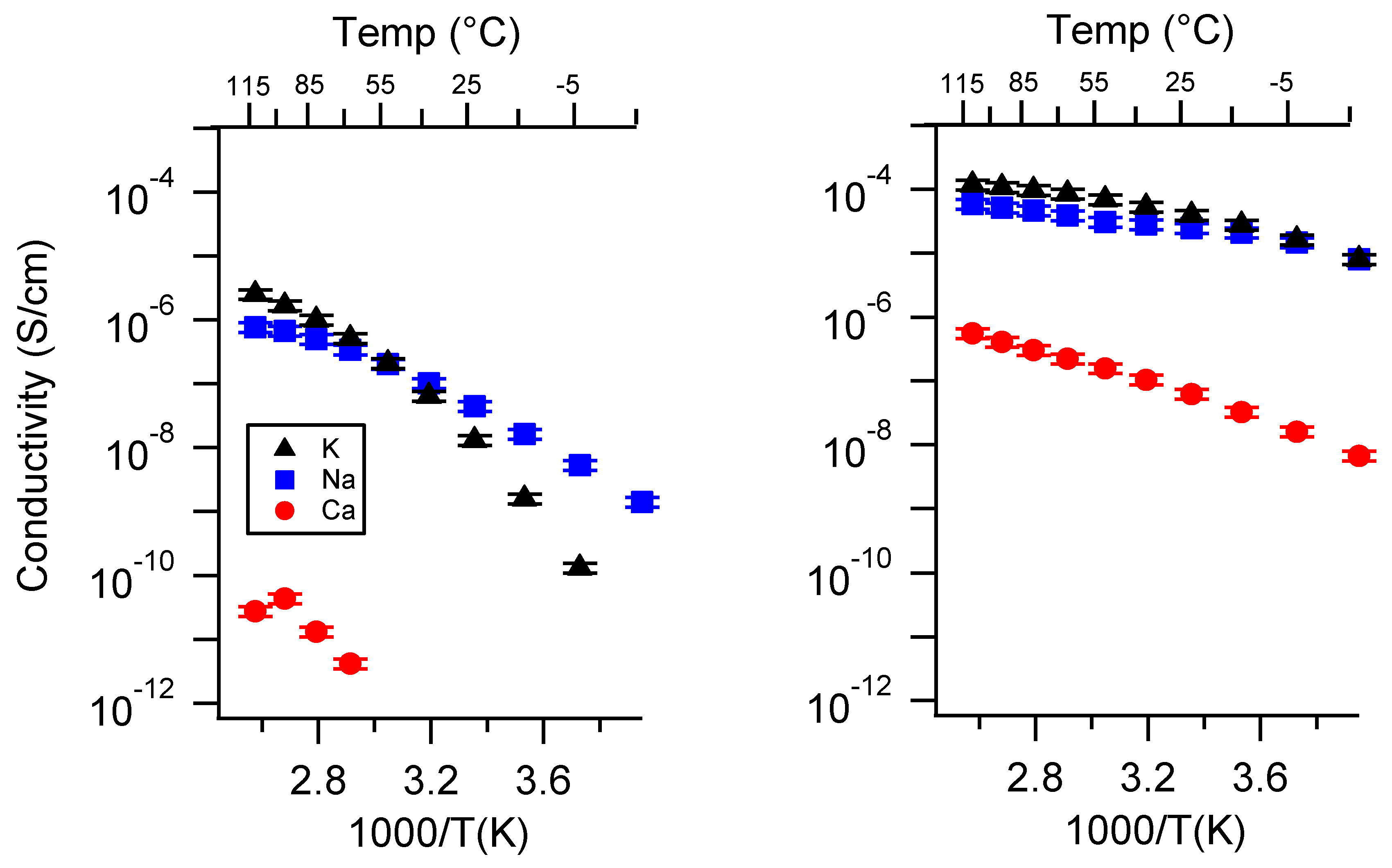
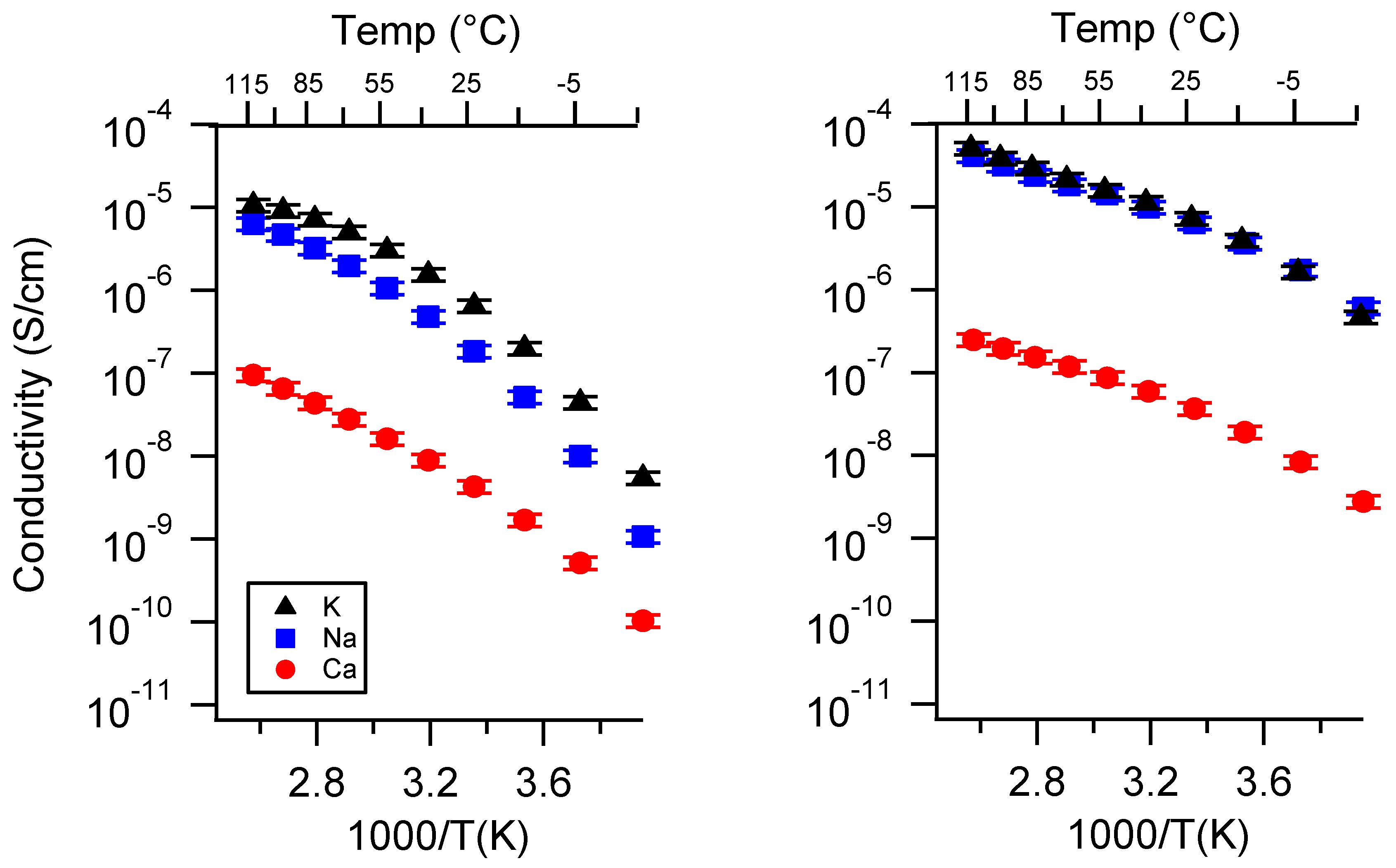
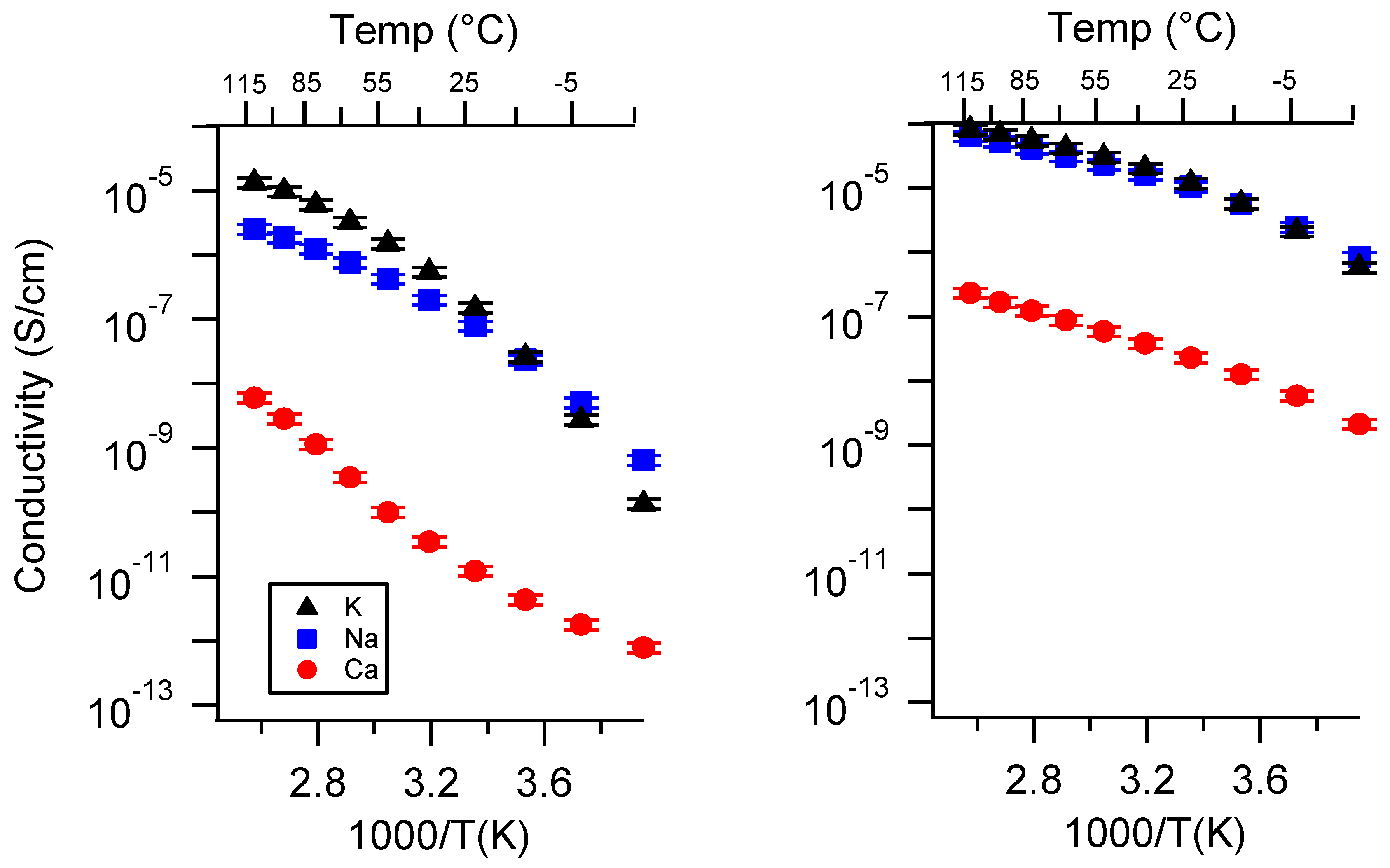
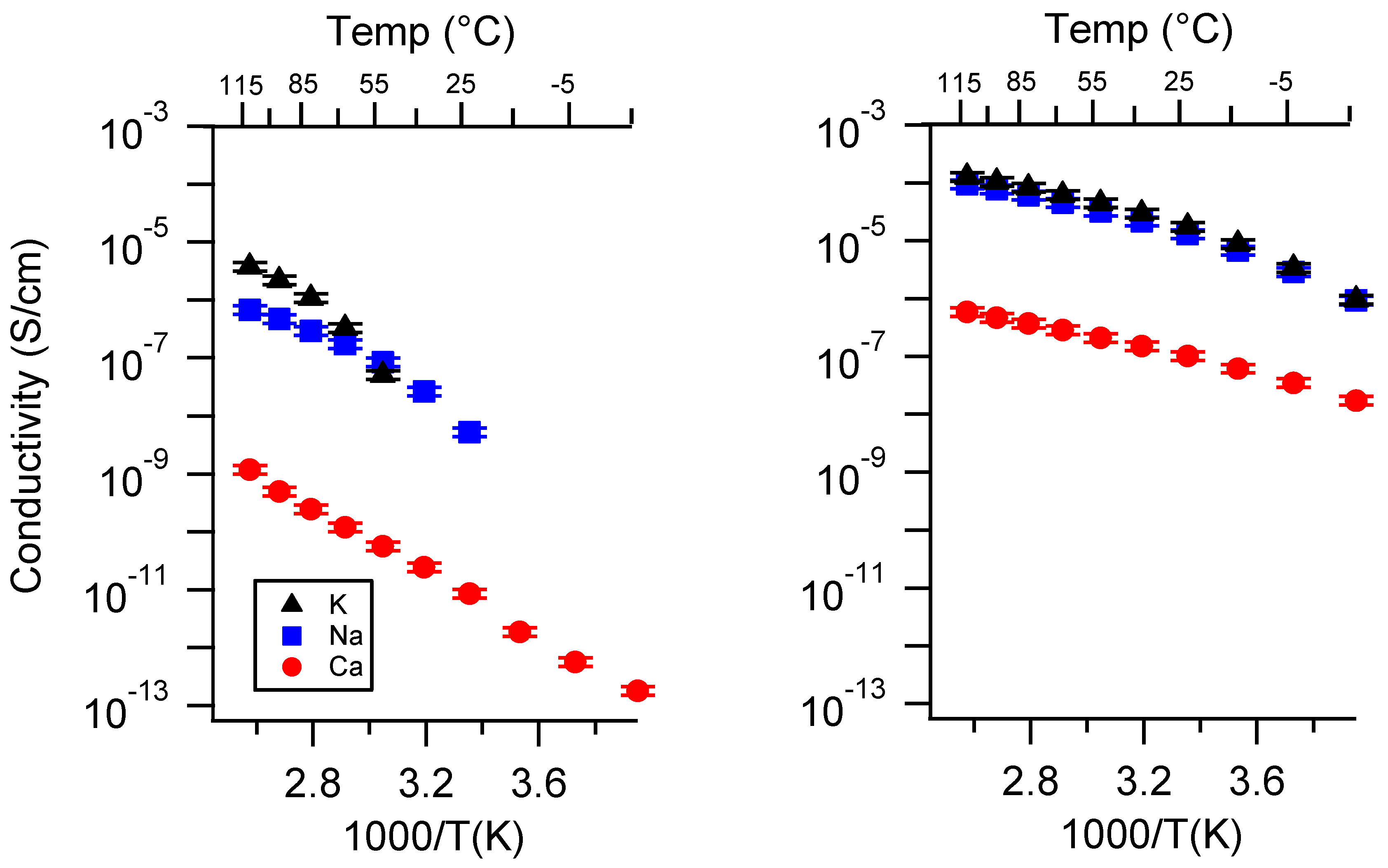
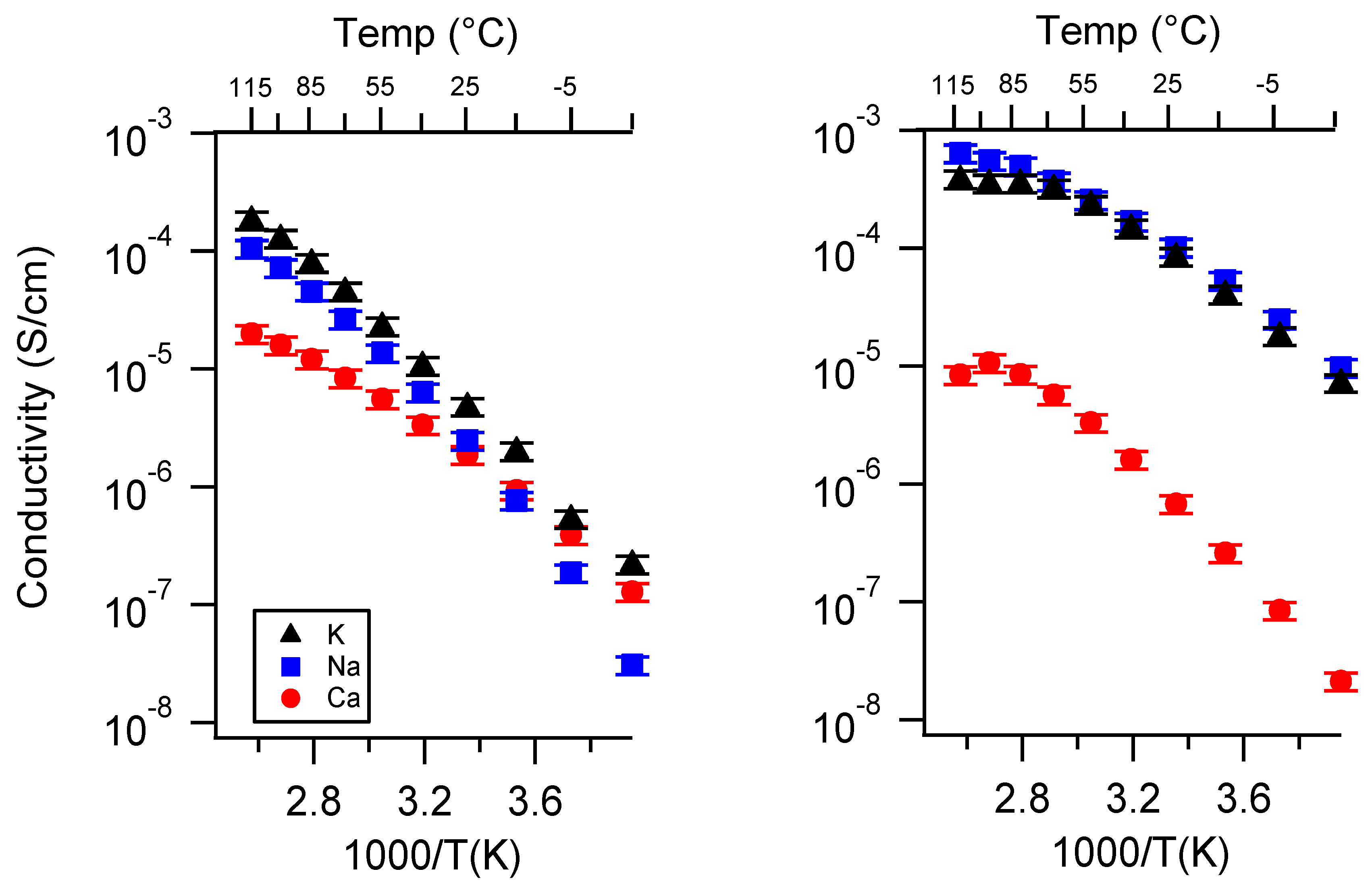
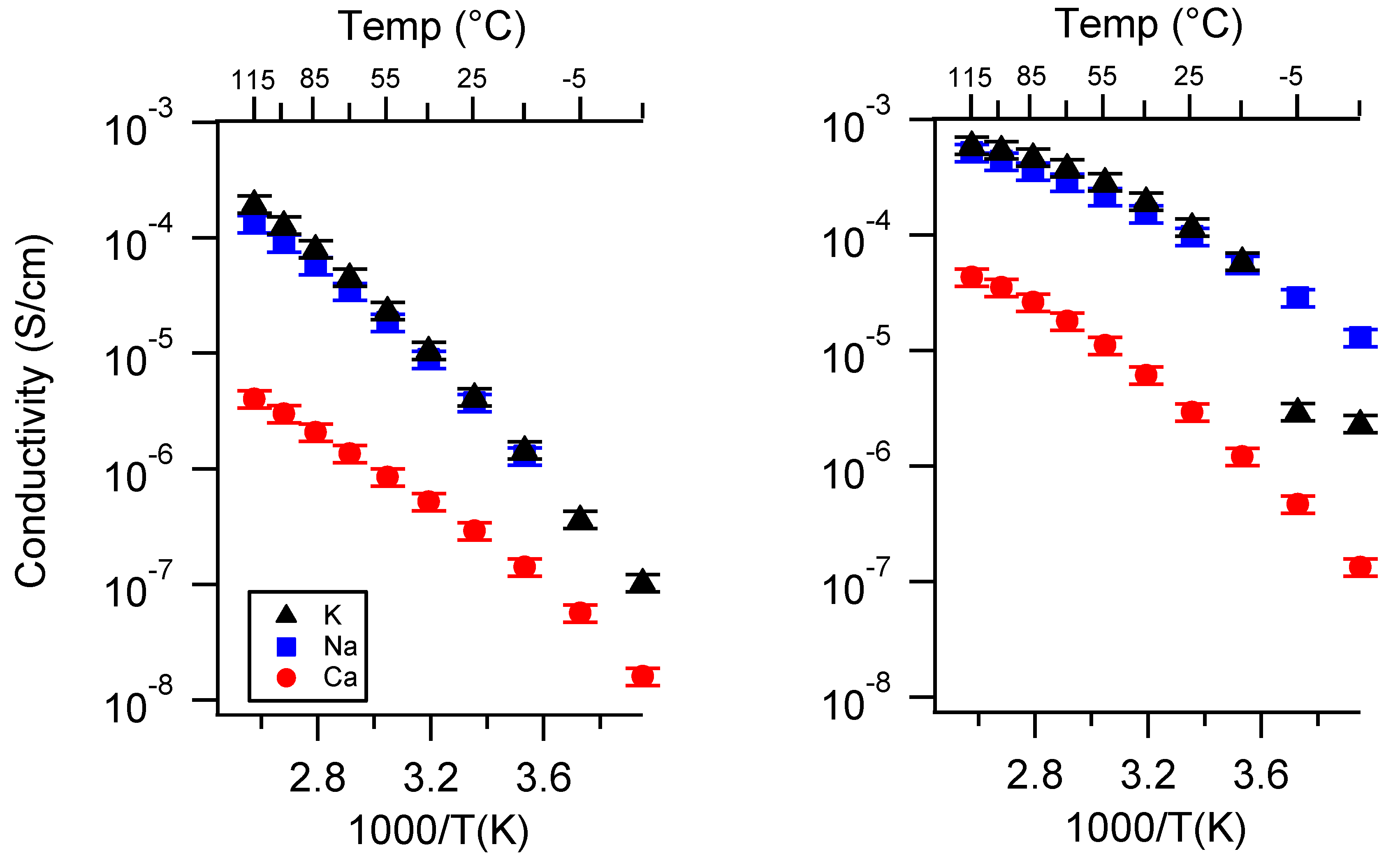
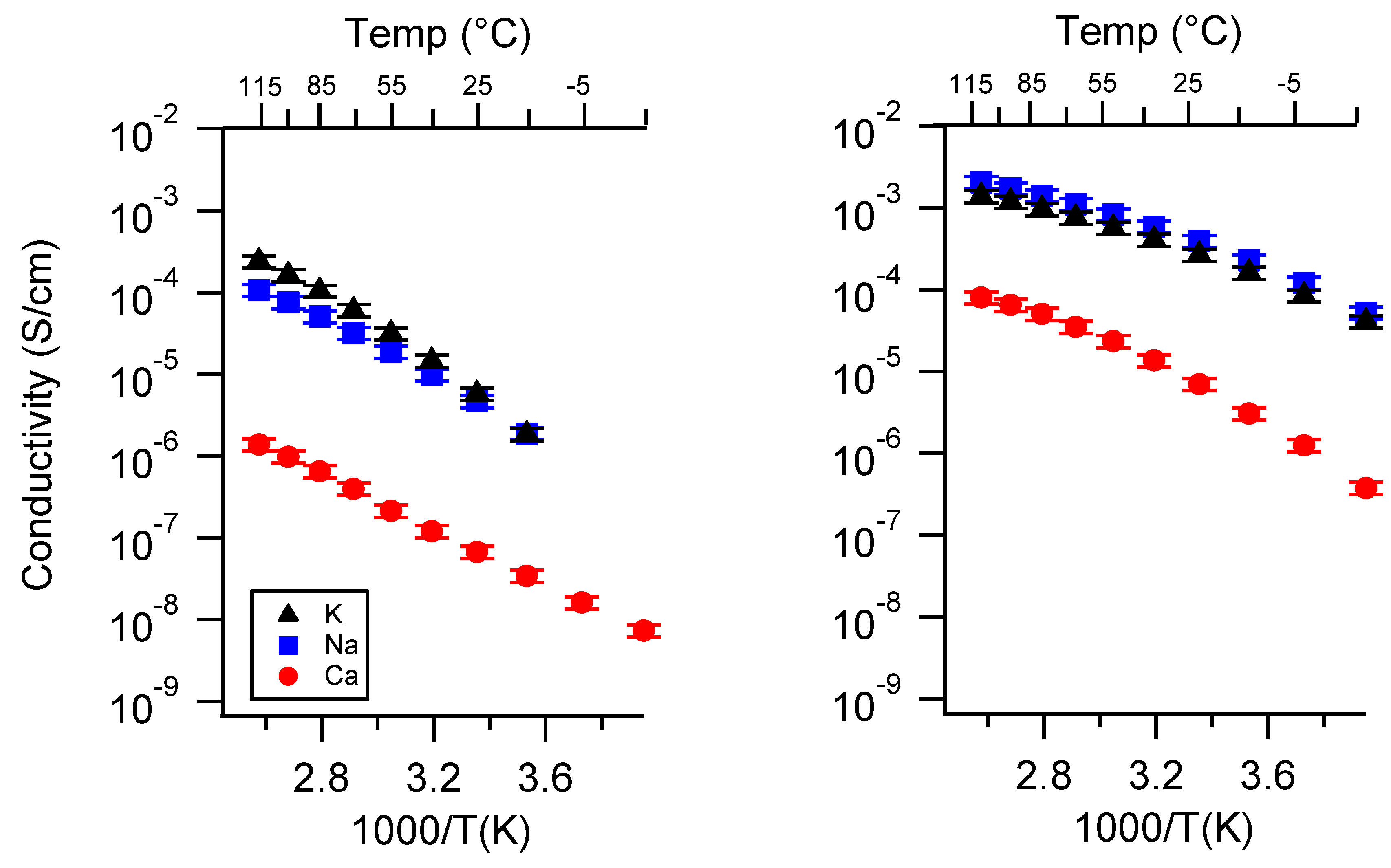
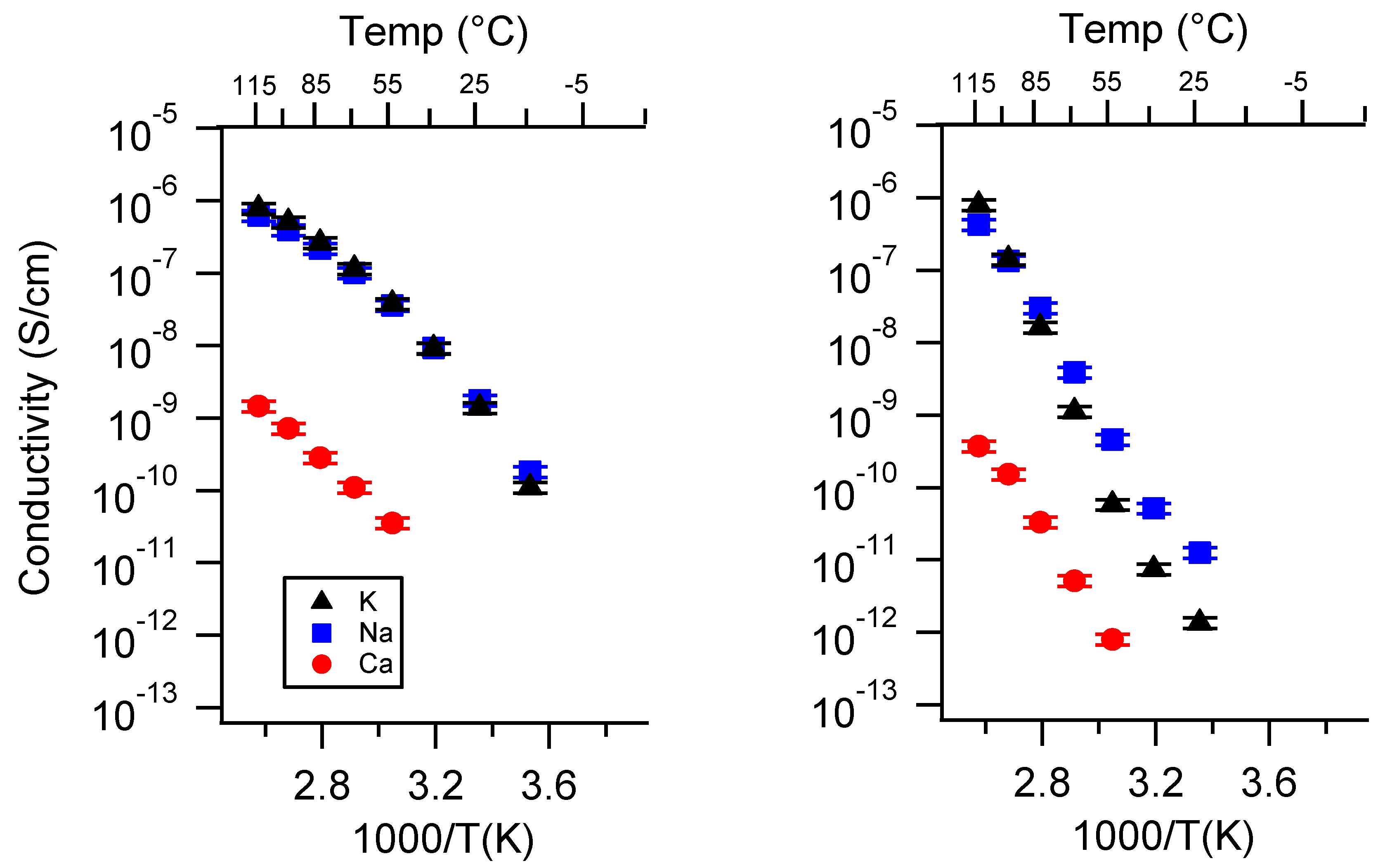
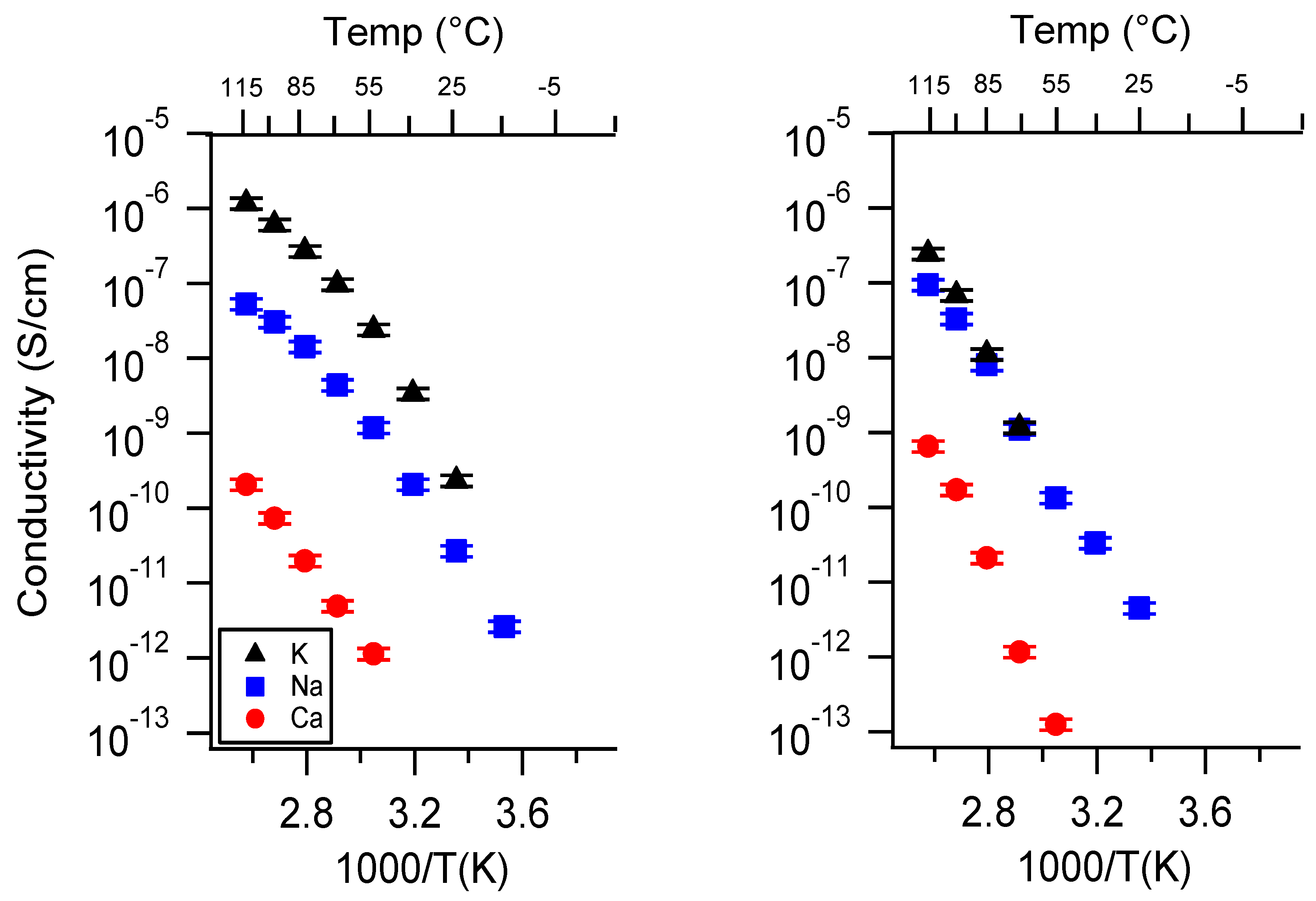
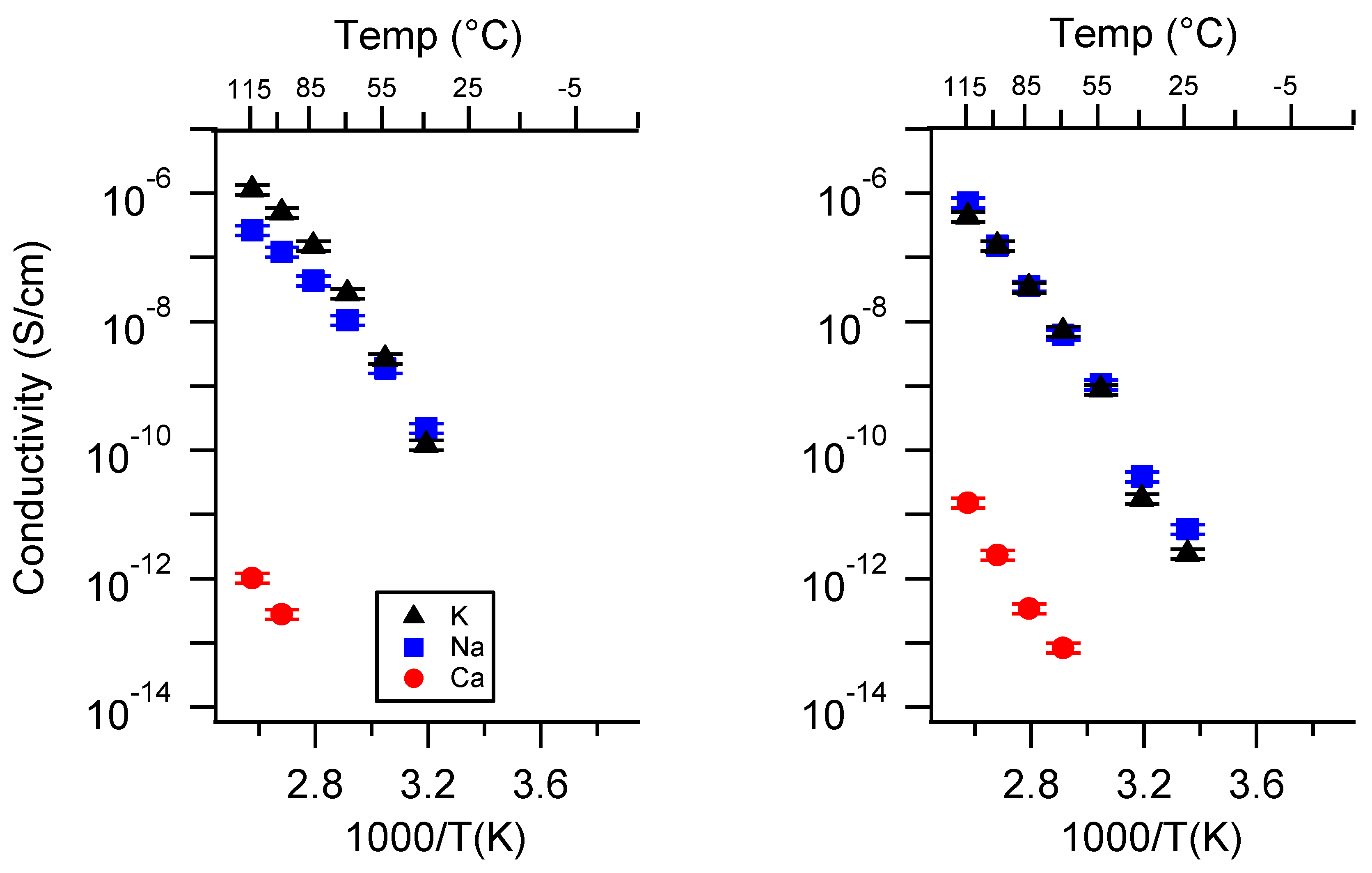
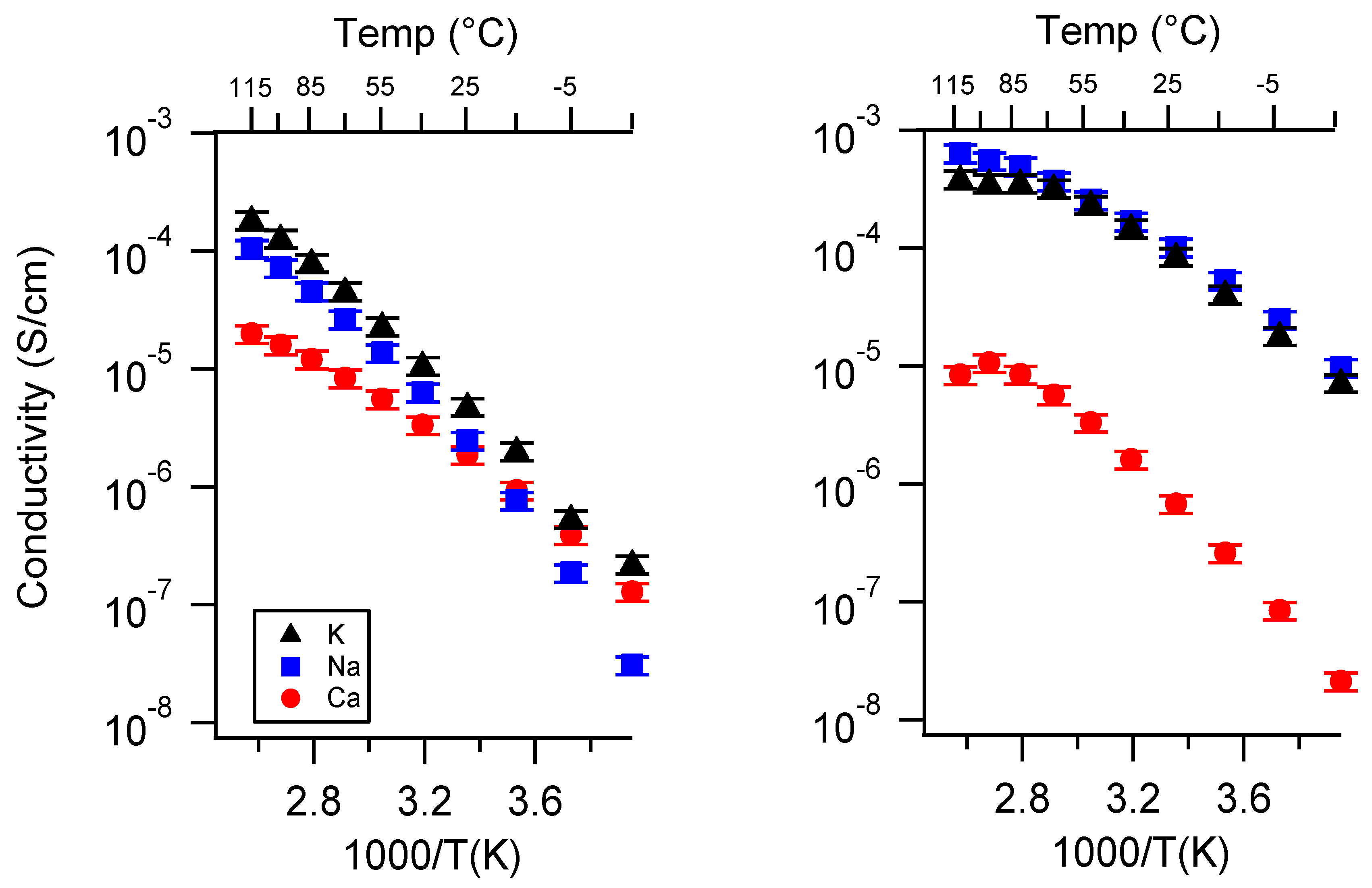
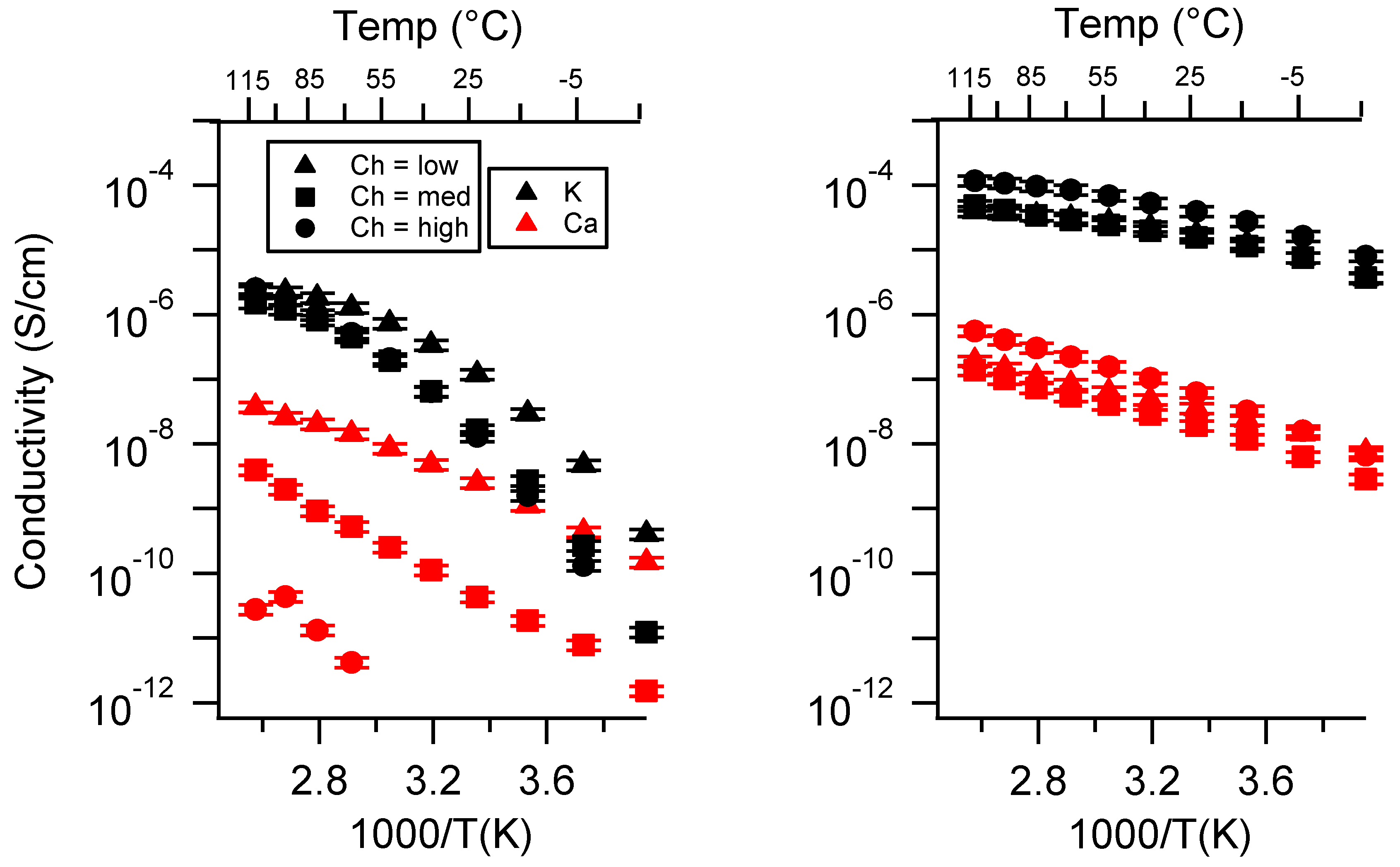
Appendix B. All SIPE Conductivity as a Function of Charge Density
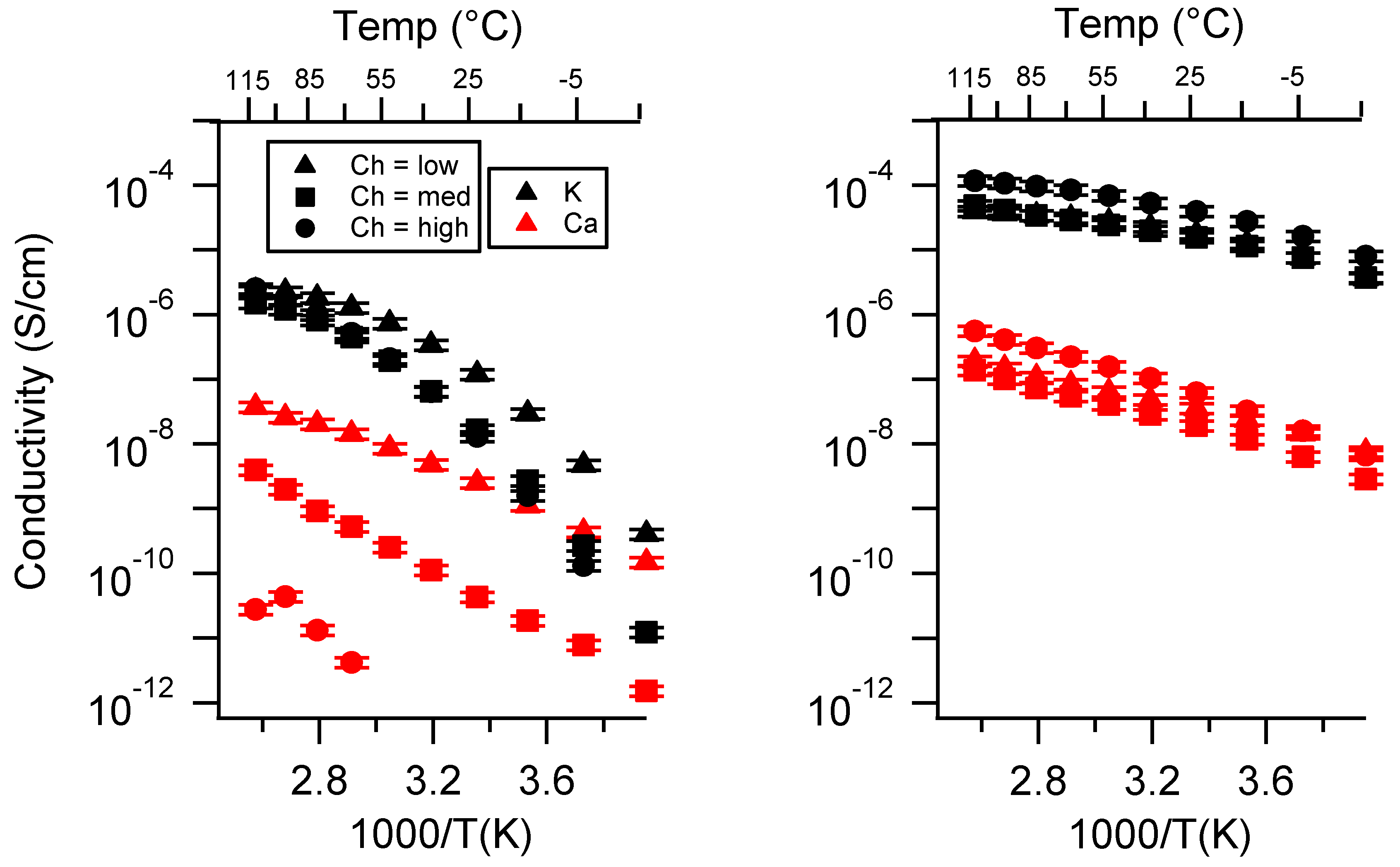
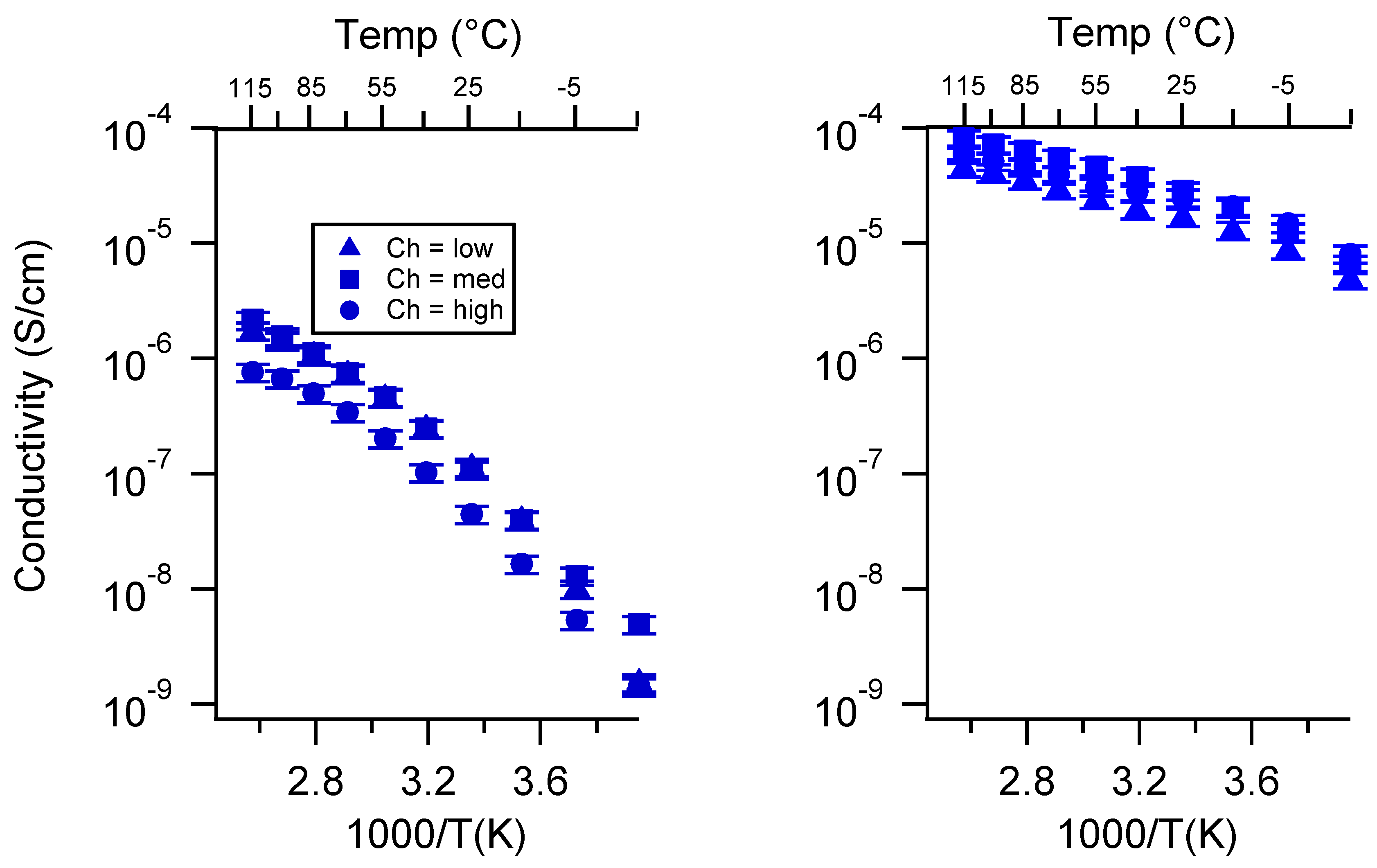

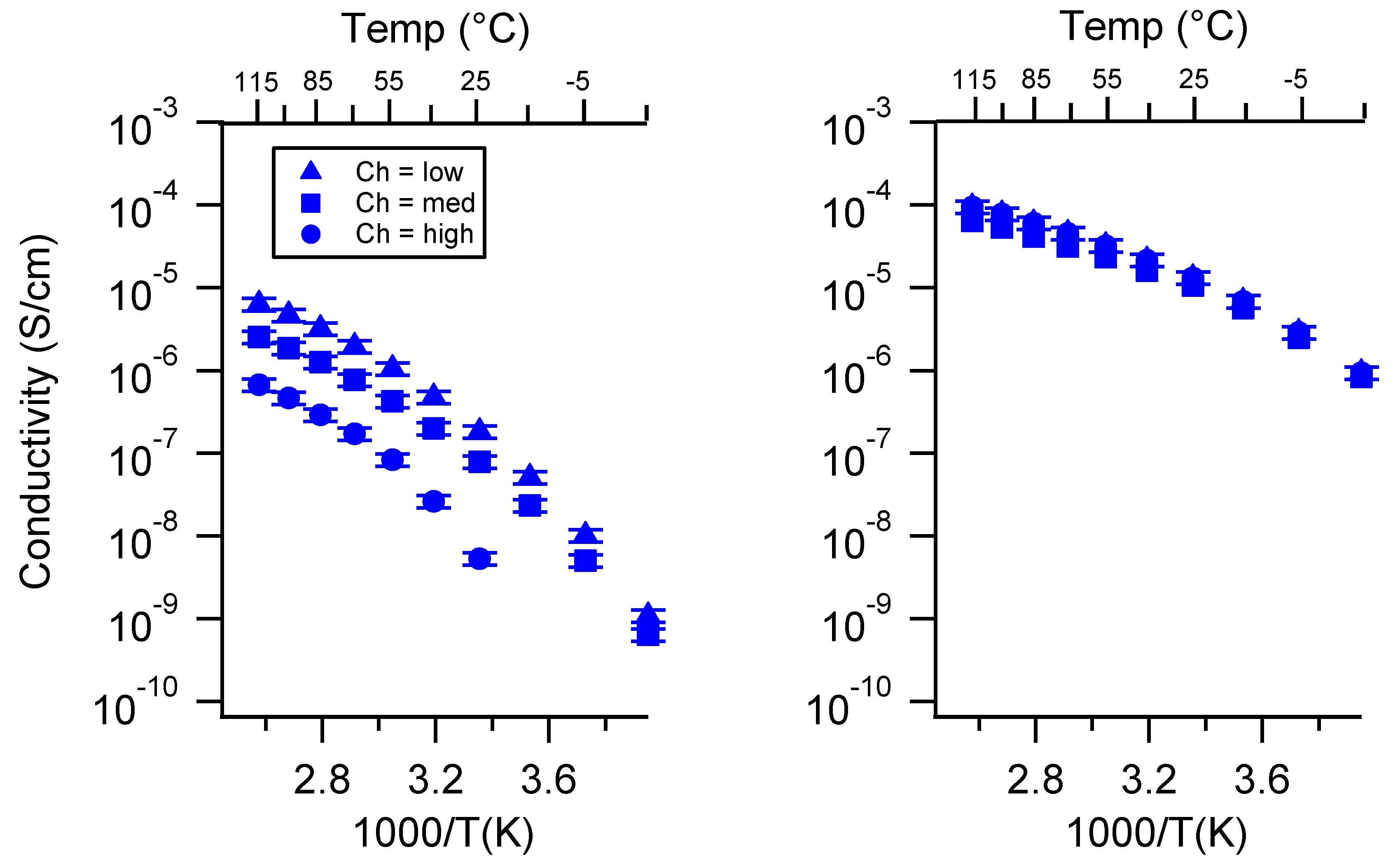

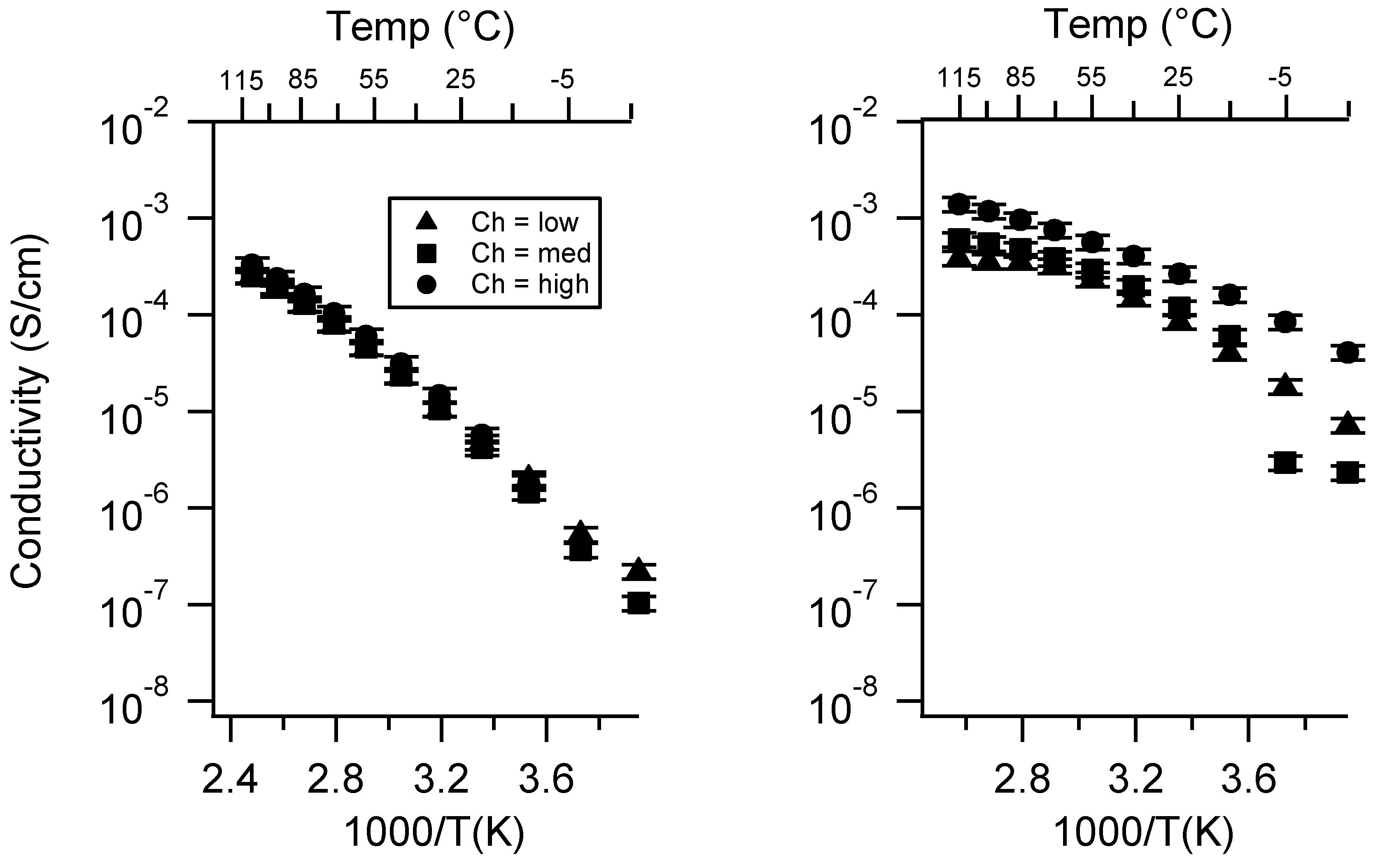
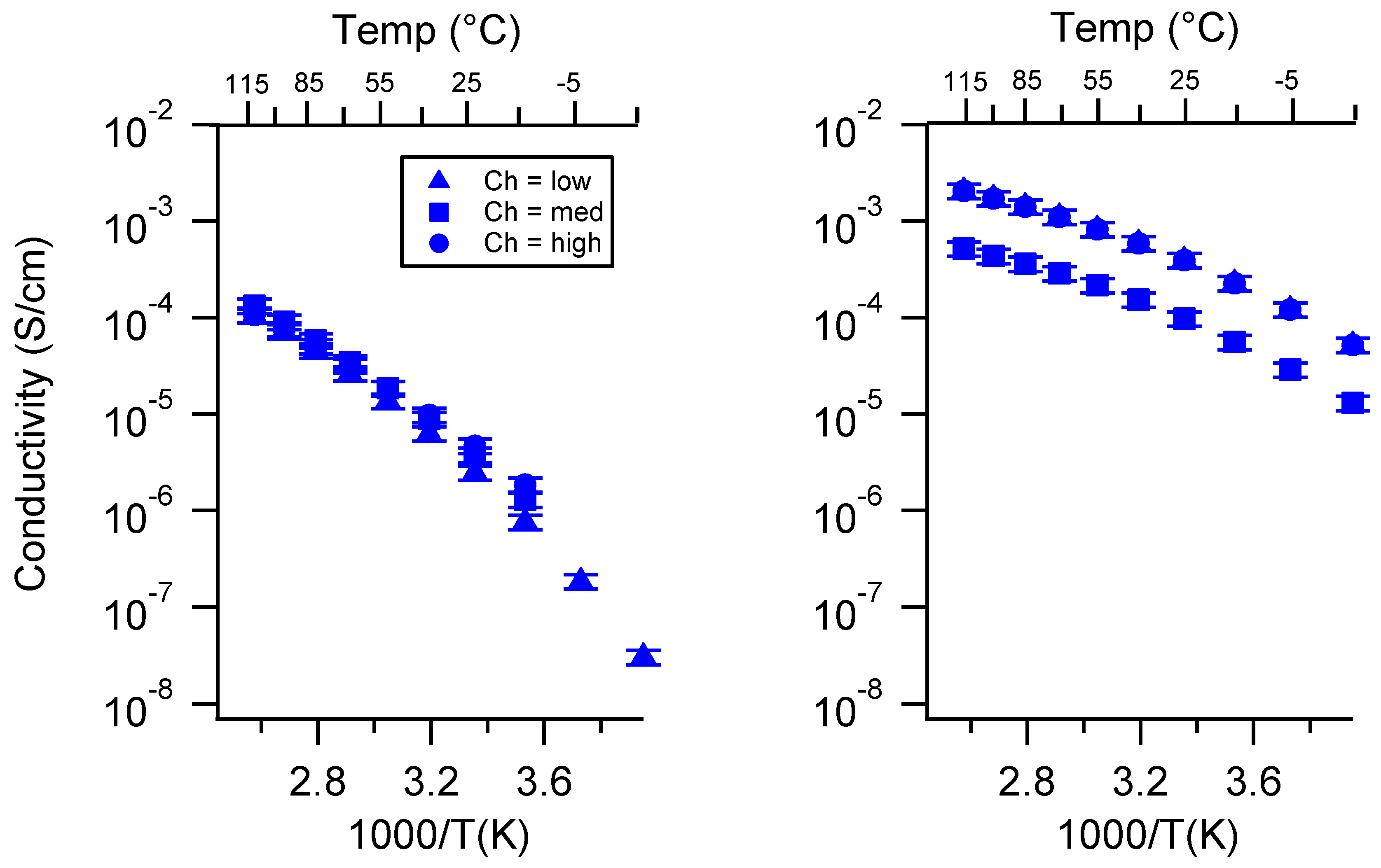
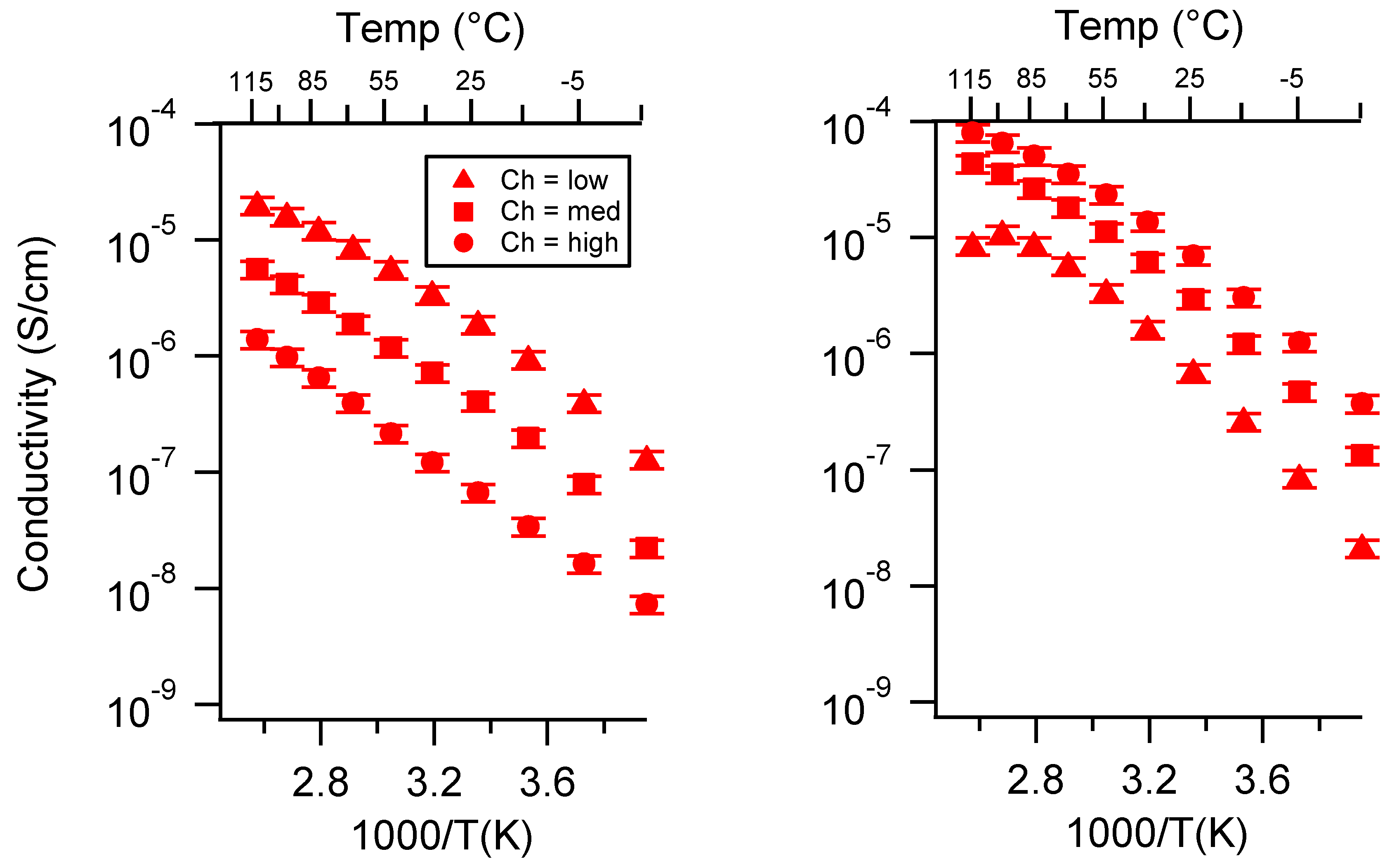
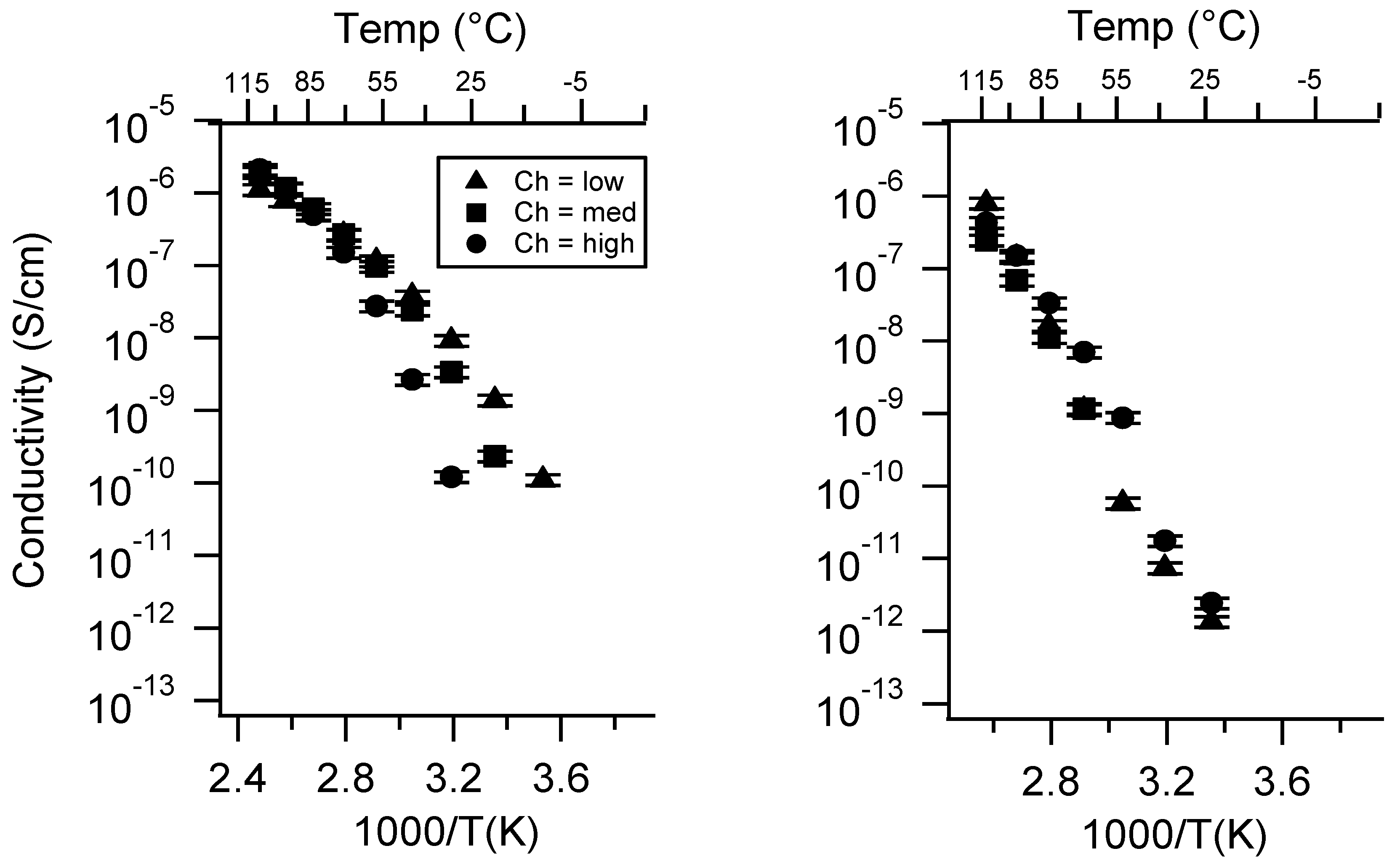
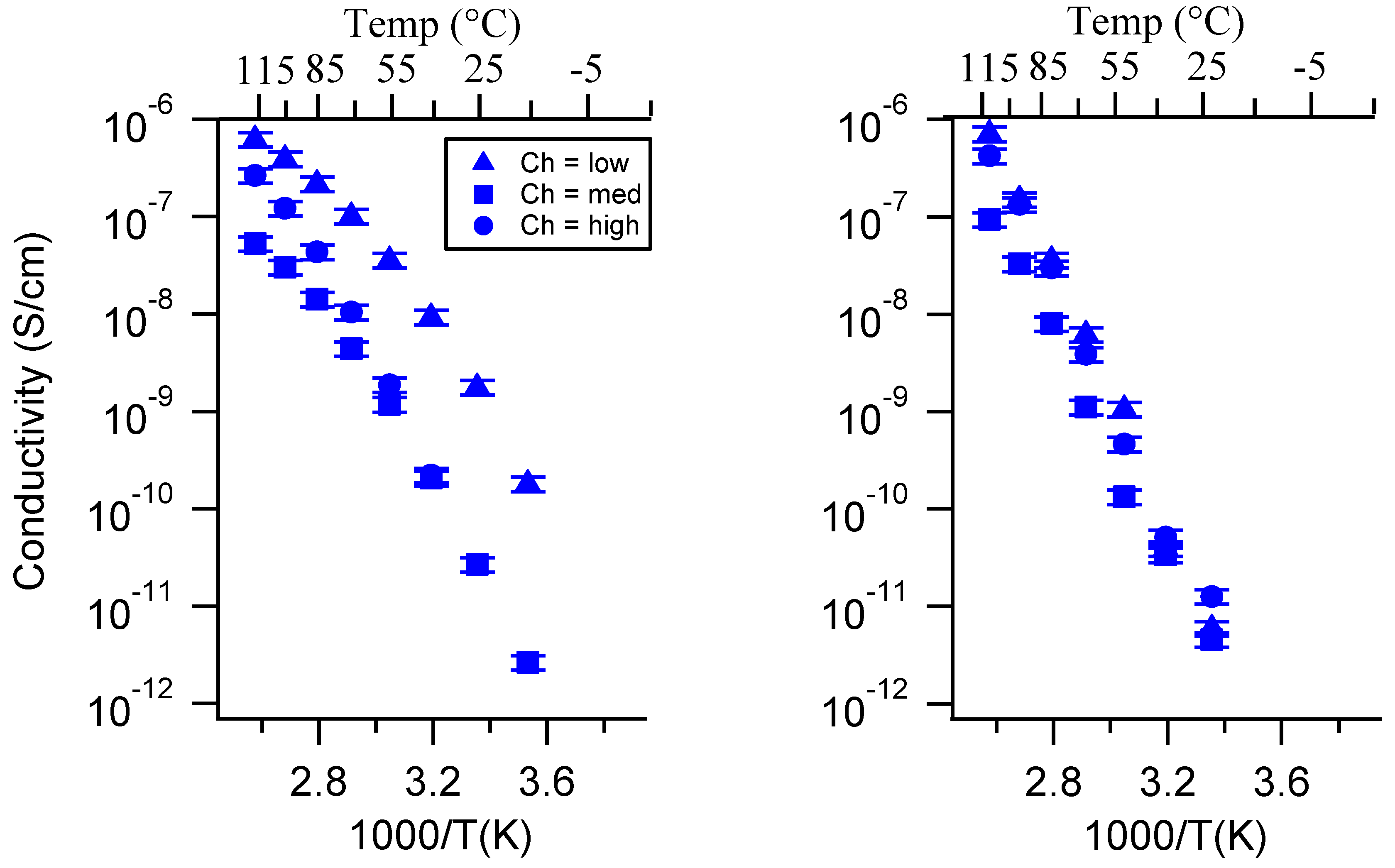
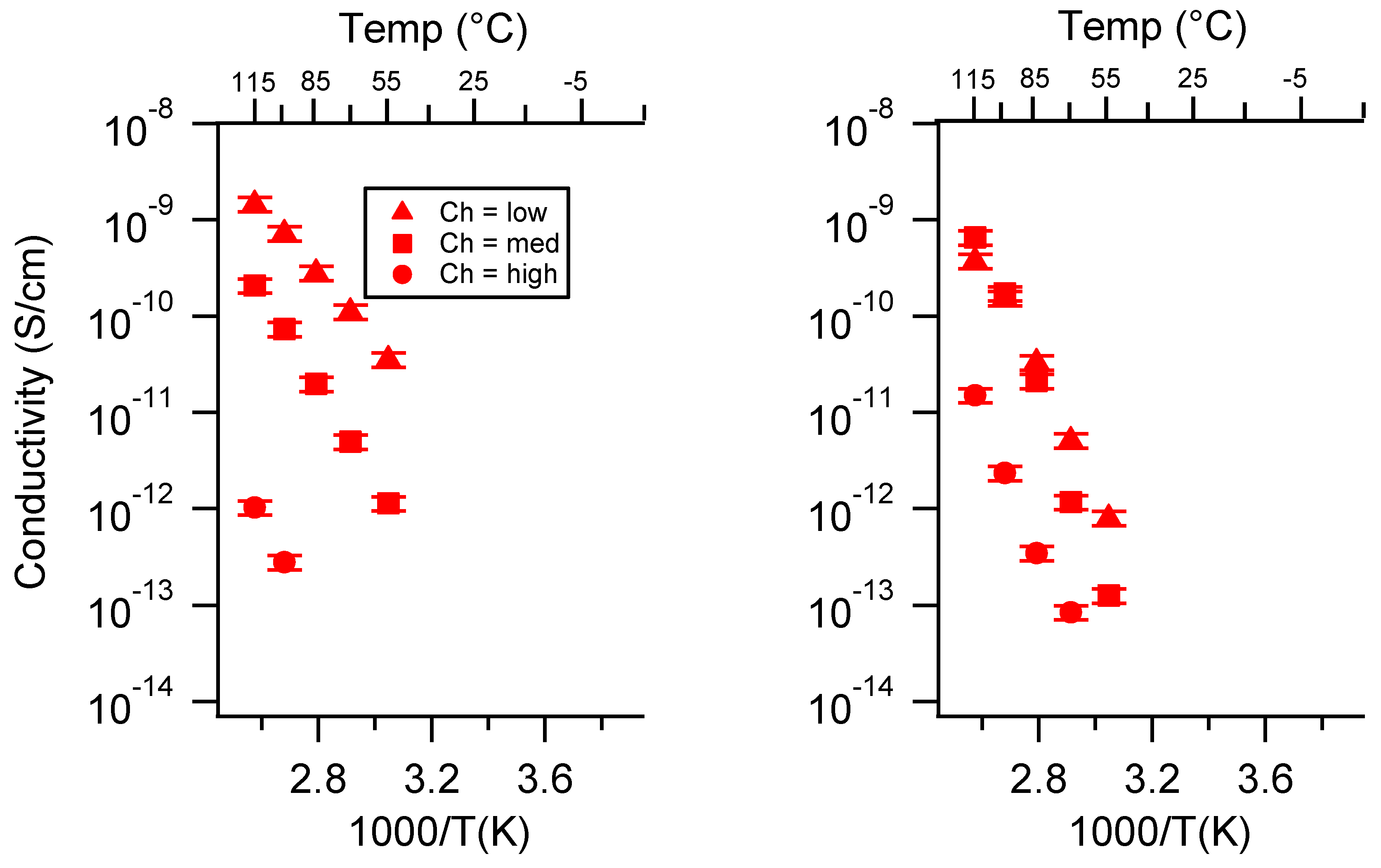
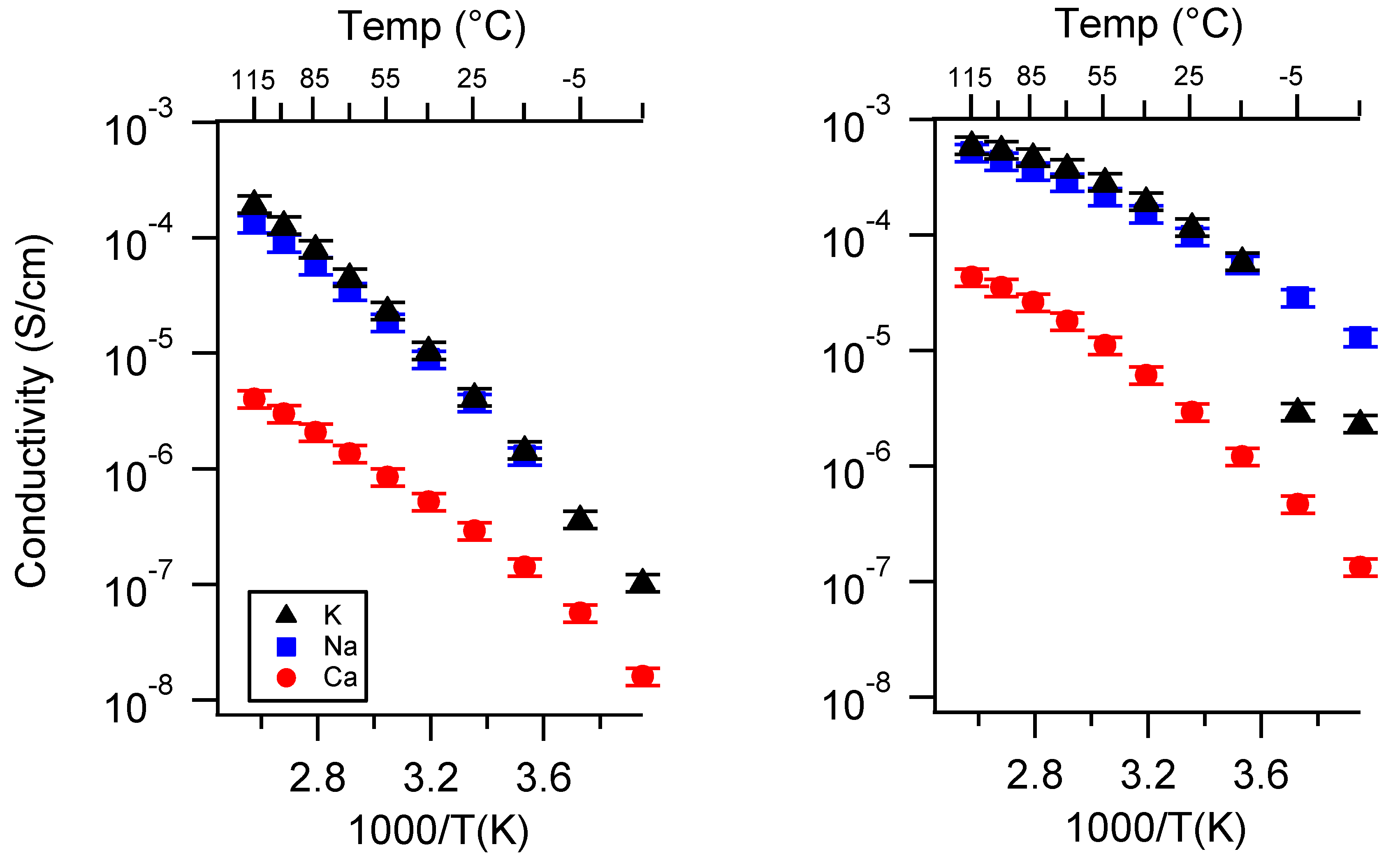
Appendix C. All SIPE Conductivity as a Function of Swelling State
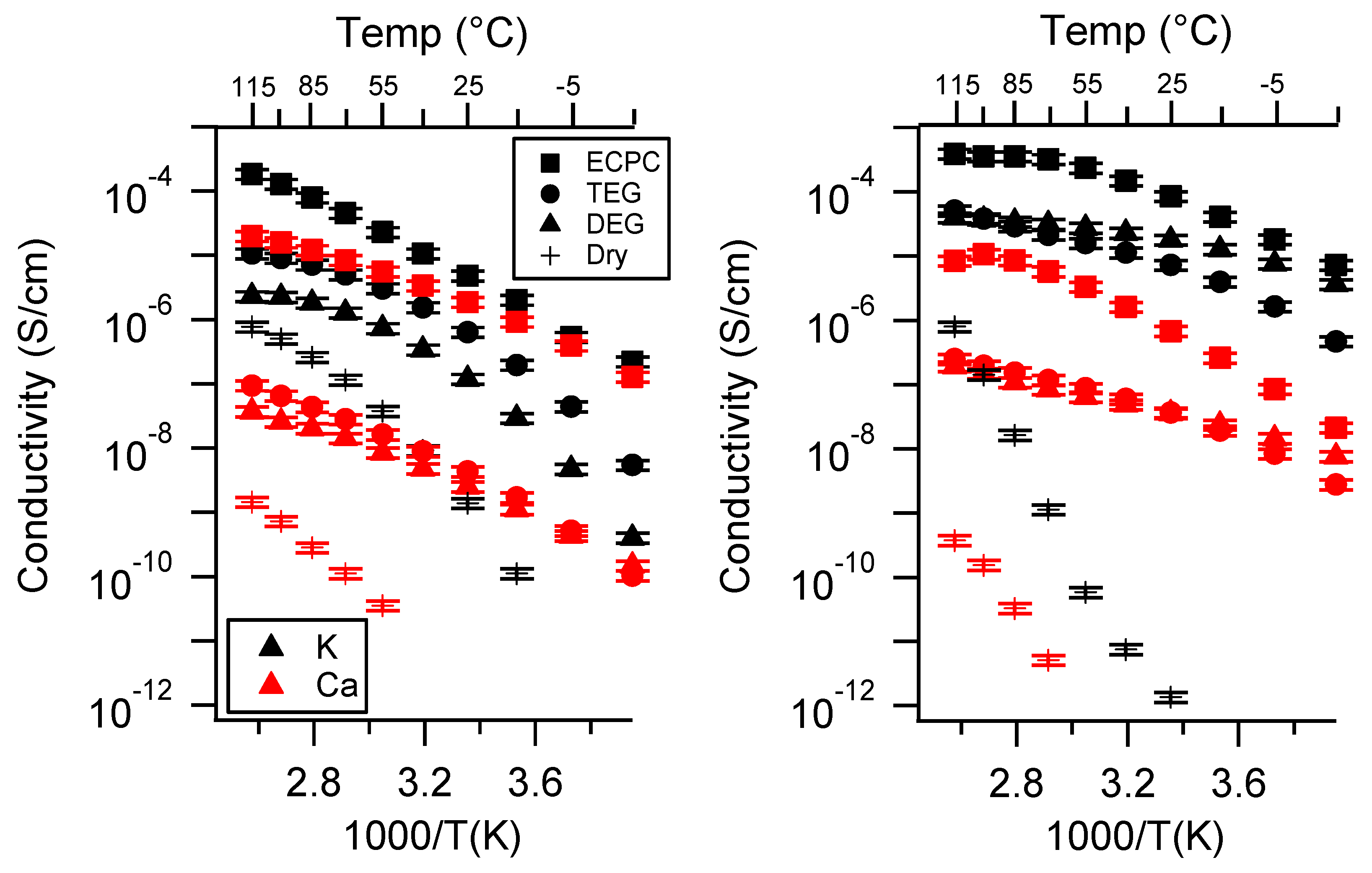
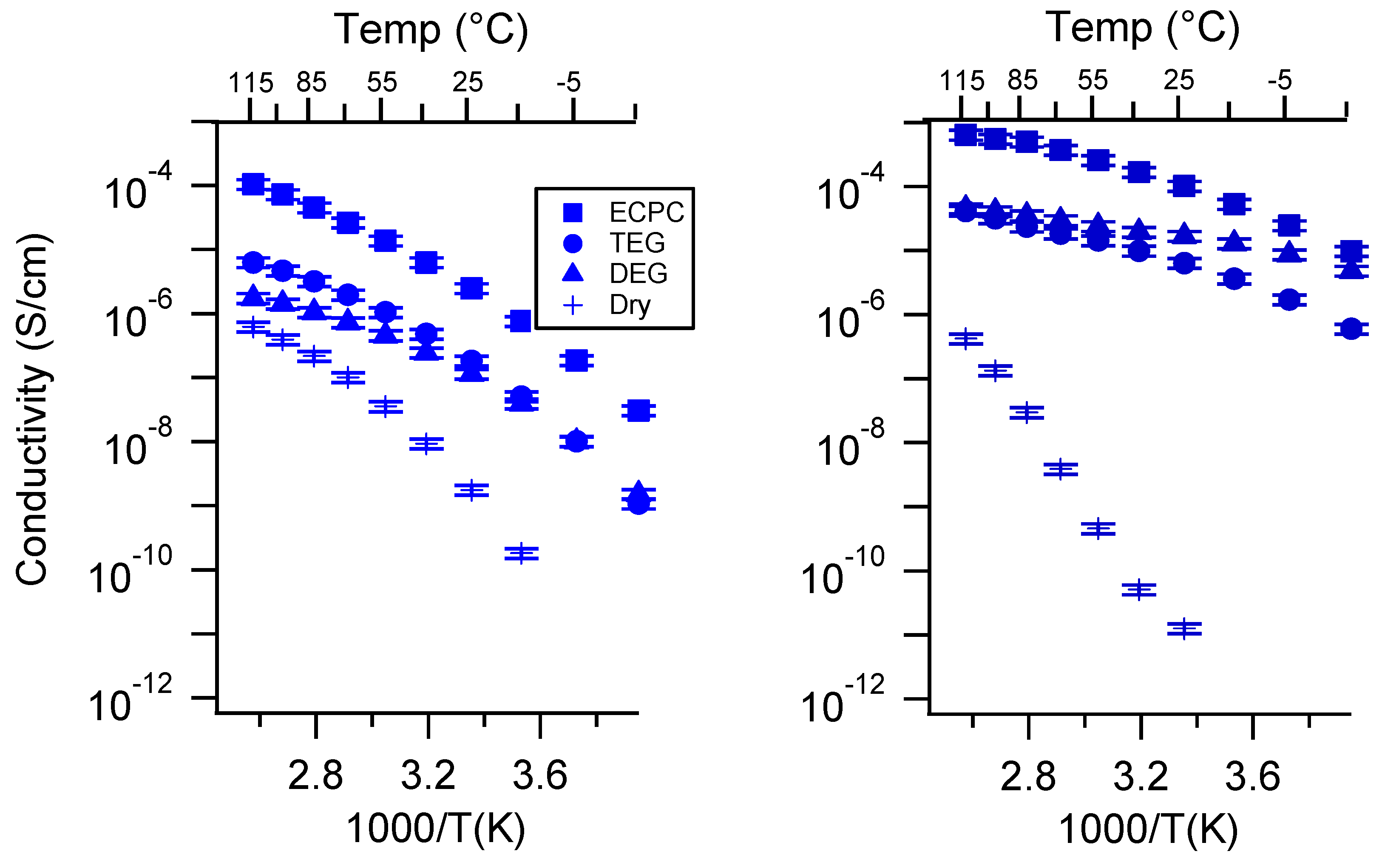
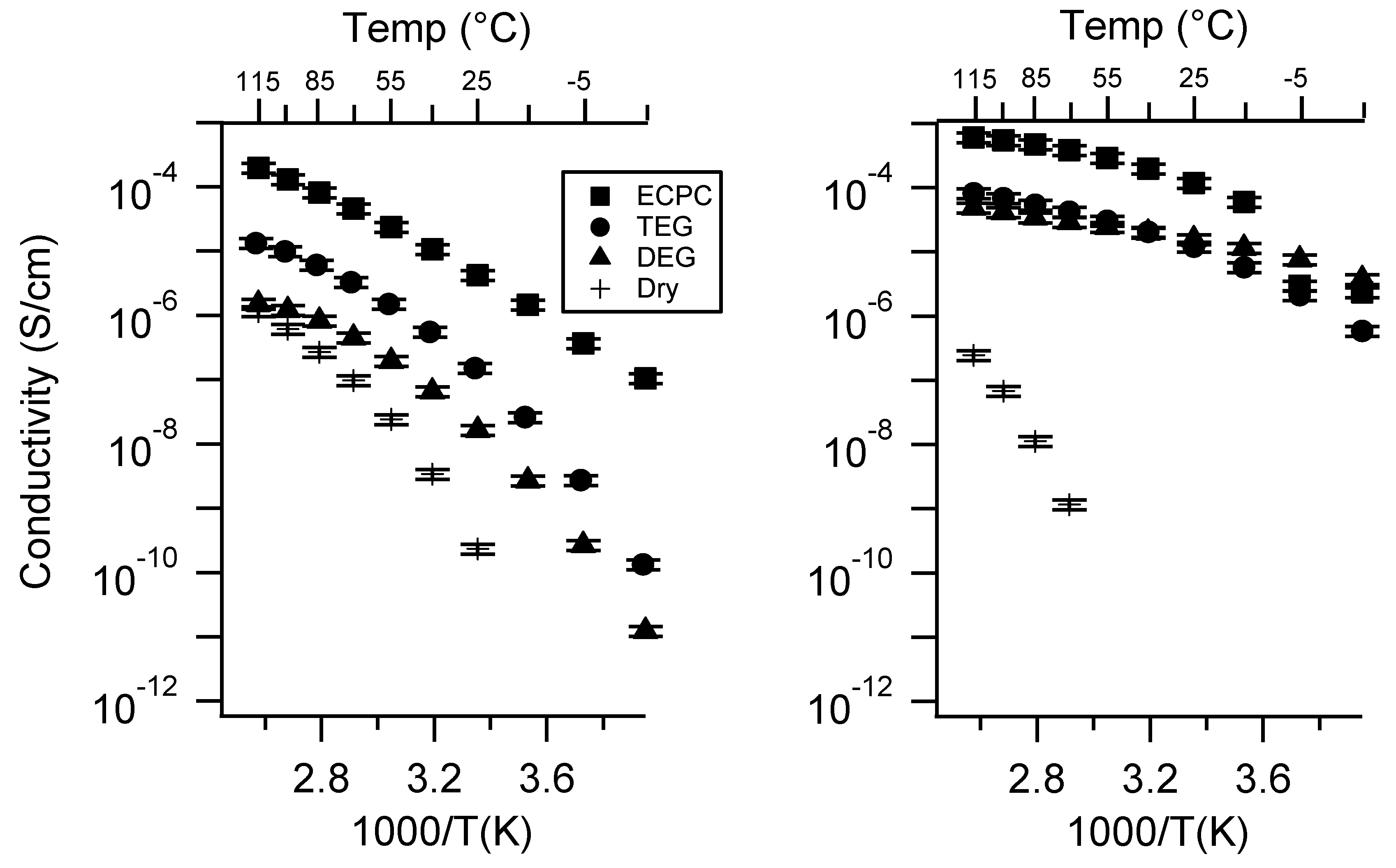
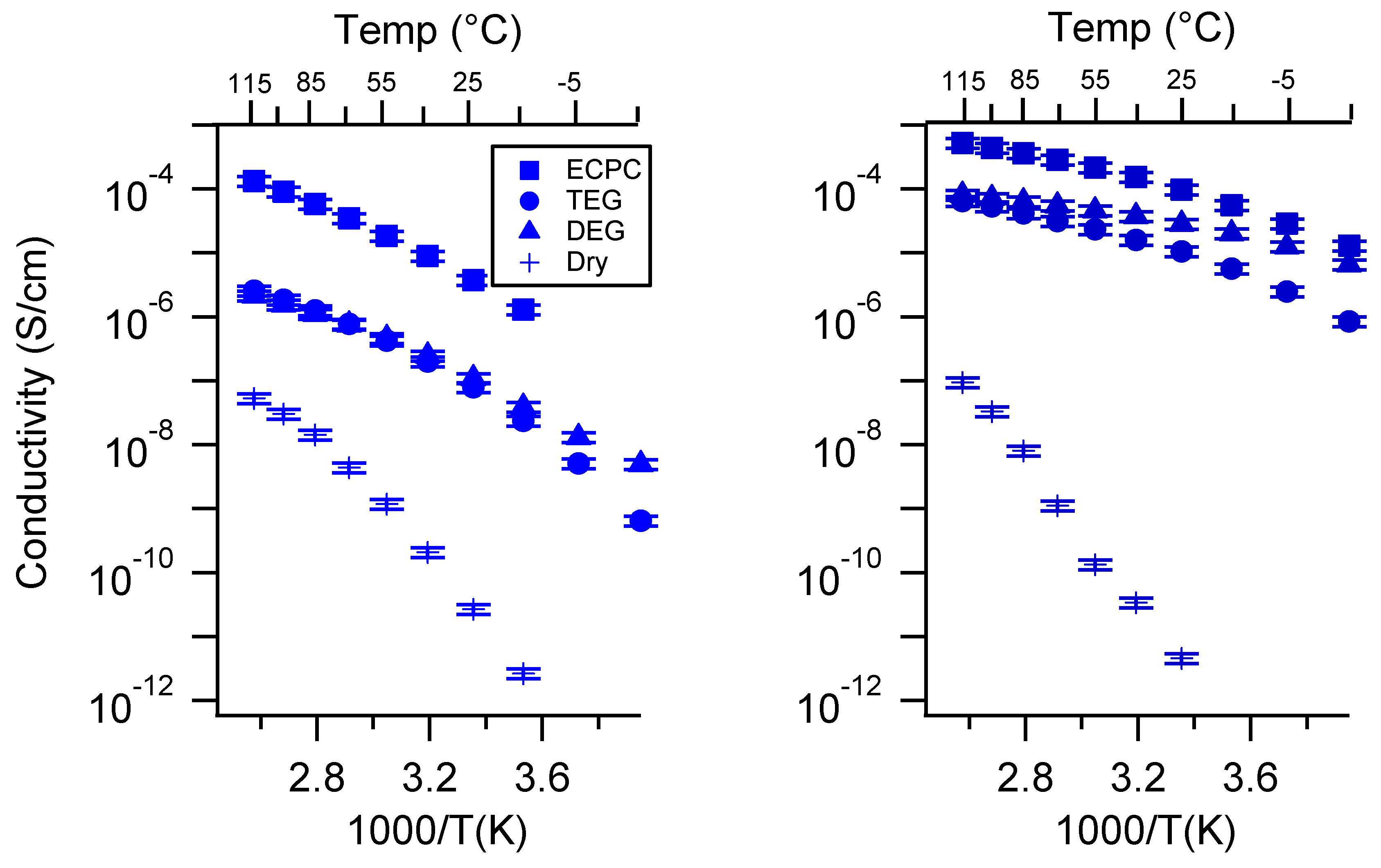
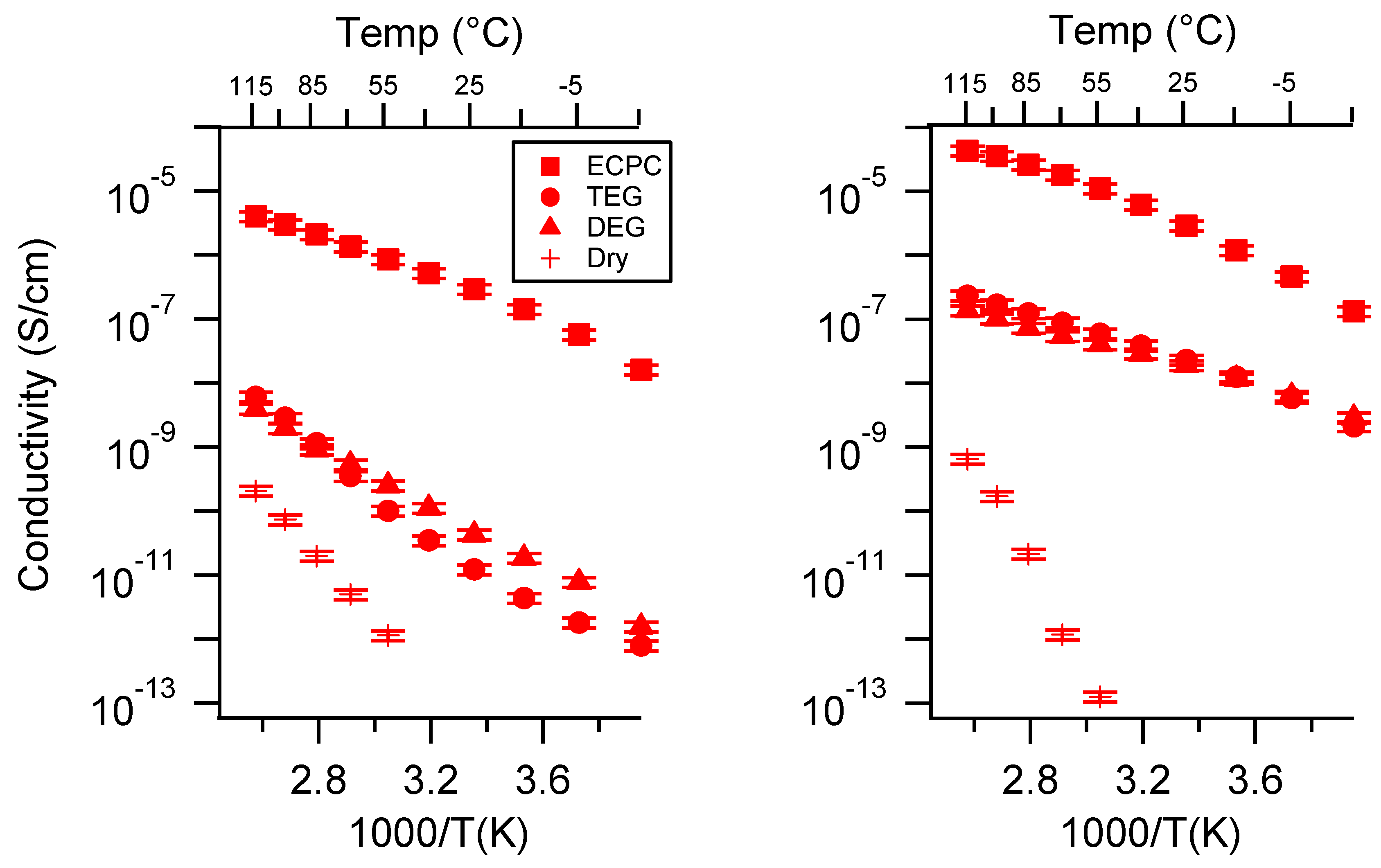
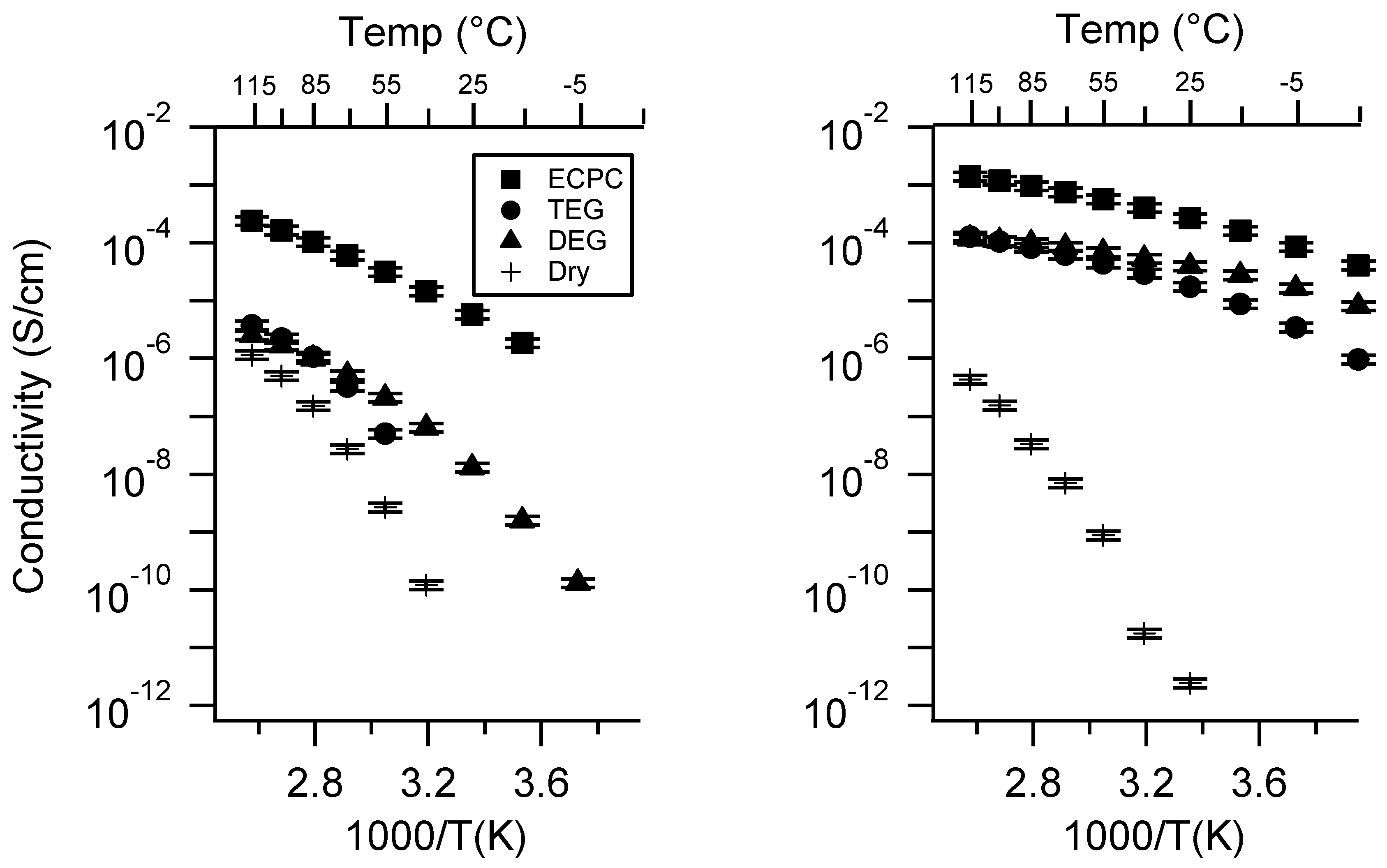
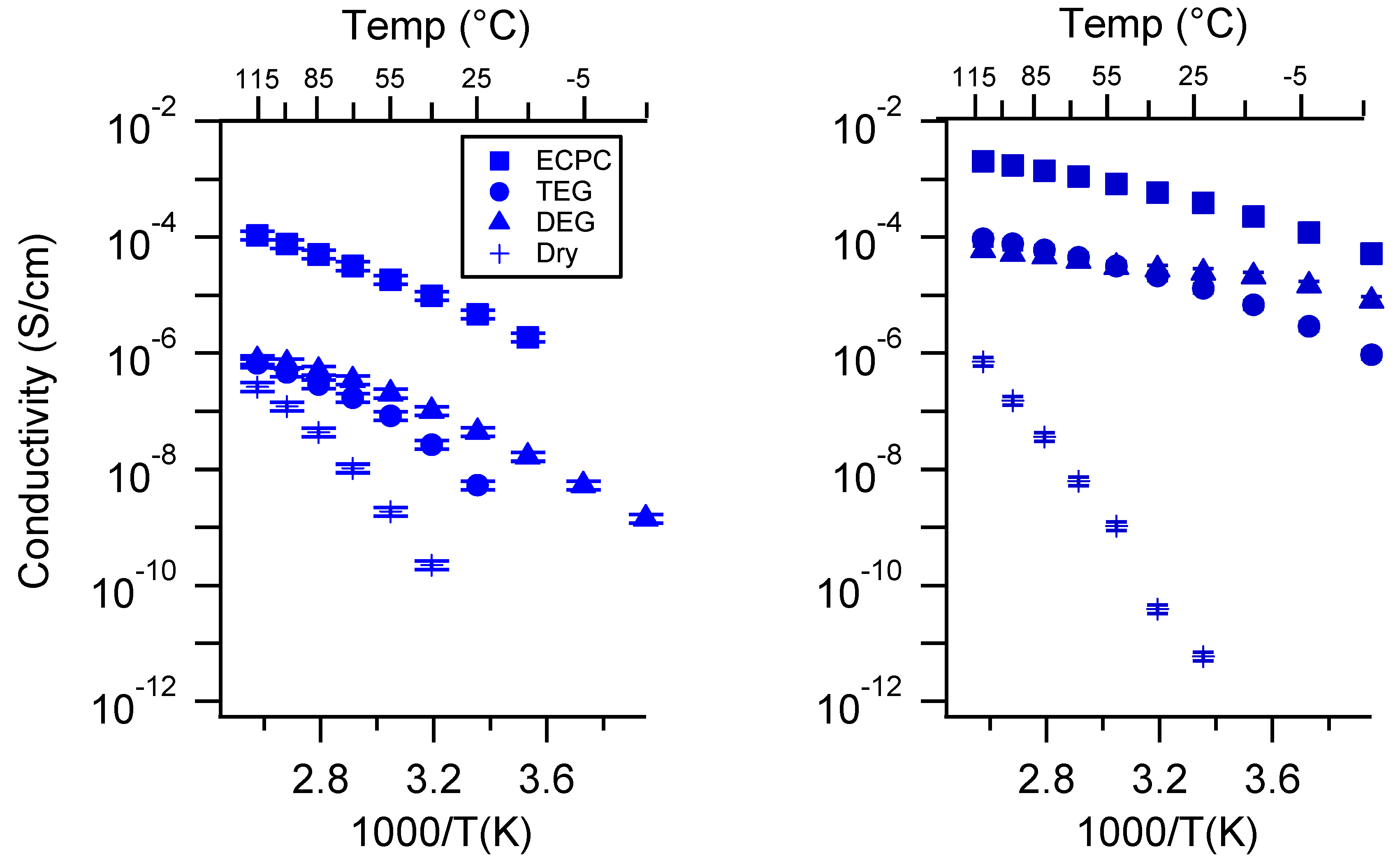

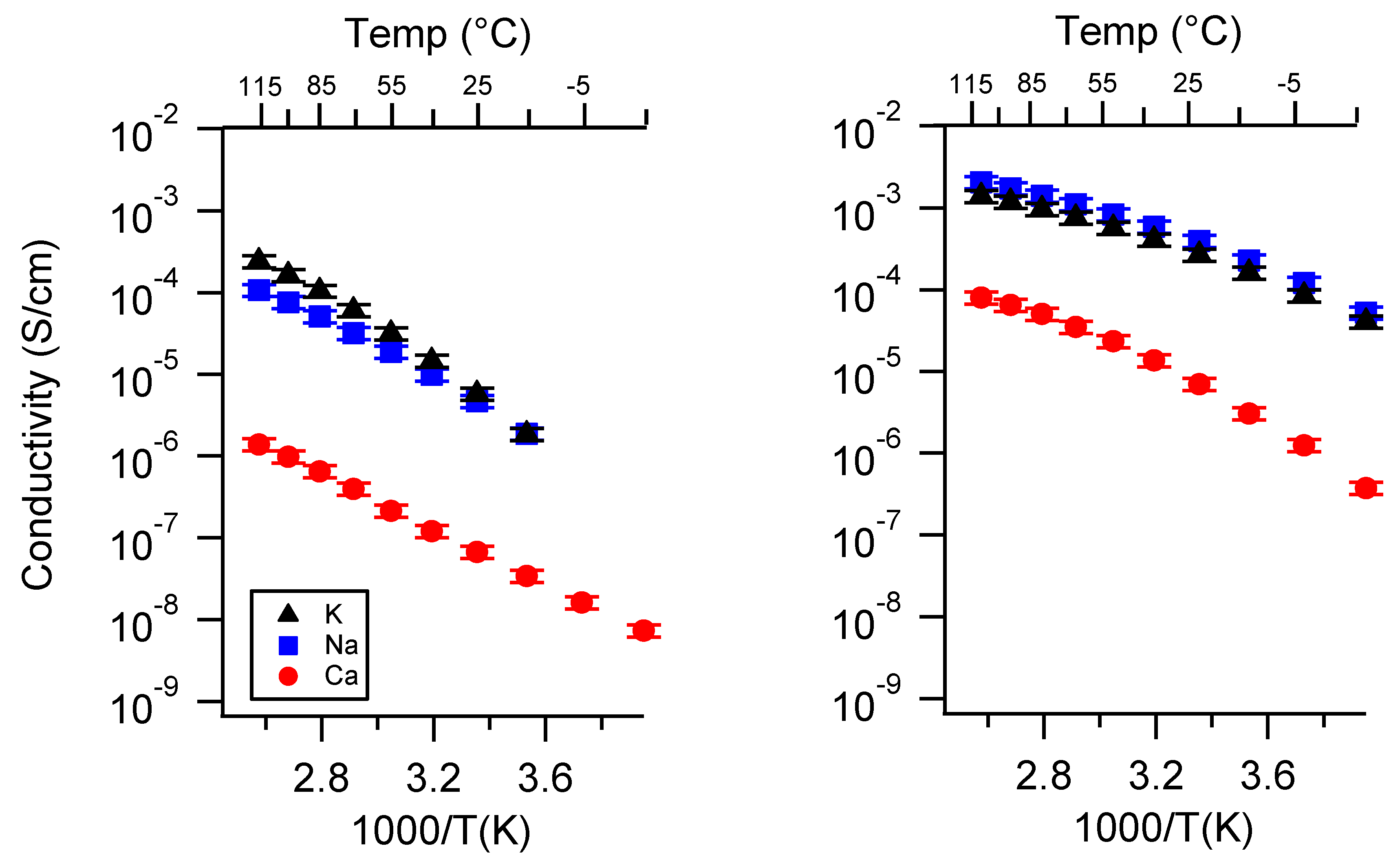
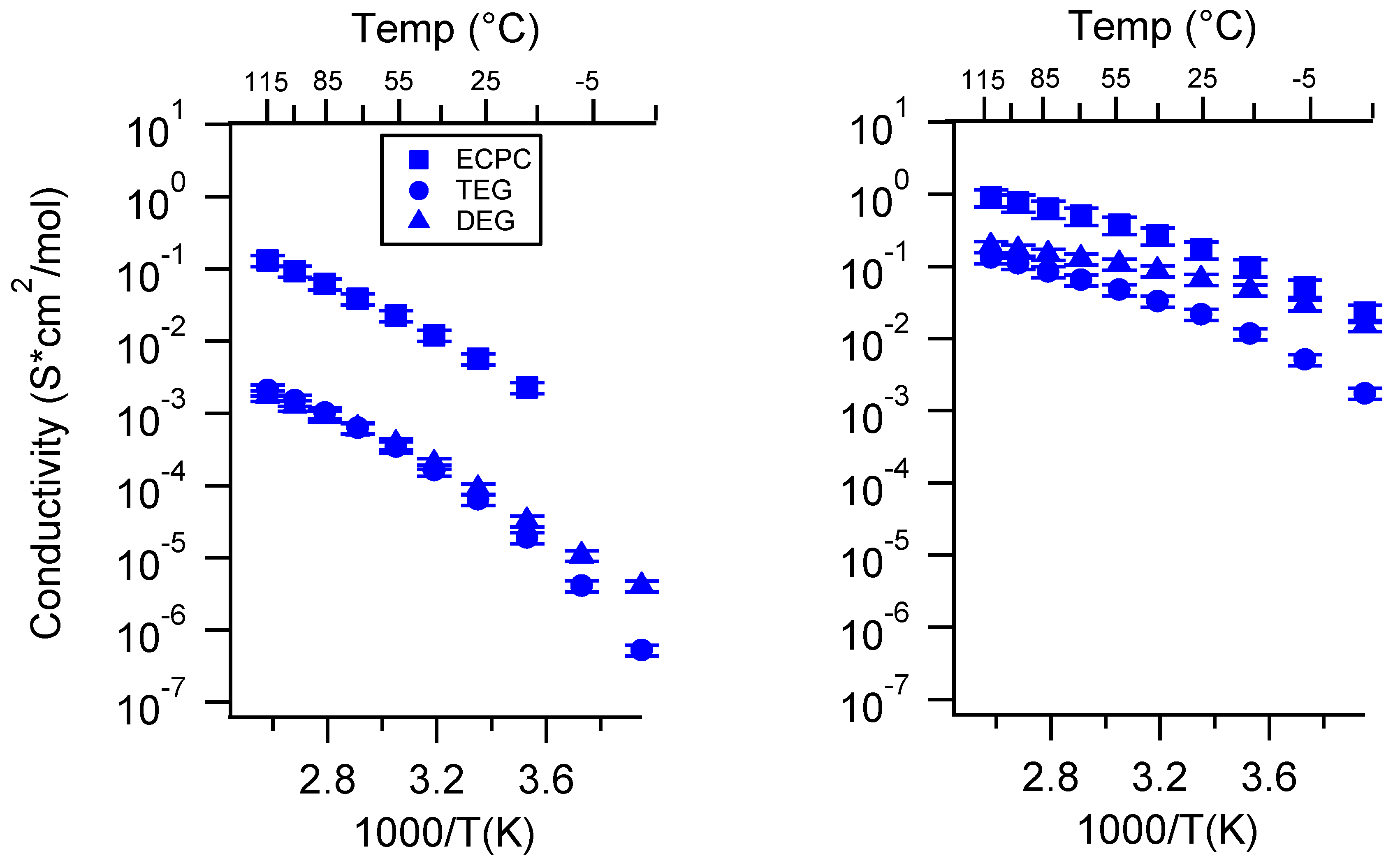
Appendix D. Volume Change with Swelling for all Other SIPEs and Select Molar Conductivity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
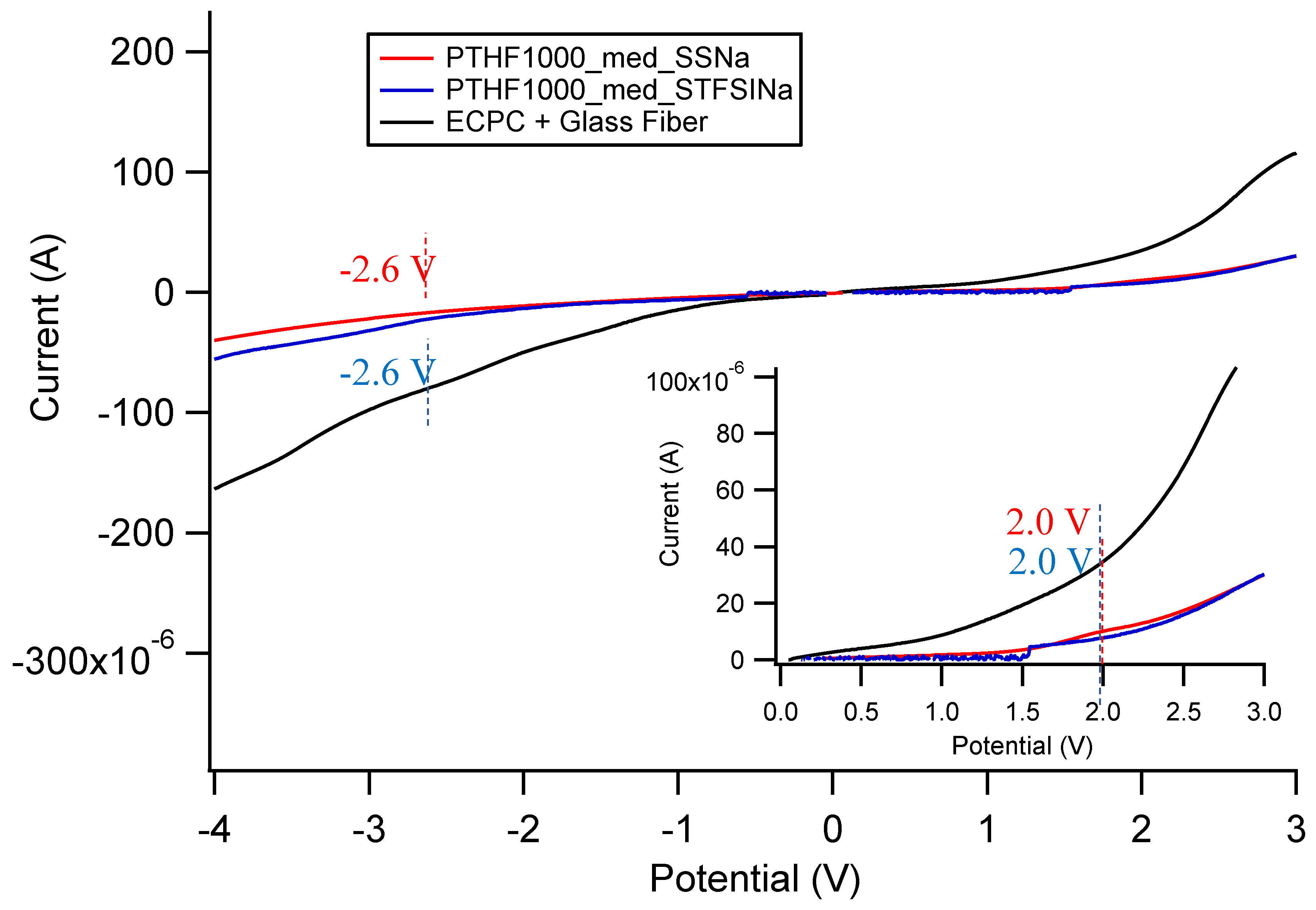{kind=link}
| Sample | Solvent | Volume Change % | Estimated Cation Concentration (mol Charge/cm3 Polymer) |
|---|---|---|---|
| PEGDMA1000_low_SSK | DEG | 5.8 ± 0.3 | 0.00093 |
| TEG | 34.9 ± 2.0 | 0.00073 | |
| EC:PC | 90.7 ± 5.2 | 0.00052 | |
| PEGDMA1000_med_SSK | DEG | 15.4 ± 0.9 | 0.00127 |
| TEG | 28.1 ± 1.6 | 0.00114 | |
| EC:PC | 92.0 ± 5.2 | 0.00076 | |
| PEGDMA1000_high_SSK | DEG | 12.2 ± 0.7 | 0.00173 |
| TEG | 20.5 ± 1.2 | 0.00162 | |
| EC:PC | 93.4 ± 5.3 | 0.00101 | |
| PEGDMA1000_low_SSNa | DEG | 37.9 ± 2.2 | 0.00071 |
| TEG | 8.3 ± 0.5 | 0.00091 | |
| EC:PC | 109.7 ± 6.3 | 0.00047 | |
| PEGDMA1000_high_SSNa | DEG | 4.6 ± 0.2 | 0.00186 |
| TEG | 6.4 ± 0.4 | 0.00183 | |
| EC:PC | 99.6 ± 5.7 | 0.00098 | |
| PEGDMA1000_low_SSCa | DEG | 0.0 | 0.00049 |
| TEG | 0.0 | 0.00049 | |
| EC:PC | 16.1 ± 0.9 | 0.00042 | |
| PEGDMA1000_med_SSCa | DEG | 0.0 | 0.00073 |
| TEG | 0.0 | 0.00073 | |
| EC:PC | 61.0 ± 3.5 | 0.00045 | |
| PEGDMA1000_high_SSCa | DEG | 0.0 | 0.00097 |
| TEG | 8.3 ± 0.5 | 0.00090 | |
| EC:PC | 20.1 ± 1.1 | 0.00081 | |
| PTHFDA1000_low_STFSIK | DEG | 99.1 ± 5.6 | 0.00042 |
| TEG | 61.8 ± 3.5 | 0.00052 | |
| EC:PC | 38.2 ± 2.2 | 0.00061 | |
| PTHFDA1000_med_STFSIK | DEG | 79.4 ± 4.5 | 0.00069 |
| TEG | 50.5 ± 2.9 | 0.00082 | |
| EC:PC | 27.0 ± 1.5 | 0.00097 | |
| PTHFDA1000_high_STFSIK | DEG | 86.5 ± 4.9 | 0.00084 |
| TEG | 86.5 ± 4.9 | 0.00084 | |
| EC:PC | 111.7 ± 6.4 | 0.00074 | |
| PTHFDA1000_low_STFSINa | DEG | 133.1 ± 7.6 | 0.00036 |
| TEG | 99.1 ± 5.6 | 0.00042 | |
| EC:PC | 65.1 ± 3.7 | 0.00051 | |
| PTHFDA1000_high_STFSINa | DEG | 109.7 ± 6.3 | 0.00075 |
| TEG | 49.7 ± 2.8 | 0.00105 | |
| EC:PC | 165.1 ± 9.4 | 0.00059 | |
| PTHFDA1000_low_STFSICa | DEG | 145.5 ± 8.3 | 0.00017 |
| TEG | 141.1 ± 8.0 | 0.00017 | |
| EC:PC | 30.6 ± 1.7 | 0.00032 | |
| PTHFDA1000_med_STFSICa | DEG | 107.8 ± 6.1 | 0.00030 |
| TEG | 107.8 ± 6.1 | 0.00030 | |
| EC:PC | 48.8 ± 2.8 | 0.00042 | |
| PTHFDA1000_high_STFSICa | DEG | 164.5 ± 9.4 | 0.00030 |
