
The Halogen Effect on the Magnetic Behaviour of Dimethylformamide Solvates in [Fe(halide-salEen)2]BPh4

 , ,
, ,  ,
,  ,
,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. General Remarks
2.2. Syntheses
2.3. Crystallography
2.4. Mössbauer Studies
2.5. DFT Calculations
3. Results
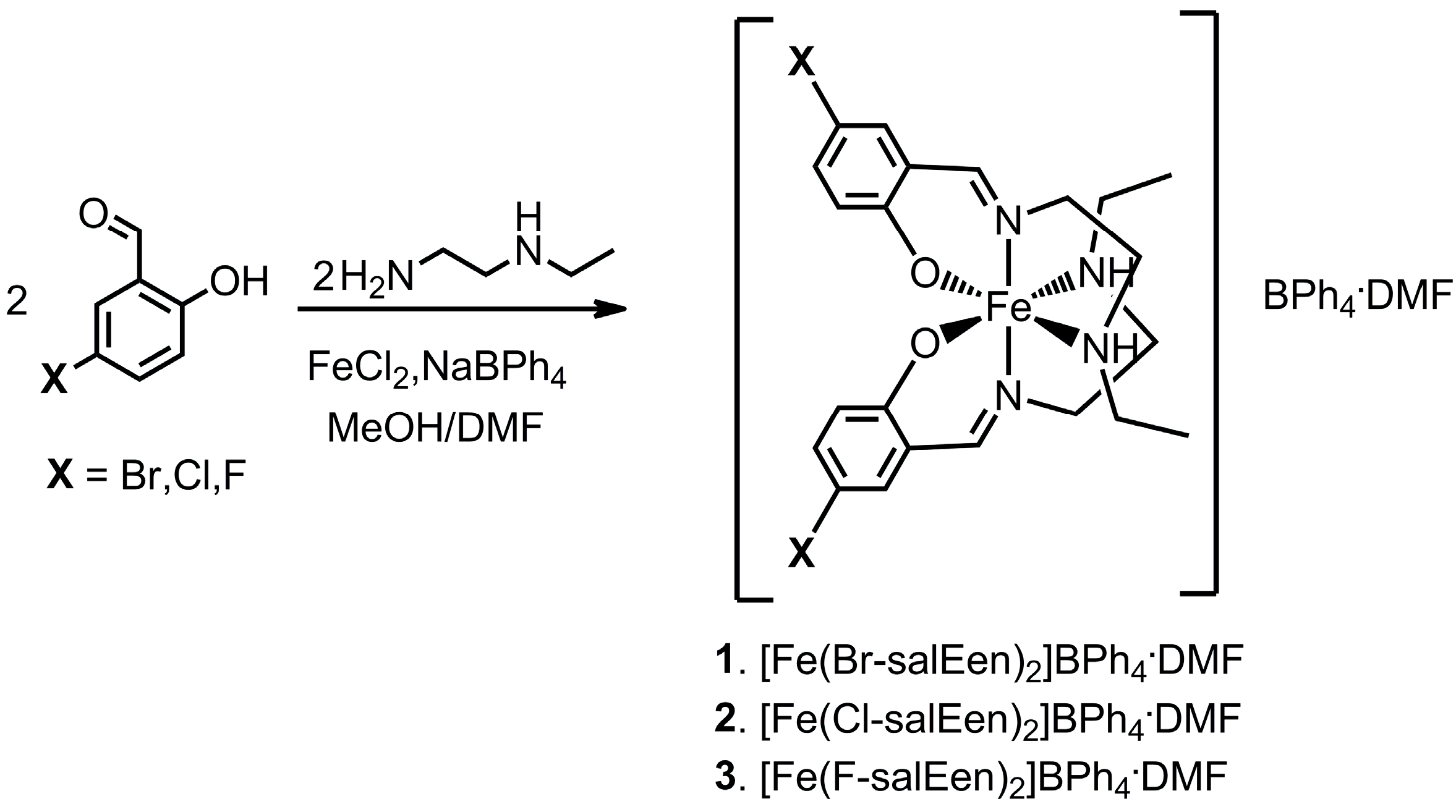
3.1. Chemical Studies
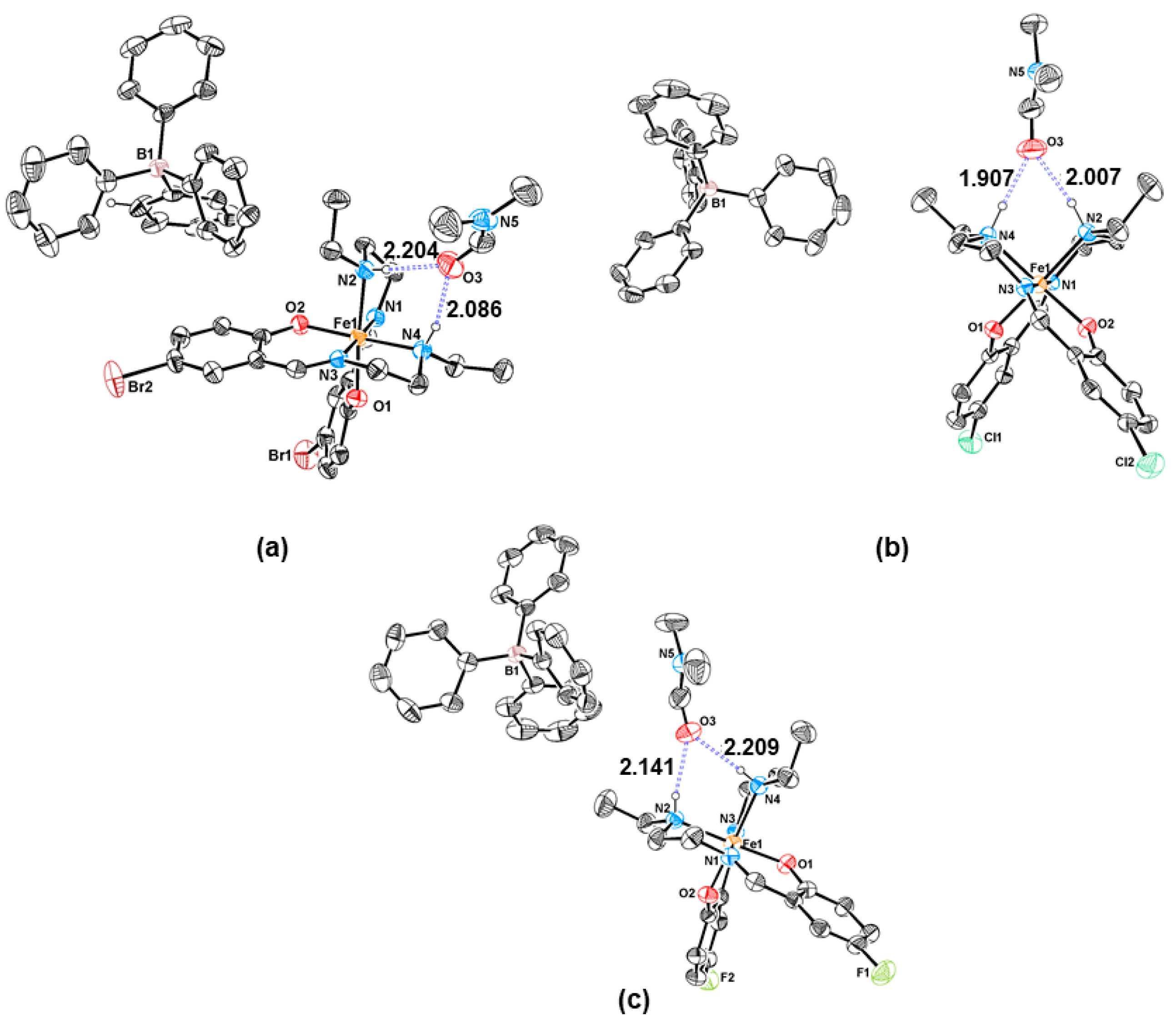
3.2. Structural Characterisation
3.3. Influence of the Halogen on the Magnetic Properties
3.4. Mössbauer Studies
3.5. Computational Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Halcrow, M.A. Spin-Crossover Materials: Properties and Applications; Halcrow, M.A., Ed.; John Wiley & Sons Ltd.: Oxford, UK, 2013; ISBN 9781119998679. [Google Scholar]
- Gütlich, P.; Goodwin, H.A. Spin Crossover-An Overall Perspective. Top. Curr. Chem. 2004, 233, 1–47. [Google Scholar]
- Martinho, P.N.; Martins, F.F.; Bandeira, N.A.G.; Calhorda, M.J. Spin Crossover in 3d Metal Centers Binding Halide-Containing Ligands: Magnetism, Structure and Computational Studies. Sustainability 2020, 12, 2512. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989; ISBN 9780195092769. [Google Scholar]
- Vela, S.; Paulsen, H. Cooperativity in Spin Crossover Systems. An Atomistic Perspective on the Devil’s Staircase. Inorg. Chem. 2018, 57, 9478–9488. [Google Scholar] [CrossRef]
- Vela, S.; Paulsen, H. Deciphering Crystal Packing Effects in the Spin Crossover of Six [FeII(2-Pic)3]Cl2 Solvatomorphs. Dalton Trans. 2019, 48, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Harding, D.J.; Phonsri, W.; Harding, P.; Gass, I.A.; Murray, K.S.; Moubaraki, B.; Cashion, J.D.; Liud, L.; Telfer, S.G. Abrupt Spin Crossover in an Iron(III) Quinolylsalicylaldimine Complex: Structural Insights and Solvent Effects. Chem. Commun. 2013, 49, 6340–6342. [Google Scholar] [CrossRef] [PubMed]
