
Transcriptome Analysis of Rhododendron liliiflorum H. Lév. Flower Colour Differences
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Materials
2.2. RNA Extraction, Library Construction and Sequencing
2.3. Sequence Filtering, Splicing and Assembly
2.4. Unigene Functional Annotation and Expression Level Analysis
2.5. Screening and Enrichment Analysis of DEGs
3. Results
3.1. Transcriptome Sequencing and Assembly
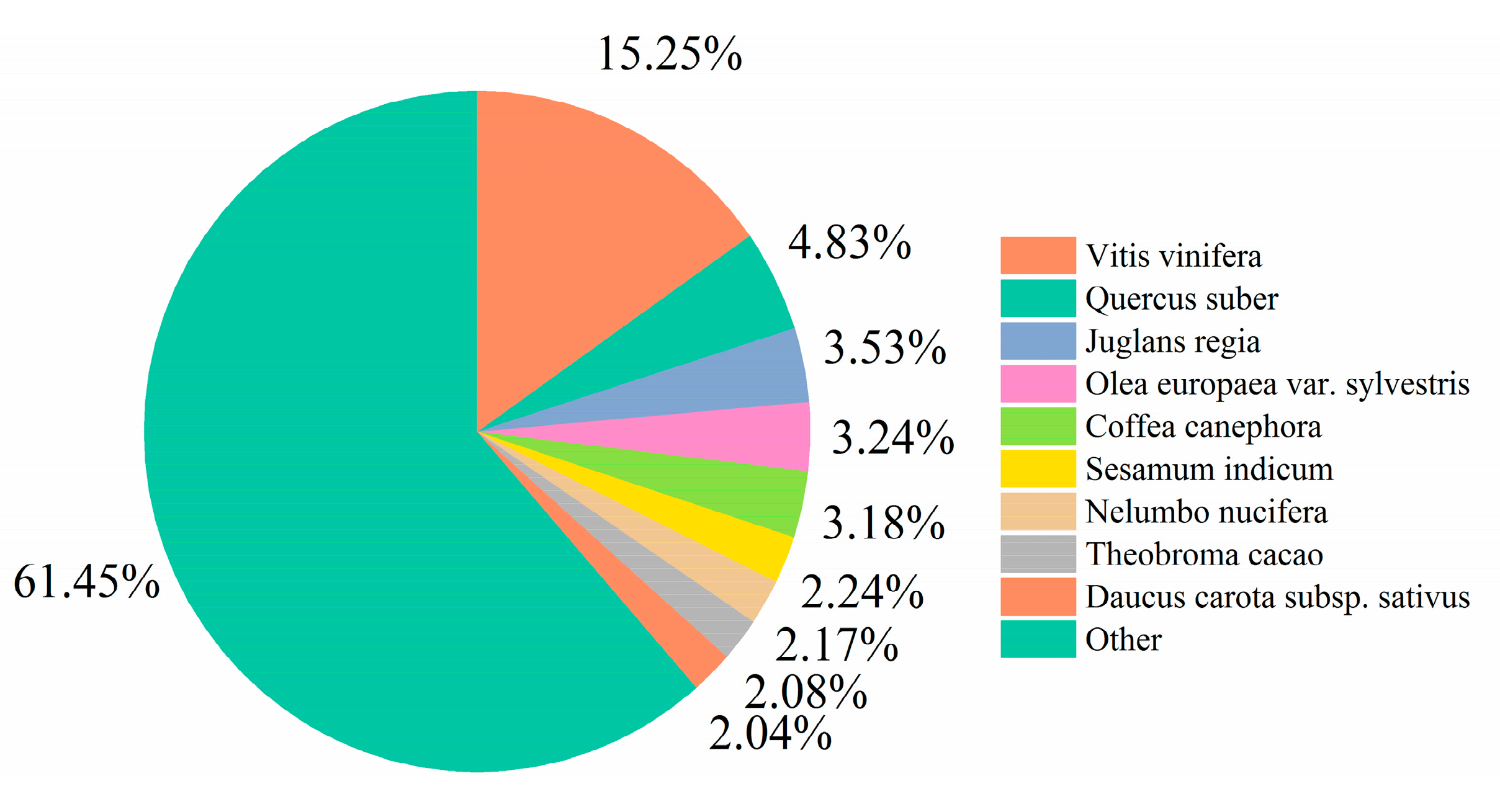
3.2. Functional Annotation Results
3.3. GO Terms and Classifications
3.4. KOG Terms and Classifications
3.5. Screening and Enrichment Analysis of DEGs in Yellow and White Parts of R. liliiflorum Petals