| TEG | 117.7 ± 6.7 | 0.00036 | |
| EC:PC | 108.7 ± 6.2 | 0.00038 |
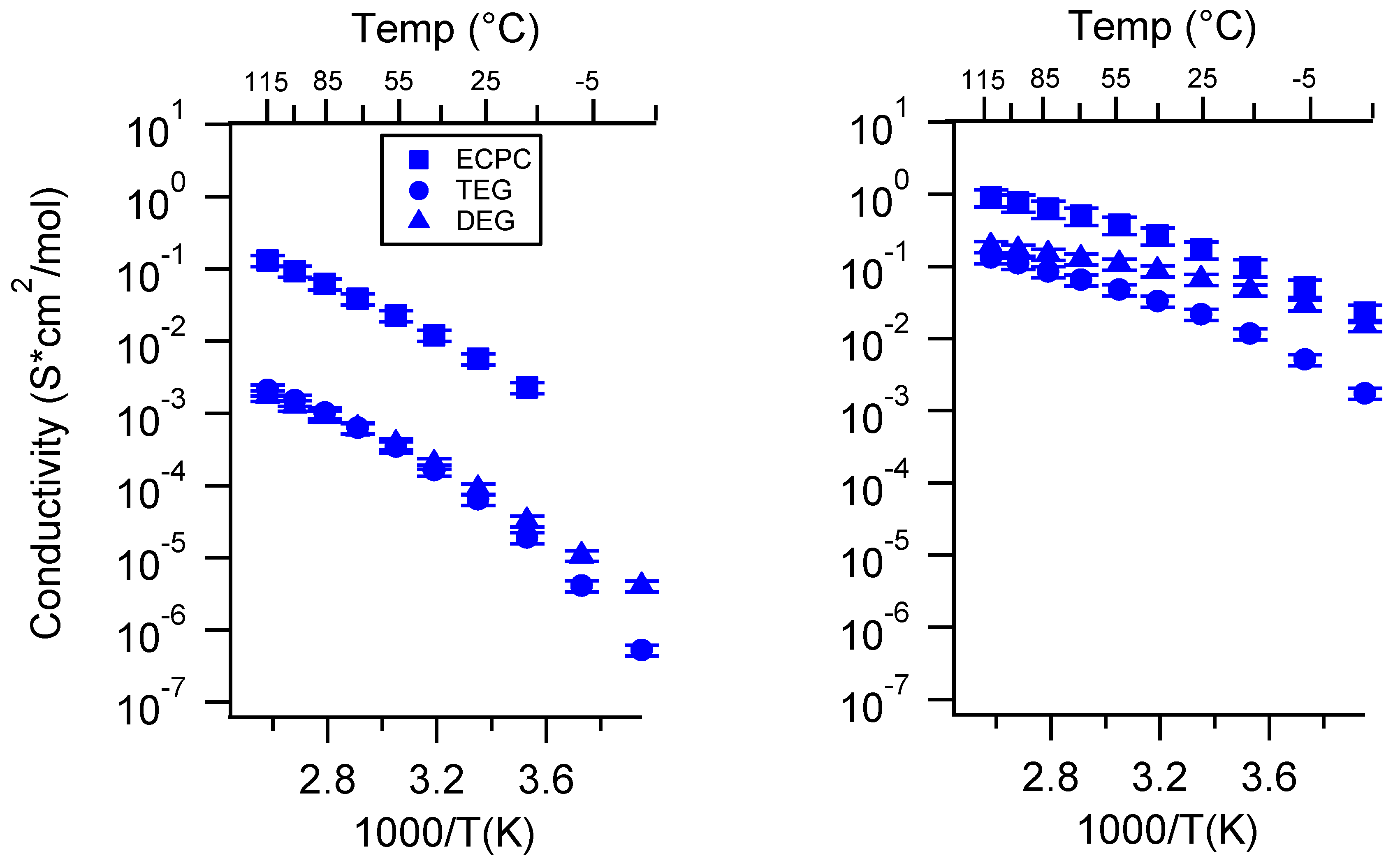
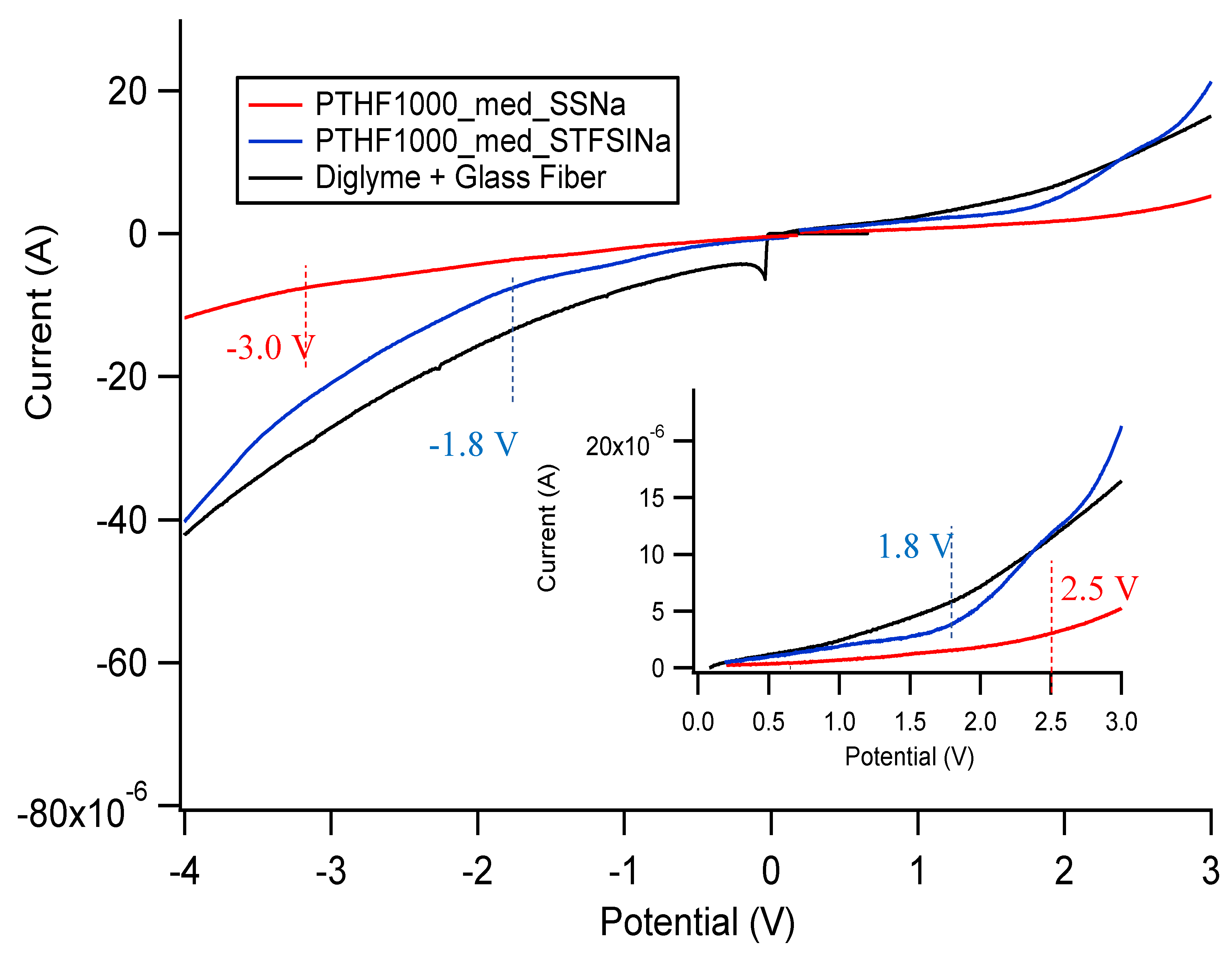
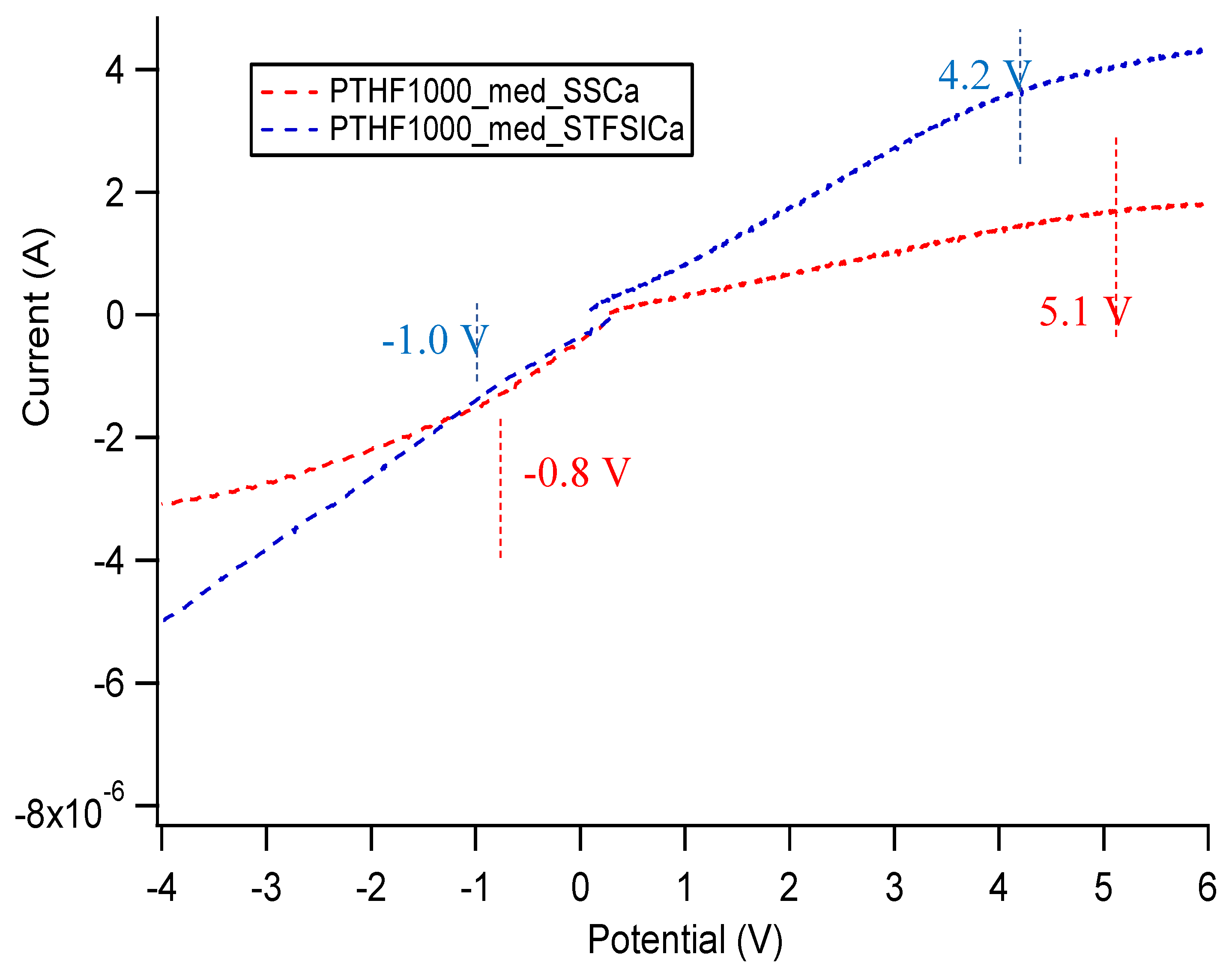
Appendix E. Electrochemical Stability Window of SIPEs

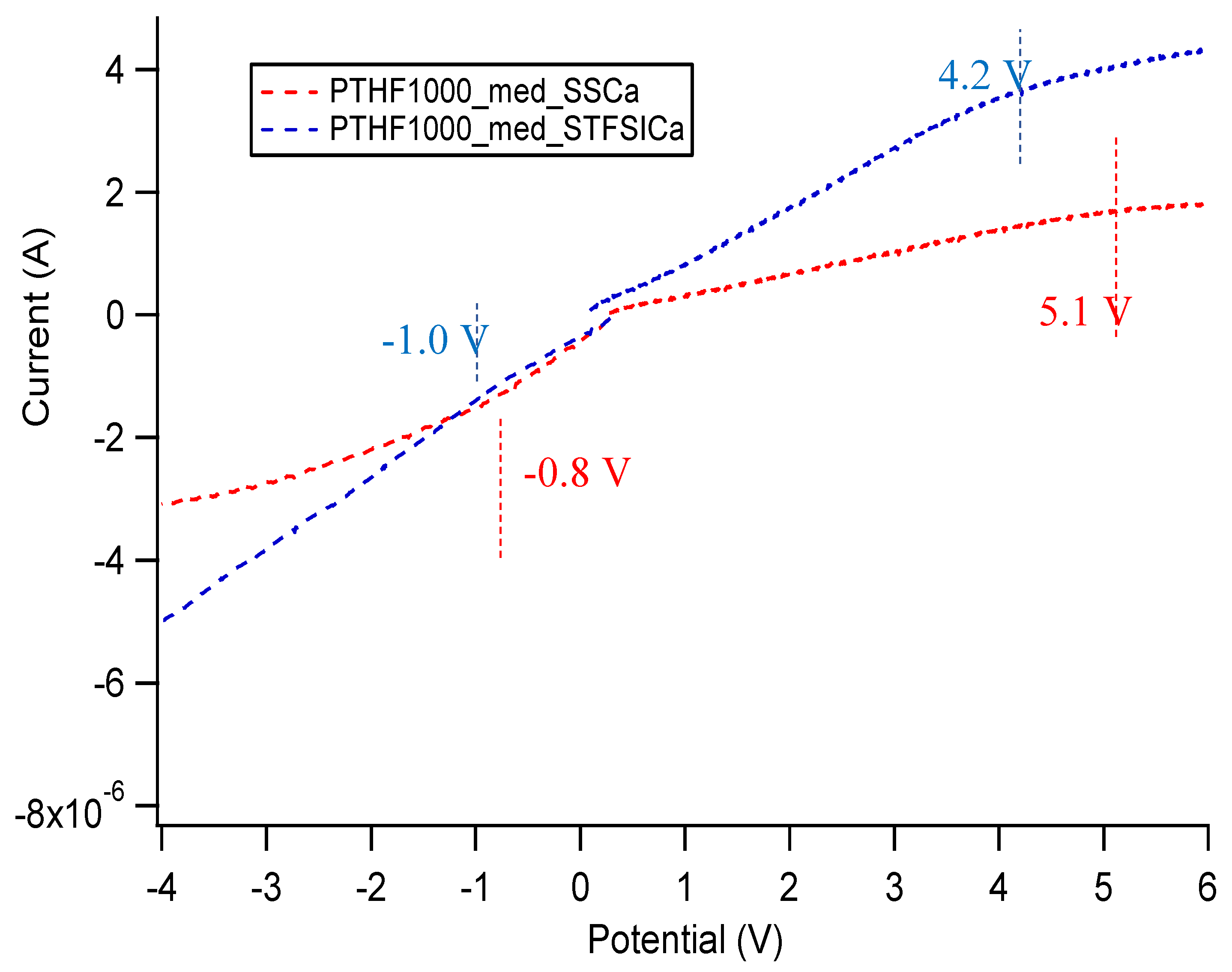

| Sample | Approximate Stability Window (V) |
|---|---|
| PTHF1000_med_SSCa DEG | 5.2 |
| PTHF1000_med_STFSICa DEG | 5.9 |
| PTHF1000_med_SSNa DEG | 5.5 |
| PTHF1000_med_STFSINa DEG | 3.6 |
| PTHF1000_med_SSNa EC/PC | 4.6 |
| PTHF1000_med_STFSINa EC/PC | 4.6 |
References
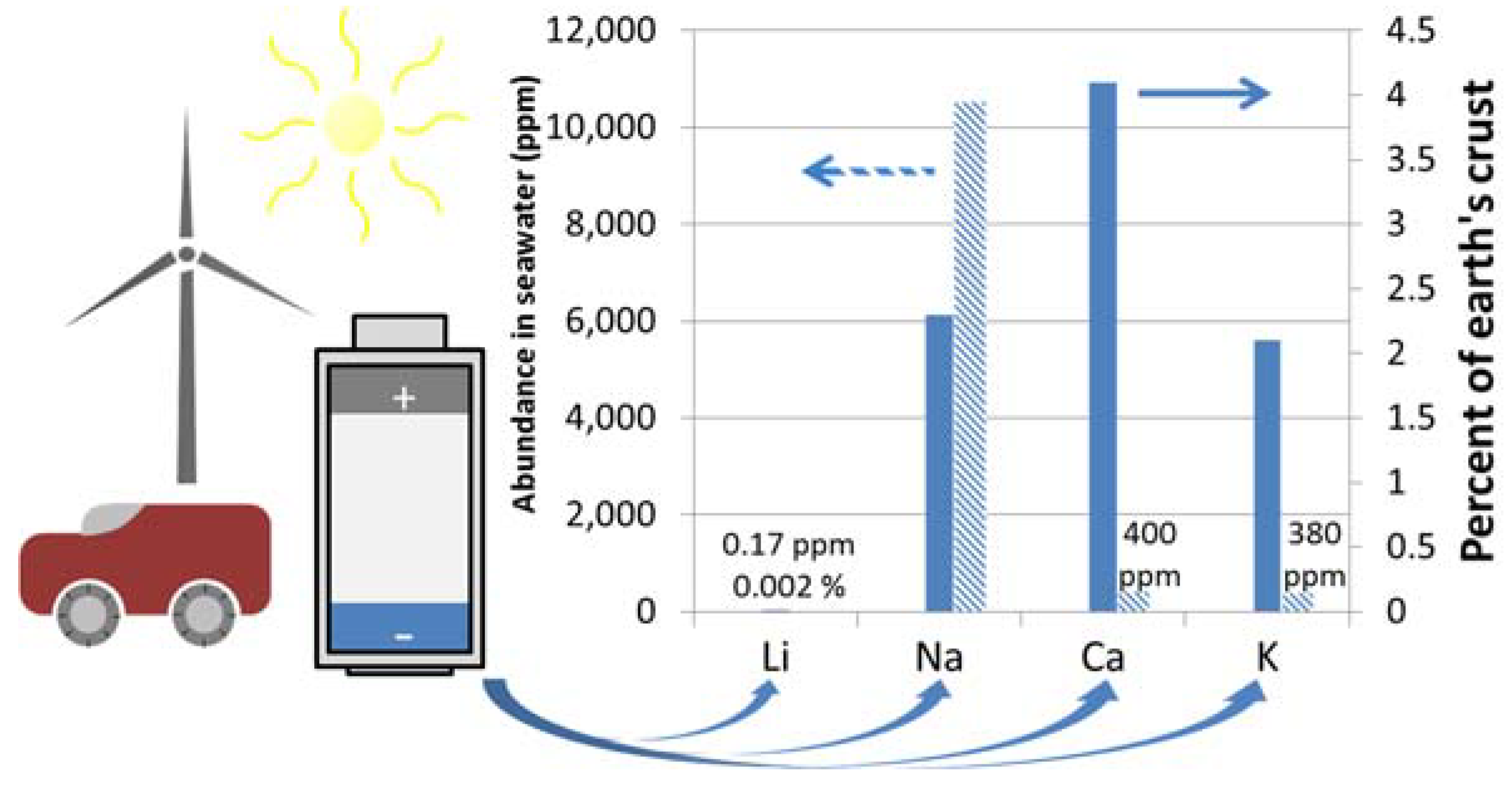
- Haynes, W.M. Abundance Of Elements In The Earth’s Crust And In The Sea. In CRC Handbook of Chemistry and Physics, 97th ed.; CRC Press: Boca Raton, FL, USA, 2016; pp. 14–17. [Google Scholar]
- Duxbury, A.; Mackenzie, F.; Byrne, R. Seawater; Encyclopædia Britannica, Inc.: Chicago, IL, USA, 2017. [Google Scholar]
- Diederichsen, K.M.; McShane, E.J.; McCloskey, B.D. Promising Routes to a High Li+ Transference Number Electrolyte for Lithium Ion Batteries. ACS Energy Lett. 2017, 2, 2563–2575. [Google Scholar] [CrossRef]
- Doyle, M.; Fuller, T.F.; Newman, J. The Importance of the Lithium Ion Transference Number in Lithium/Polymer Cells. Electrochim. Acta 1994, 39, 2073–2081. [Google Scholar] [CrossRef]
- Every, H.; Forsyth, M.; MacFarlane, D.R. Plasticized Single Conductung Polyelectrolytes Based on Poly(AMPS). Ionics 1996, 2, 53–62. [Google Scholar] [CrossRef]
- Doyle, M.; Lewittes, M.E.; Roelofs, M.G.; Perusich, S.A.; Lowrey, R.E. Relationship between Conductivity of Perfluorinated Ionomeric Membranes and Nonaqueous Solvent Properties. J. Membr. Sci. 2001, 184, 257–273. [Google Scholar] [CrossRef]
- Kreuer, K.D.; Wohlfarth, A.; De Araujo, C.C.; Fuchs, A.; Maier, J. Single Alkaline-Ion (Li+, Na+) Conductors by Ion Exchange of Proton-Conducting Ionomers and Polyelectrolytes. Chem. Phys. Chem. 2011, 12, 2558–2560. [Google Scholar] [CrossRef]
- Pourcelly, G.; Oikonomou, A.; Gavach, C.; Hurwitz, H.D. Influence of the Water Content on the Kinetics of Counter-Ion Transport in Perfluorosulphonic Membranes. J. Electroanal. Chem. 1990, 287, 43–59. [Google Scholar] [CrossRef]
- Cao, C.; Wang, H.; Liu, W.; Liao, X.; Li, L. Nafion Membranes as Electrolyte and Separator for Sodium-Ion Battery. Int. J. Hydrog. Energy 2014, 39, 16110–16115. [Google Scholar] [CrossRef]
- Sanginov, E.A.; Kayumov, R.R.; Shmygleva, L.V.; Lesnichaya, V.A.; Karelin, A.I.; Dobrovolsky, Y.A. Study of the Transport of Alkali Metal Ions in a Nonaqueous Polymer Electrolyte Based on Nafion. Solid State Ion. 2017, 300, 26–31. [Google Scholar] [CrossRef]
- Cao, C.; Liu, W.; Tan, L.; Liao, X.; Li, L. Sodium-Ion Batteries Using Ion Exchange Membranes as Electrolytes and Separators. Chem. Commun. 2013, 49, 11740–11742. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, H.; Chai, J.; Liu, T.; Hu, R.; Zhang, Z.; Li, G.; Cui, G. A Novel Single-Ion Conducting Gel Polymer Electrolyte Based on Polymeric Sodium Tartaric Acid Borate for Elevated-Temperature Sodium Metal Batteries. Solid State Ion. 2019, 337, 140–146. [Google Scholar] [CrossRef]
- Pan, Q.; Li, Z.; Zhang, W.; Zeng, D.; Sun, Y.; Cheng, H. Single Ion Conducting Sodium Ion Batteries Enabled by a Sodium Ion Exchanged Poly(Bis(4-Carbonyl Benzene Sulfonyl)Imide-Co-2,5-Diamino Benzesulfonic Acid) Polymer Electrolyte. Solid State Ion. 2017, 300, 60–66. [Google Scholar] [CrossRef]
- Villaluenga, I.; Bogle, X.; Greenbaum, S.; Gil De Muro, I.; Rojo, T.; Armand, M. Cation Only Conduction in New Polymer-SiO2 Nanohybrids: Na + Electrolytes. J. Mater. Chem. A 2013, 1, 8348–8352. [Google Scholar] [CrossRef]