- Phonsri, W.; Harding, D.J.; Harding, P.; Murray, K.S.; Moubaraki, B.; Gass, I.A.; Cashion, J.D.; Jameson, G.N.L.; Adams, H. Stepped Spin Crossover in Fe(III) Halogen Substituted Quinolylsalicylaldimine Complexes. Dalton Trans. 2014, 43, 17509–17518. [Google Scholar] [CrossRef]
- Harding, D.J.; Phonsri, W.; Harding, P.; Murray, K.S.; Moubaraki, B.; Jameson, G.N.L. Abrupt Two-Step and Symmetry Breaking Spin Crossover in an Iron(III) Complex: An Exceptionally Wide [LS–HS] Plateau. Dalton Trans. 2015, 44, 15079–15082. [Google Scholar] [CrossRef]
- Li, Z.Y.; Dai, J.W.; Gagnon, K.J.; Cai, H.L.; Yamamoto, T.; Einaga, Y.; Zhao, H.H.; Kanegawa, S.; Sato, O.; Dunbar, K.R.; et al. A Neutral Fe(III) Compound Exhibiting a Two-Step Spin Transition and Dielectric Anomalies. Dalton Trans. 2013, 42, 14685–14688. [Google Scholar] [CrossRef]
- Zelentsov, V.V.; Mokshin, V.M.; Sobolev, S.S.; Shipilov, V.I. The Influence of Crystallization Water upon Magnetic Properties of Fe(III) Thiosemicarbazones. Dokl. Akad. Nauk. SSSR 1984, 277, 900. [Google Scholar]
- Floquet, S.; Guillou, N.; Négrier, P.; Rivière, E.; Boillot, M.L. The Crystallographic Phase Transition for a Ferric Thiosemicarbazone Spin Crossover Complex Studied by X-Ray Powder Diffraction. New J. Chem. 2006, 30, 1621–1627. [Google Scholar] [CrossRef]
- Kang, S.; Shiota, Y.; Kariyazaki, A.; Kanegawa, S.; Yoshizawa, K.; Sato, O. Heterometallic FeIII/K Coordination Polymer with a Wide Thermal Hysteretic Spin Transition at Room Temperature. Chem. Eur. J. 2016, 22, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Phonsri, W.; Macedo, D.S.; Davies, C.G.; Jameson, G.N.L.; Moubaraki, B.; Murray, K.S. Heteroleptic Iron(III) Schiff Base Spin Crossover Complexes: Halogen Substitution, Solvent Loss and Crystallite Size Effects. Dalton Trans. 2017, 46, 7020–7029. [Google Scholar] [CrossRef] [PubMed]