- Dou, S.; Zhang, S.; Klein, R.J.; Runt, J.; Colby, R.H. Synthesis and Characterization of Poly(Ethylene Glycol)-Based Single-Ion Conductors. Chem. Mater. 2006, 18, 4288–4295. [Google Scholar] [CrossRef]
- Benrabah, D.; Sylla, S.; Alloin, F.; Sanchez, J.Y.; Armand, M. Perfluorosulfonate-Polyether Based Single Ion Conductors. Electrochim. Acta 1995, 40, 2259–2264. [Google Scholar] [CrossRef]
- Yamazaki, K.; Tomoo, S. Single-Ion Conduction in UV-Crosslinked Films of Poly(Urethane-Co- (Alkali-Metal Methacrylates)). Int. J. Syst. Bacteriol. 1995, 40, 565–571. [Google Scholar]
- Elmore, C.T.; Seidler, M.E.; Ford, H.O.; Merrill, L.C.; Upadhyay, S.P.; Schneider, W.F.; Schaefer, J.L. Ion Transport in Solvent-Free, Crosslinked, Single-Ion Conducting Polymer Electrolytes for Post-Lithium Ion Batteries. Batteries 2018, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.W.; Colby, R.H. Linear Viscoelasticity and Cation Conduction in Polyurethane Sulfonate Ionomers with Ions in the Soft Segment-Single Phase Systems. Macromolecules 2018, 51, 2757–2766. [Google Scholar] [CrossRef]
- Sinha, K.; Maranas, J. Does Ion Aggregation Impact Polymer Dynamics and Conductivity in PEO-Based Single Ion Conductors? Macromolecules 2014, 47, 2718–2726. [Google Scholar] [CrossRef]
- Doan, K.E.; Ratner, M.A.; Shriver, D.F. Synthesis and Electrical Response of Single-Ion Conducting Network Polymers Based on Sodium Poly(Tetraalkoxyaluminates). Chem. Mater. 1991, 3, 418–423. [Google Scholar] [CrossRef]
- Tsuchida, E.; Kobayashi, N.; Ohno, H. Single-Ion Conduction in Poly[(Oligo(Oxyethylene) Methacrylate)-Co-(Alkali-Metal Methacrylates)]. Macromolecules 1988, 21, 96–100. [Google Scholar] [CrossRef]
- Kim, H.T.; Park, J.K. Effects of Cations on Ionic States of Poly(Oligo-Oxyethylene Methacrylate-Co-Alkali Metal Acrylamidocaproate) Single-Ion Conductor. Solid State Ion. 1997, 98, 237–244. [Google Scholar] [CrossRef]
- Fumkawa, T.; Yoneya, K.; Takahashi, Y.; Ito, K.; Ohno, H. Correlation between Ionic and Dipolar Motions in a Single-Ion Conducting Polymer P[MEO9-MAM]. Electrochim. Acta 2000, 45, 1443–1448. [Google Scholar]
- Wang, J.H.H.; Yang, C.H.C.; Masser, H.; Shiau, H.S.; O’Reilly, M.V.; Winey, K.I.; Runt, J.; Painter, P.C.; Colby, R.H. Ion States and Transport in Styrenesulfonate Methacrylic PEO9 Random Copolymer Ionomers. Macromolecules 2015, 48, 7273–7285. [Google Scholar] [CrossRef]
- Rank, C.; Yan, L.; Mecking, S.; Winey, K.I. Periodic Polyethylene Sulfonates from Polyesterification: Bulk and Nanoparticle Morphologies and Ionic Conductivities. Macromolecules 2019, 52, 8466–8475. [Google Scholar] [CrossRef]
- Yan, L.; Rank, C.; Mecking, S.; Winey, K.I. Gyroid and Other Ordered Morphologies in Single-Ion Conducting Polymers and Their Impact on Ion Conductivity. J. Am. Chem. Soc. 2019. [Google Scholar] [CrossRef]
- Li, J.; Zhu, H.; Wang, X.; Armand, M.; Macfarlane, D.R.; Forsyth, M. Synthesis of Sodium Poly[4-Styrenesulfonyl(Trifluoromethylsulfonyl)Imide]-Co-Ethylacrylate] Solid Polymer Electrolytes. Electrochim. Acta 2015, 175, 232–239. [Google Scholar] [CrossRef]
- Noor, S.A.M.; Sun, J.; Macfarlane, D.R.; Armand, M.; Gunzelmann, D.; Forsyth, M. Decoupled Ion Conduction in Poly(2-Acrylamido-2-Methyl-1-Propane-Sulfonic Acid) Homopolymers. J. Mater. Chem. A 2014, 2, 17934–17943. [Google Scholar] [CrossRef]
- Yue, X.; He, Q.; Lim, H.D.; Liu, P. Hierarchical Structural Designs of Ion Exchange Membranes for Flow Batteries. J. Mater. Chem. A 2019, 7, 5794–5802. [Google Scholar] [CrossRef]
- Guo, D.; Wang, J.; Lei, J. Synthesis and Characterization of an Acrylate-Copolymer-Based Antistatic Agent Composed of a Single-Ion Conductive Polymer Electrolyte. J. Appl. Polym. Sci. 2011, 119, 2674–2682. [Google Scholar] [CrossRef]
- Wang, Y.P.; Feng, H.Y.; Fu, Z.S.; Tsuchida, E.; Takeoka, S.; Ohta, T. Ion Dissociation and Conduction of Nafion/Modified Oligo(Oxyethylene) Composite Films. Polym. Adv. Technol. 1991, 2, 295–299. [Google Scholar] [CrossRef]
- Ren, X.; Lau, K.C.; Yu, M.; Bi, X.; Kreidler, E.; Curtiss, L.A.; Wu, Y. Understanding Side Reactions in K-O2 Batteries for Improved Cycle Life. ACS Appl. Mater. Interfaces 2014, 6, 19299–19307. [Google Scholar] [CrossRef] [PubMed]
- Genier, F.S.; Burdin, C.V.; Biria, S.; Hosein, I.D. A Novel Calcium-Ion Solid Polymer Electrolyte Based on Crosslinked Poly(Ethylene Glycol) Diacrylate. J. Power Sources 2019, 414, 302–307. [Google Scholar] [CrossRef]
- Wang, J.; Genier, F.S.; Li, H.; Biria, S.; Hosein, I.D. A Solid Polymer Electrolyte from Cross-Linked Polytetrahydrofuran for Calcium Ion Conduction. ACS Appl. Polym. Mater. 2019, 1, 1837–1844. [Google Scholar] [CrossRef]
- Bouchet, R.; Maria, S.; Meziane, R.; Aboulaich, A.; Lienafa, L.; Bonnet, J.P.; Phan, T.N.T.; Bertin, D.; Gigmes, D.; Devaux, D.; et al. Single-Ion BAB Triblock Copolymers as Highly Efficient Electrolytes for Lithium-Metal Batteries. Nat. Mater. 2013, 12, 452–457. [Google Scholar] [CrossRef]
- Ramanujapuram, A.; Gordon, D.; Magasinski, A.; Ward, B.; Nitta, N.; Huang, C.; Yushin, G. Degradation and Stabilization of Lithium Cobalt Oxide in Aqueous Electrolytes. Energy Environ. Sci. 2016, 9, 1841–1848. [Google Scholar] [CrossRef]
- Vaalma, C.; Buchholz, D.; Passerini, S. Non-Aqueous Potassium-Ion Batteries: A Review. Curr. Opin. Electrochem. 2018, 9, 41–48. [Google Scholar] [CrossRef]
- Arroyo-De Dompablo, M.E.; Ponrouch, A.; Johansson, P.; Palacín, M.R. Achievements, Challenges, and Prospects of Calcium Batteries. Chem. Rev. 2019. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Myung, S.T.; Sun, Y.K. Recent Progress in Rechargeable Potassium Batteries. Adv. Funct. Mater. 2018, 28, 1802938. [Google Scholar] [CrossRef]
- Che, H.; Chen, S.; Xie, Y.; Wang, H.; Amine, K.; Liao, X.Z.; Ma, Z.F. Electrolyte Design Strategies and Research Progress for Room-Temperature Sodium-Ion Batteries. Energy Environ. Sci. 2017, 10, 1075–1101. [Google Scholar] [CrossRef]
- Bommier, C.; Ji, X. Electrolytes, SEI Formation, and Binders: A Review of Nonelectrode Factors for Sodium-Ion Battery Anodes. Small 2018, 14, 1703576. [Google Scholar] [CrossRef]
- Chen, L.; Fiore, M.; Wang, J.E.; Ruffo, R.; Kim, D.-K.; Longoni, G. Readiness Level of Sodium-Ion Battery Technology: A Materials Review. Adv. Sustain. Syst. 2018, 2, 1700153. [Google Scholar] [CrossRef]
- Merrill, L.C.; Ford, H.O.; Schaefer, J.L. Application of Single-Ion Conducting Gel Polymer Electrolytes in Magnesium Batteries. ACS Appl. Energy Mater. 2019, 2, 6355–6363. [Google Scholar] [CrossRef]
- Ford, H.O.; Merrill, L.C.; He, P.; Upadhyay, S.P.; Schaefer, J.L. Cross-Linked Ionomer Gel Separators for Polysulfide Shuttle Mitigation in Magnesium-Sulfur Batteries: Elucidation of Structure-Property Relationships. Macromolecules 2018, 51, 8629–8636. [Google Scholar] [CrossRef]
- Ford, H.O.; Park, B.; Jiang, J.; Seidler, M.E.; Schaefer, J.L. Enhanced Li + Conduction within Single-Ion Conducting Polymer Gel Electrolytes via Reduced Cation- Polymer Interaction. ACS Materials Lett. 2020. [Google Scholar] [CrossRef]