- Martinho, P.N.; Vicente, A.I.; Realista, S.; Saraiva, M.S.; Melato, A.I.; Brandão, P.; Ferreira, L.P.; Carvalho, M.d.D. Solution and Solid State Properties of Fe(III) Complexes Bearing N-Ethyl-N-(2-Aminoethyl)Salicylaldiminate Ligands. J. Organomet. Chem. 2014, 760, 48–54. [Google Scholar] [CrossRef]
- Vicente, A.I.; Joseph, A.; Ferreira, L.P.; de Deus Carvalho, M.; Rodrigues, V.H.N.; Duttine, M.; Diogo, H.P.; Minas da Piedade, M.E.; Calhorda, M.J.; Martinho, P.N.; et al. Dynamic Spin Interchange in a Tridentate Fe( III ) Schiff-Base Compound. Chem. Sci. 2016, 7, 4251–4258. [Google Scholar] [CrossRef] [PubMed]
- Vicente, A.I.; Ferreira, L.P.; Carvalho, M.D.D.; Rodrigues, V.H.N.; Dîrtu, M.M.; Garcia, Y.; Calhorda, M.J.; Martinho, P.N. Selecting the Spin Crossover Profile with Controlled Crystallization of Mononuclear Fe(III) Polymorphs. Dalton Trans. 2018, 47, 7013–7019. [Google Scholar] [CrossRef]
- Martins, F.F.; Joseph, A.; Diogo, H.P.; Minas da Piedade, M.E.; Ferreira, L.P.; Carvalho, M.D.; Barroso, S.; Romão, M.J.; Calhorda, M.J.; Martinho, P.N. Irreversible Magnetic Behaviour Caused by the Thermosalient Phenomenon in an Iron(III) Spin Crossover Complex. Eur. J. Inorg. Chem. 2018, 2018, 2976–2983. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt Graphical User Interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- MacRae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, M.I. (Ed.) International Tables for Crystallography, Volume A, 6th Edition, Space-group Symmetry; John Wiley & Sons Ltd.: Oxford, UK, 2017; ISBN 9780470974230. [Google Scholar]
- Fonseca Guerra, C.; Snijders, J.G.; te Velde, G.; Baerends, E.J. Towards an Order-N DFT Method. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar] [CrossRef]
- van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- ADF2013, SCM; Theoretical Chemistry. Vrije Universiteit: Amsterdam, The Netherlands. Available online: http://scm.com (accessed on 13 July 2021).