- Klein, R.J.; Zhang, S.; Dou, S.; Jones, B.H.; Colby, R.H.; Runt, J. Modeling Electrode Polarization in Dielectric Spectroscopy: Ion Mobility and Mobile Ion Concentration of Single-Ion Polymer Electrolytes. J. Chem. Phys. 2006, 124, 144903. [Google Scholar] [CrossRef]
- Seki, S.; Susan, M.A.B.H.; Kaneko, T.; Tokuda, H.; Noda, A.; Watanabe, M. Distinct Difference in Ionic Transport Behavior in Polymer Electrolytes Depending on the Matrix Polymers and Incorporated Salts. J. Phys. Chem. B 2005, 109, 3886–3892. [Google Scholar] [CrossRef]
- Petrowsky, M.; Frech, R. Temperature Dependence of Ion Transport: The Compensated Arrhenius Equation. J. Phys. Chem. B 2009, 113, 5996–6000. [Google Scholar] [CrossRef]
- Marcus, Y. Ionic Radii in Aqueous Solutions. Chem. Rev. 1988, 88, 1475–1498. [Google Scholar] [CrossRef]
- Mähler, J.; Persson, I. A Study of the Hydration of the Alkali Metal Ions in Aqueous Solution. Inorg. Chem. 2012, 51, 425–438. [Google Scholar] [CrossRef]
- Hall, L.M.; Seitz, M.E.; Winey, K.I.; Opper, K.L.; Wagener, K.B.; Stevens, M.J.; Frischknecht, A.L. Ionic Aggregate Structure in Ionomer Melts: Effect of Molecular Architecture on Aggregates and the Ionomer Peak. J. Am. Chem. Soc. 2012, 134, 574–587. [Google Scholar] [CrossRef]
- Zheng, Q.; Pesko, D.M.; Savoie, B.M.; Timachova, K.; Hasan, A.L.; Smith, M.C.; Miller, T.F.; Coates, G.W.; Balsara, N.P. Optimizing Ion Transport in Polyether-Based Electrolytes for Lithium Batteries. Macromolecules 2018, 51, 2847–2858. [Google Scholar] [CrossRef] [Green Version]
- Pesko, D.M.; Webb, M.A.; Jung, Y.; Zheng, Q.; Miller, T.F.; Coates, G.W.; Balsara, N.P. Universal Relationship between Conductivity and Solvation-Site Connectivity in Ether-Based Polymer Electrolytes. Macromolecules 2016, 49, 5244–5255. [Google Scholar] [CrossRef]
- Webb, M.A.; Jung, Y.; Pesko, D.M.; Savoie, B.M.; Yamamoto, U.; Coates, G.W.; Balsara, N.P.; Wang, Z.G.; Miller, T.F. Systematic Computational and Experimental Investigation of Lithium-Ion Transport Mechanisms in Polyester-Based Polymer Electrolytes. ACS Cent. Sci. 2015, 1, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarusso, D.J.; Cooper, S.L. Microstructure of Ionomers: Interpretation of Small-Angle X-Ray Scattering Data. Macromolecules 1983, 16, 1871–1880. [Google Scholar] [CrossRef]
- Li, Y.; Peiffer, D.G.; Chu, B. Long-Range Inhomogeneities in Sulfonated Polystyrene Ionomers. Macromolecules 1993, 26, 4006–4012. [Google Scholar] [CrossRef]
- Elliott, J.A.; Hanna, S.; Elliott, A.M.S.; Cooley, G.E. Interpretation of the Small-Angle X-Ray Scattering from Swollen and Oriented Perfluorinated Ionomer Membranes. Macromolecules 2000, 33, 4161–4171. [Google Scholar] [CrossRef]
- Gebel, G.; Diat, O. Neutron and X-Ray Scattering: Suitable Tools for Studying Lonomer Membranes. Fuel Cells 2005, 5, 261–276. [Google Scholar] [CrossRef]
- Hamley, I.W.; Castelletto, V. Small-Angle Scattering of Block Copolymers in the Melt, Solution and Crystal States. Prog. Polym. Sci. 2004, 29, 909–948. [Google Scholar]
- Meziane, R.; Bonnet, J.P.; Courty, M.; Djellab, K.; Armand, M. Single-Ion Polymer Electrolytes Based on a Delocalized Polyanion for Lithium Batteries. Electrochim. Acta 2011, 57, 14–19. [Google Scholar] [CrossRef]

| Chemistry | Conductivity (S/cm) | Solvent | Ref |
|---|---|---|---|
| Poly(tetrahydrofuran) diacrylate-x-4-styrenesulfonyl (trifluoromethylsulfonyl) imide (PTHFDA-STFSI) sodium exchanged | 2.4 × 10−5 (25 °C) | DEG | This work |
| 1.3 × 10−5 (25 °C) | TEG | ||
| 3.9 × 10−4 (25 °C) | EC:PC | ||
| 5.9 × 10−12 (25 °C) | Dry | ||
| Poly(2-acrylamido-2-methyl-propane- 1-sulphonic acid) (PAMPS) | 10−2 (20 °C) * | 50H2O/SO3 | [5] |
| 10−5 (20 °C) * | Dimethyl Sulfoxide (DMSO) | ||
| PFSA (Nafion 117) sodium exchanged | 1.8 × 10−2 (23 °C) | Water | [6] |
| 3.2 × 10−4 (23 °C) | Triethyl phosphate | ||
| 3.8 × 10−3 (23 °C) | N-methyl Formamide | ||
| 5.1 × 10−4 (23 °C) | g-butyrolactone | ||
| 5.0 ×10−4(23 °C) | DMSO | ||
| Fully sulfonated poly phenylene-sulfone | 1 × 10−3 (25 °C) * | DMSO | [7] |
| 2 × 10−3 (70 °C) * | |||
| PFSA (Nafion 117) sodium exchanged | 10−3 (RT) * | 14H2O/SO3 | [8] |
| 10−6 (RT) * | 6H2O/SO3 | ||
| PFSA (Nafion 115) sodium exchanged | 3.52 × 10−4 (RT) | EC/PC 1:1 | [9] |
| 1.52 × 10−3 (70 °C) | |||
| PFSA (Nafion 115) sodium exchanged | 3.5 × 10−4 (30 °C) | DMSO | [10] |
| Perfluorinated sulfonic sodium membrane (PFSA) Nafion-sodium exchanged | 2.5 × 10−4 (25 °C) | EC/PC 1:1 | [11] |
| 1.5 × 10−3 (70 °C) | |||
| 1.5 × 10−4 (25 °C) | |||
| 1.1 × 10−3 (70 °C) | |||
| Poly(sodium tartaric acid borate) blend poly(vinylene carbonate) | 1 × 10−4 (RT) * | EC/DEC 1:1 | [12] |
| poly(bis(4-carbonyl benzene sulfonyl)imide-co-2.5-diamino benzesulfonic acid) (NaPA) + PVDF/HFP | 0.91 × 10−4 (20 °C) | EC/DMC 1:1 | [13] |
| 4.1 × 10−4 (80 °C) | |||
| SiO2 particles grafted with Poly(ethylene glycol) (PEG) and NaSTFSI dispersed in PEO | 10−5 (RT) * | Poly(ethylene glycol) dimethyl ether (PEGDME) 250 g/mol | [14] |
| 10−4 (75 °C) * | |||
| PEG900 and dimethyl 5-sulfoisophthalate sodium salt | 1.07 × 10−6 (25 °C) | Dry | [15] |
| N,N-Diallyl-l-amido-tetrafluoroethanesulfonate (DaaRFSO3Na) | 4 × 10−7 (25 °C) * | Dry | [16] |
| Poly(ethylene glycol-co-2,4-tolyldiisocyanate-co-(alkali-metal methacrylates)) (PEG-PU-AMMA) | 3 × 10−7 (25 °C) * | Dry | [17] |
| 9 × 10−6 (80 °C) * | |||
| Poly(ethylene glycol) diacrylate (PEGDA, 1500 g/mol)-co-STFSI Na | 2.0 × 10−7 (25 °C) | Dry | [18] |
| 5.0 × 10−6 (85 °C) | |||
| Polyurethane sulfonate | 10−7 (25 °C) * | Dry | [19] |
| PEO-co-benzenesulfonate | 10−7 (25 °C) * | Dry | [20] |
| Network Polymers Based on Sodium Poly(tetraalkoxyaluminates) with Poly(ethylene oxide) (PEO) blend | 10−7 (50 °C) * | Dry | [21] |
| 10−6 (80 °C) * | |||
| Poly[ (oligo(oxyethylene) methacrylate)-co-(alkali-metal methacrylates)] | 8 × 10−8 (25 °C) * | Dry | [22] |
| Poly(oligo-oxyethylenemethacrylate-co-alkali metal acrylamidocaproate) | 6.9 × 10−8 (RT) | Dry | [23] |
| Nona-oxyethylene Methacrylate-co-alkali-metal methacrylate | 10−8 (25 °C) * | Dry | [24] |
| Methacrylic PEO-co-sodium stytene sulfonate (PEO9M-r-NaSS) | 10−8 (RT) * | Dry | [25] |
| 10−7 (80 °C)* | |||
| Polyethylene-like 48 carbon sodium dimethylsulfosuccinate (PES48Na) Precise ionomer | 10−10 (100 °C) * | Dry | [26] |
| 10−6 (160 °C) * | |||
| Polyethylene-like sulfonated precise ionomer–gyroid phase | 10−11 (80 °C) * | Dry | [27] |
| 10−7 (120 °C) * | |||
| Sodium Poly(4-styrenesulfonyl (trifluoromethylsulfonyl)imide) (NaPSTFSI) NaPSTFSI Poly ethyl acrylate blends | 10−14 to 10−11 (40 °C) * | Dry | [28] |
| 10−12 to 10−9 (40 °C) * | Dry | ||
| Poly(2-acrylamido-2-methyl-1-propane-sulfonic acid sodium salt) homopolymer | 10−11 (130 °C) * | Dry | [29] |
| Chemistry | Conductivity (S/cm) | Solvent | Ref |
|---|---|---|---|
| Poly(tetrahydrofuran) diacrylate-x-4-styrenesulfonyl (trifluoromethylsulfonyl) imide (PTHFDA-STFSI) potassium exchanged | 4.0 × 10−5 (25 °C) | DEG | This work |
| 1.8 × 10−5 (25 °C) | TEG | ||
| 2.7 × 10−4 (25 °C) | EC:PC | ||
| 2.4 × 10−12 (25 °C) | Dry | ||
| PFSA (Nafion 117) potassium exchanged | 1.4 × 10−2 (23 °C) | Water | [6] |
| 1.4 × 10−4 (23 °C) | Triethyl phosphate | ||