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Reiher, M.; Salomon, O.; Hess, B.A. Reparameterization of Hybrid Functionals Based on Energy Differences of States of Different Multiplicity. Theor. Chem. Acc. 2001, 107, 48–55. [Google Scholar] [CrossRef]
- Salomon, O.; Reiher, M.; Hess, B.A. Assertion and Validation of the Performance of the B3LYP* Functional for the First Transition Metal Row and the G2 Test Set. J. Chem. Phys. 2002, 117, 4729–4737. [Google Scholar] [CrossRef]
- van Lenthe, E.; Ehlers, A.; Baerends, E.-J. Geometry Optimizations in the Zero Order Regular Approximation for Relativistic Effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J. Optimized Slater-Type Basis Sets for the Elements 1-118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Buongiorno Nardelli, M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced Capabilities for Materials Modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed]
- Garrity, K.F.; Bennett, J.W.; Rabe, K.M.; Vanderbilt, D. Pseudopotentials for High-Throughput DFT Calculations. Comput. Mater. Sci. 2014, 81, 446–452. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Johnson, E.R. A Density-Functional Model of the Dispersion Interaction. J. Chem. Phys. 2005, 123, 154101. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, B.D.P.; Vicente, A.I.; Ward, J.B.J.; Sebastião, P.J.; Chávez, F.V.; Barroso, S.; Carvalho, A.; Keely, S.J.; Martinho, P.N.; Calhorda, M.J. Fe(III) SalEen Derived Schiff Base Complexes as Potential Contrast Agents. Inorg. Chim. Acta 2015, 432, 258–266. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | |
|---|---|---|---|
| Formula | C49H55BBr2FeN5O3 | C52H62BCl2FeN6O4 | C52H62BF2FeN6O4 |
| M | 988.46 | 972.65 | 939.74 |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 |
| T (K) | 296(2) | 296(2) | 296(2) |
| Crystal system | Orthorhombic | Monoclinic | Monoclinic |
| Space group [25] | P 212121 | P 21/n | P 21/n |
| a (Å) | 15.0641(11) | 14.557(3) | 14.758(11) |
| b (Å) | 15.9098(10) | 18.870(4) | 19.005(16) |
| c (Å) | 19.9825(12) | 19.498(4) | 19.688(17) |
| β (°) | 90 | 103.416(6) | 106.19(2) |
| V (Å3) | 4789.1(5) | 5209.5(19) | 5303(7) |
| Z | 4 | 4 | 4 |
| ρcalc (g.cm−3) | 1.371 | 1.147 | 1.086 |
| µ (mm−1) | 2.029 | 0.434 | 0.331 |
| Crystal size | 0.18 × 0.12 × 0.12 | 0.20 × 0.20 × 0.10 | 0.40 × 0.20 × 0.16 |
| Crystal colour | Brown | Red | Red |
| Crystal description | Block | Prism | Prism |
| θmax (°) | 25.048 | 25.854 | 25.475 |
| total data | 36794 | 114,491 | 86,088 |
| unique data | 8434 | 9993 | 9745 |
| Rint | 0.0674 | 0.1397 | 0.1497 |
| R [I>2σ(I)] | 0.0409 | 0.0684 | 0.0612 |
| Rw | 0.0887 | 0.1946 | 0.1789 |
| GooF (all data) | 1.053 | 1.032 | 1.054 |
| ρmin ρmax | −0.497 0.427 | −0.316 0.417 | −0.430 0.318 |
| 1 | 1 (150 K) | 2 | 3 | 3 (150 K) | |
|---|---|---|---|---|---|