| 4.1 × 10−3 (23 °C) | N-methylFormamide | ||
| 2.3 × 10−5 (23 °C) | g-butyrolactone | ||
| 8.0 × 10−4 (23 °C) | DMSO | ||
| PFSA (Nafion 117) potassium exchanged | 8.0 × 10−3 (RT) | Water, fully hydrated | [30] |
| PFSA (Nafion 117) potassium exchanged | 10−3 (RT) * | 9H2O/SO3 | [8] |
| 10−6 (RT) * | 6H2O/SO3 | ||
| PFSA (Nafion 115) potassium exchanged | 3.3 × 10−4 (30 °C) | DMSO | [10] |
| Acrylic acid and methoxy poly(ethylene glycol)s coordinating K+ | 6 × 10−6 (30 °C) * | 10% RH | [31] |
| N,N-Diallyl-l-amido-tetrafluoroethanesulfonate (DaaRFSO3K) | 3 × 10−6 (25 °C) * | Dry | [16] |
| PFSA (Nafion) potassium exchanged | 10−6 (100 °C) * | Dry | [32] |
| Poly(ethylene glycol)-co-2,4-tolyl diisocyanate-co-(alkali-metal methacrylates))(PEG-PU-AMMA) | 6 × 10−7 (25 °C) * | Dry | [17] |
| 1 × 10−5 (80 °C) * | |||
| Poly(oligo-oxyethylenemethacrylate-co-alkali metal acrylamidocaproate) | 2 × 10−7 (RT) * | Dry | [23] |
| Poly(oligo(oxyethylene) methacrylate)-co-(alkali-metal methacrylates) | 2 × 10−7 (25 °C) * | Dry | [22] |
| Nona-oxyethylene methacrylate and alkali-metal methacrylate-K | 10−7 (25 °C) * | Dry | [24] |
| PEGDA(1500 g/mol)-co-STFSI K | 9 × 10−8 (25 °C) | Dry | [18] |
| 5 × 10−6 (85 °C) | |||
| PFSA (Nafion) 211 potassium exchanged | Unreported | Water | [33] |
| Chemistry | Conductivity (S/cm) | Solvent | Ref |
|---|---|---|---|
| Poly(tetrahydrofuran) diacrylate-x-4-styrenesulfonyl (trifluoromethylsulfonyl) imide (PTHFDA-STFSI) calcium exchanged | 6.2 × 10−7 (25 °C) | DEG | This work |
| 1.0 × 10−7 (25 °C) | TEG | ||
| 7.0 × 10−6 (25 °C) | EC:PC | ||
| 3.5 × 10−13 (85 °C) | Dry | ||
| PFSA (Nafion 117) calcium exchanged | 9.1 × 10−3 (23 °C) | Water | [6] |
| 1.0 × 10−6 (23 °C) | Triethyl phosphate | ||
| 2.7 × 10−3 (23 °C) | N-methyl Formamide | ||
| 1.1 × 10−5 (23 °C) | g-butyrolactone | ||
| 3.5 × 10−4(23 °C) | DMSO | ||
| PFSA (Nafion 117) calcium exchanged | 10−3 (RT) * | 14H2O/SO3 | [8] |
| 10−6 (RT) * | 8H2O/SO3 | ||
| PEGDA 1500 g/mol -co-STFSI Ca | 2.0 × 10−9 (25 °C) | Dry | [18] |
| 9.0 × 10−8 (85 °C) | |||
| PEGDA 575 g/mol + Ca(NO3)2 4H2O | 5 × 10−6 (25 °C) * | Dry | [34] ‡ |
| Poly(tetrahydrofuran) (PTHF)-Epoxy + Ca(NO3)2 | 2 × 10−4 (25 °C) * | Dry | [35] ‡ |
| Sample | Cation | Charge Density × 104 (mol ch/g Monomer) | Solvent | Conductivity (S/cm) 25 °C | Conductivity (S/cm) 85 °C |
|---|---|---|---|---|---|
| PEGDMA1000-SS | K | 8 (low) | DEG | 1.2 × 10‒7 | 1.8 × 10‒6 |
| TEG | 6.4 × 10‒7 | 7.1 × 10‒6 | |||
| EC:PC | 4.8 × 10‒6 | 8.0 × 10‒5 | |||
| Dry | 1.4 × 10‒9 | 2.6 × 10‒7 | |||
| 12 (med) | DEG | 1.7 × 10‒8 | 8.2 × 10‒7 | ||
| TEG | 1.5 × 10‒7 | 6.1 × 10‒6 | |||
| EC:PC | 4.2 × 10‒6 | 8.1 × 10‒5 | |||
| Dry | 2.4 × 10‒10 | 2.7 × 10‒7 | |||
| 16 (high) | DEG | 1.3 × 10‒8 | 9.9 × 10‒7 | ||
| TEG | 1.1 × 10‒6 | ||||
| EC:PC | 5.7 × 10‒6 | 1.0 × 10‒4 | |||
| Dry | 1.5 × 10‒7 | ||||
| PEGDMA1000-SS | Na | 8 (low) | DEG | 1.1 × 10‒7 | 1.1 × 10‒7 |
| TEG | 1.8 × 10‒7 | 3.2 × 10‒6 | |||
| EC:PC | 2.5 × 10‒6 | 4.6 × 10‒5 | |||
| Dry | 1.8 × 10‒9 | 2.2 × 10‒7 | |||
| 12 (med) | DEG | 1.1 × 10‒7 | 1.1 × 10‒6 | ||
| TEG | 7.9 × 10‒8 | 1.3 × 10‒6 | |||
| EC:PC | 3.8 × 10‒6 | 5.8 × 10‒5 | |||
| Dry | 2.7 × 10‒11 | 1.4 × 10‒8 | |||
| 16 (high) | DEG | 4.4 × 10‒8 | 5.0 × 10‒7 | ||
| TEG | 5.3 × 10‒9 | 2.9 × 10‒7 | |||
| EC:PC | 4.7 × 10‒6 | 5.1 × 10‒5 | |||
| Dry | 4.4 × 10‒8 | ||||
| PEGDMA1000-SS | Ca | 8 (low) | DEG | 2.5 × 10‒9 | 2.0 × 10‒8 |
| TEG | 4.3 × 10‒9 | 4.4 × 10‒8 | |||
| EC:PC | 1.9 × 10‒6 | 1.2 × 10−5 | |||
| Dry | 2.8 × 10−10 | ||||
| 12 (med) | DEG | 4.3 × 10‒11 | 9.2 × 10−10 | ||
| TEG | 1.2 × 10‒11 | 1.2 × 10−9 | |||
| EC:PC | 2.9 × 10‒7 | 2.1 × 10−6 | |||
| Dry | 2.0 × 10−11 | ||||
| 16 (high) | DEG | 1.3 × 10−11 | |||
| TEG | 8.5 × 10‒12 | 2.5 × 10−10 | |||
| EC:PC | 6.7 × 10−8 | 6.5 × 10−7 | |||
| Dry | 2.8 × 10−13 (100 °C) | ||||
| PTHFDA-STFSI | K | 7 (low) | DEG | 1.8 × 10−5 | 3.4 × 10−5 |
| TEG | 7.3 × 10−6 | 2.9 × 10−5 | |||
| EC:PC | 8.5 × 10−5 | 3.6 × 10−4 | |||
| Dry | 1.4 × 10−12 | 1.6 × 10−8 | |||
| 10 (med) | DEG | 1.6 × 10−5 | 3.4 × 10−5 | ||
| TEG | 1.2 × 10−5 | 5.4 × 10−5 | |||
| EC:PC | 1.2 × 10−4 | 4.7 × 10−4 | |||
| Dry | 1.1 × 10−8 | ||||
| 13 (high) | DEG | 4.0 × 10−5 | 9.8 × 10−5 | ||
| TEG | 1.8 × 10−5 | 8.3 × 10−5 | |||
| EC:PC | 2.7 × 10−4 | 9.7 × 10−4 | |||
| Dry | 2.4 × 10−12 | 3.4 × 10−8 | |||
| PTHFDA-STFSI | Na | 7 (low) | DEG | 1.7 × 10−5 | 3.5 × 10−5 |
| TEG | 6.4 × 10−6 | 2.4 × 10−6 | |||
| EC:PC | 1.0 × 10−4 | 5.0 × 10−4 | |||
| Dry | 1.3 × 10−11 | 3.0 × 10−8 | |||
| 10 (med) | DEG | 2.8 × 10−5 | 6.3 × 10−5 | ||
| TEG | 1.1 × 10−5 | 4.1 × 10−5 | |||
| EC:PC | 9.8 × 10−5 | 3.6 × 10−4 | |||
| Dry | 4.6 × 10−12 | 8.0 × 10−9 | |||
| 13 (high) | DEG | 2.5 × 10−5 | 4.6 × 10−5 | ||
| TEG | 1.3 × 10−5 | 6.0 × 10−5 | |||
| EC:PC | 3.9 × 10−4 | 1.4 × 10−3 | |||
| Dry | 5.9 × 10−12 | 3.6 × 10−8 | |||
| PTHFDA-STFSI | Ca | 7 (low) | DEG | 3.5 × 10−8 | 1.1 × 10−7 |
| TEG | 3.7 × 10−8 | 1.5 × 10−7 | |||
| EC:PC | 6.8 × 10−7 | 8.5 × 10−6 | |||
| Dry | 3.3 × 10−11 | ||||
| 10 (med) | DEG | 1.9 × 10−8 | 7.3 × 10−8 | ||
| TEG | 2.3 × 10−8 | 1.2 × 10−7 | |||
| EC:PC | 2.9 × 10−6 | 2.6 × 10−5 | |||
| Dry | 2.1 × 10−11 | ||||
| 13 (high) | DEG | 6.2 × 10−8 | 3.1 × 10−7 | ||
| TEG | 1.0 × 10−7 | 3.7 × 10−7 | |||
| EC:PC | 7.0 × 10−6 | 5.1 × 10−5 | |||
| Dry | 3.5 × 10−13 |
| Sample | Solvent | Mass Change % | Volume Change % | Cation Conc. (mol Charge/cm3 Polymer) |
|---|---|---|---|---|
| PEGDMA1000_med_SSNa | DEG | 24 ± 3.6 | 24 ± 2.6 | 0.00121 |
| PTHFDA1000_med_STFSINa | DEG | 164 ± 14 | 175 ± 2.3 | 0.00043 |
| PEGDMA1000_med_SSNa | TEG | 21 ± 5.7 | 23 ± 4.3 | 0.00122 |
| PTHFDA1000_med_STFSINa | TEG | 142 ± 7.8 | 157 ± 4.8 | 0.00048 |
| PEGDMA1000_med_SSNa | EC:PC | 97 ± 9.1 | 91 ± 4.9 | 0.00082 |
| PTHFDA1000_med_STFSINa | EC:PC | 169 ± 14.9 | 135 ± 19.8 | 0.00057 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ford, H.O.; Cui, C.; Schaefer, J.L. Comparison of Single-Ion Conducting Polymer Gel Electrolytes for Sodium, Potassium, and Calcium Batteries: Influence of Polymer Chemistry, Cation Identity, Charge Density, and Solvent on Conductivity. Batteries 2020, 6, 11. https://doi.org/10.3390/batteries6010011
Ford HO, Cui C, Schaefer JL. Comparison of Single-Ion Conducting Polymer Gel Electrolytes for Sodium, Potassium, and Calcium Batteries: Influence of Polymer Chemistry, Cation Identity, Charge Density, and Solvent on Conductivity. Batteries. 2020; 6(1):11. https://doi.org/10.3390/batteries6010011
Chicago/Turabian StyleFord, Hunter O., Chuanchuan Cui, and Jennifer L. Schaefer. 2020. "Comparison of Single-Ion Conducting Polymer Gel Electrolytes for Sodium, Potassium, and Calcium Batteries: Influence of Polymer Chemistry, Cation Identity, Charge Density, and Solvent on Conductivity" Batteries 6, no. 1: 11. https://doi.org/10.3390/batteries6010011
APA StyleFord, H. O., Cui, C., & Schaefer, J. L. (2020). Comparison of Single-Ion Conducting Polymer Gel Electrolytes for Sodium, Potassium, and Calcium Batteries: Influence of Polymer Chemistry, Cation Identity, Charge Density, and Solvent on Conductivity. Batteries, 6(1), 11. https://doi.org/10.3390/batteries6010011