| Fe–O | 1.880(3) | 1.867(3) | 1.866(2) | 1.883(3) | 1.856(5) |
| 1.870(3) | 1.878(3) | 1.866(3) | 1.891(3) | 1.873(5) | |
| Fe–Nim | 1.923(4) | 1.919(4) | 1.926(3) | 1.950(3) | 1.897(6) |
| 1.930(4) | 1.917(4) | 1.925(3) | 1.948(3) | 1.896(6) | |
| Fe–Nam | 2.051(5) | 2.021(4) | 2.052(3) | 2.061(3) | 2.028(6) |
| 2.020(5) | 2.049(4) | 2.031(3) | 2.083(3) | 2.019(6) |
| Complex | Temperature | Sites | δ (mms−1) | ΔEQ (mms−1) | Γ/2 (mms−1) | Fraction (%) |
|---|---|---|---|---|---|---|
| 1 (Br) | 77 K | Fe(III) LS | 0.22(1) | 2.93 (1) | 0.30(2) | 100 |
| 2 (Cl) | Room temperature | Fe(III) HS | 0.12(1) | 0.59(3) | 0.14(3) | 57 |
| Fe(III) LS | 0.12(5) | 2.57(1) | 0.22(7) | 43 |
| Complex | Bond | LS | 150 K | RT | HS |
|---|---|---|---|---|---|
| 1 (Br) | Fe–O | 1.919, 1.922 | 1.867, 1.878 | 1.870, 1.880 | 1.944, 1.948 |
| Fe–Nim | 1.982, 1.986 | 1.919, 1.917 | 1.930, 1.923 | 2.149, 2.163 | |
| Fe–Nam | 2.128, 2.128 | 2.021, 2.049 | 2.020, 2.050 | 2.296, 2.296 | |
| 2 (Cl) | Fe–O | 1.918,1.922 | - | 1.866.1.866 | 1.974, 1.979 |
| Fe–Nim | 1.979,1.980 | - | 1.926.1.925 | 2.174, 2.176 | |
| Fe–Nam | 2.120,2.131 | - | 2.052,2.031 | 2.305, 2.313 | |
| 3 (F) | Fe–O | 1.914, 1.917 | 1.856, 1.872 | 1.883, 1.891 | 1.963, 1.975 |
| Fe–Nim | 1.977, 1.978 | 1.897, 1.897 | 1.950, 1.949 | 2.159, 2.185 | |
| Fe–Nam | 2.121, 2.129 | 2.028, 2.019 | 2.061, 2.083 | 2.274, 2.316 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques, R.T.; Martins, F.F.; Bekiş, D.F.; Vicente, A.I.; Ferreira, L.P.; Gomes, C.S.B.; Barroso, S.; Kumar, V.; Garcia, Y.; Bandeira, N.A.G.; et al. The Halogen Effect on the Magnetic Behaviour of Dimethylformamide Solvates in [Fe(halide-salEen)2]BPh4. Magnetochemistry 2022, 8, 162. https://doi.org/10.3390/magnetochemistry8120162
Marques RT, Martins FF, Bekiş DF, Vicente AI, Ferreira LP, Gomes CSB, Barroso S, Kumar V, Garcia Y, Bandeira NAG, et al. The Halogen Effect on the Magnetic Behaviour of Dimethylformamide Solvates in [Fe(halide-salEen)2]BPh4. Magnetochemistry. 2022; 8(12):162. https://doi.org/10.3390/magnetochemistry8120162
Chicago/Turabian StyleMarques, Rafaela T., Frederico F. Martins, Deniz F. Bekiş, Ana I. Vicente, Liliana P. Ferreira, Clara S. B. Gomes, Sónia Barroso, Varun Kumar, Yann Garcia, Nuno A. G. Bandeira, and et al. 2022. "The Halogen Effect on the Magnetic Behaviour of Dimethylformamide Solvates in [Fe(halide-salEen)2]BPh4" Magnetochemistry 8, no. 12: 162. https://doi.org/10.3390/magnetochemistry8120162
APA StyleMarques, R. T., Martins, F. F., Bekiş, D. F., Vicente, A. I., Ferreira, L. P., Gomes, C. S. B., Barroso, S., Kumar, V., Garcia, Y., Bandeira, N. A. G., Calhorda, M. J., & Martinho, P. N. (2022). The Halogen Effect on the Magnetic Behaviour of Dimethylformamide Solvates in [Fe(halide-salEen)2]BPh4. Magnetochemistry, 8(12), 162. https://doi.org/10.3390/magnetochemistry8120162