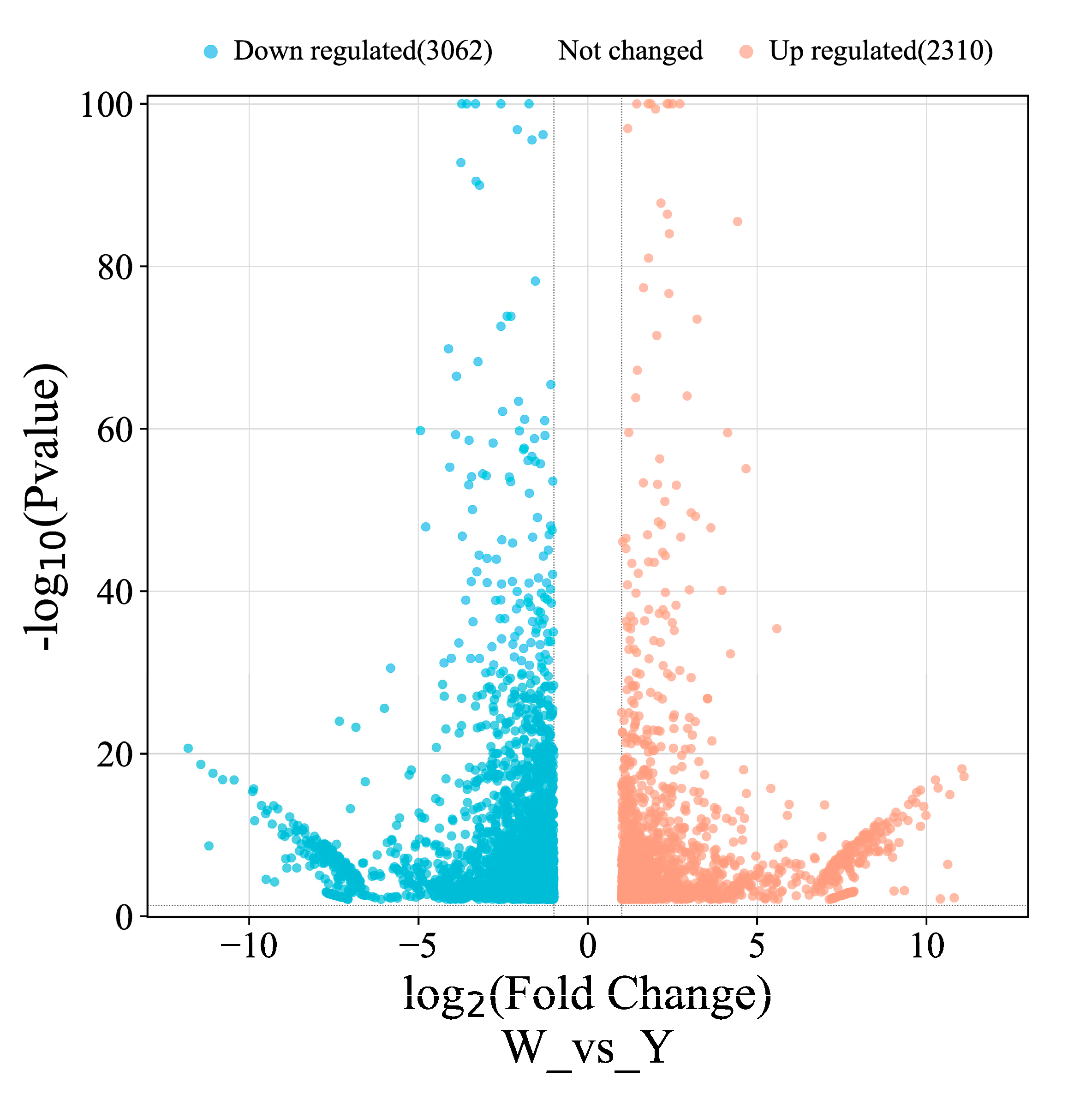
3.5.1. Screening of DEGs
3.5.2. GO Enrichment of DEGs
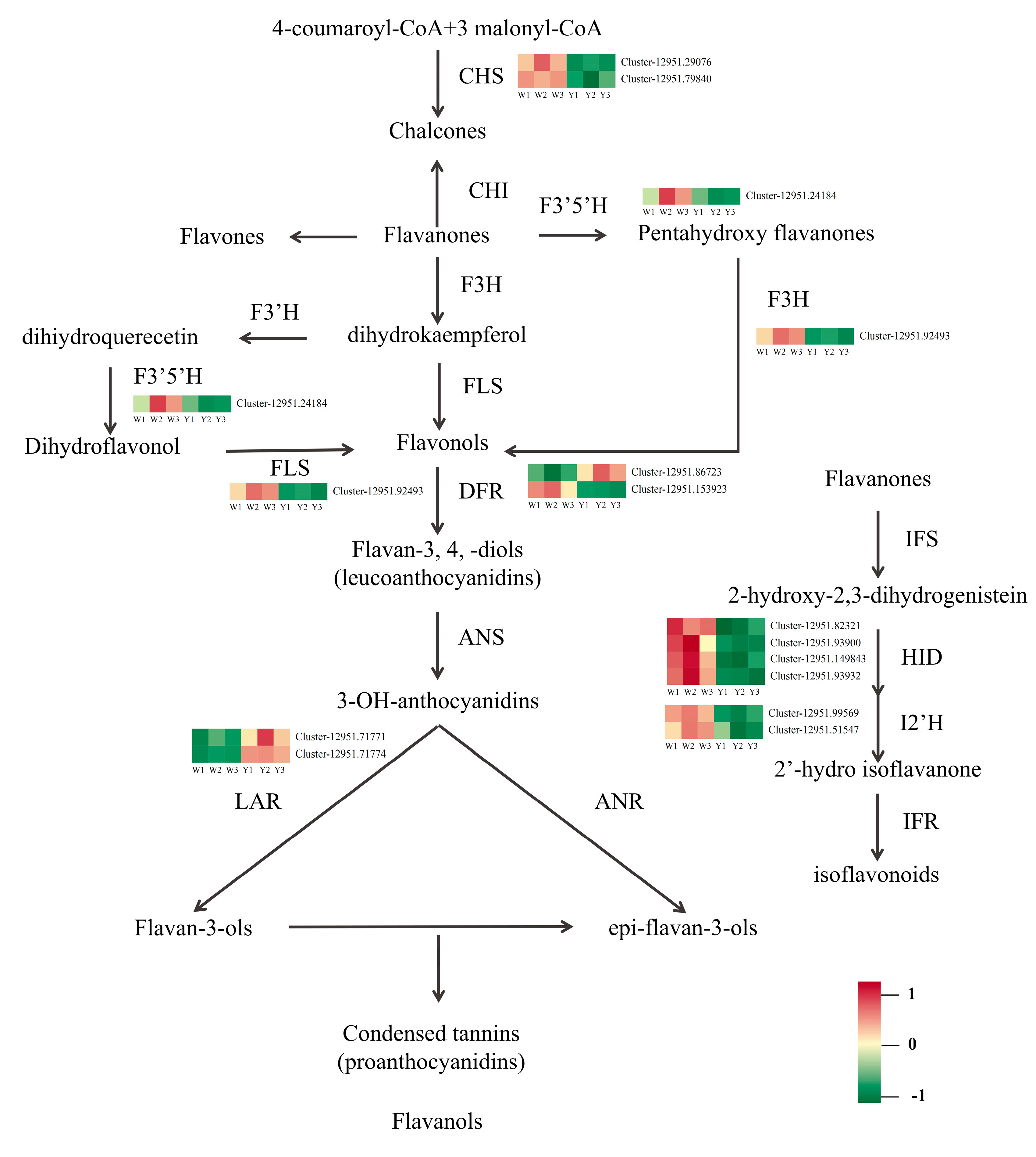
3.5.3. KEGG Enrichment Analysis of DEGs
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bradshaw, H.D.; Schemske, D.W. Allele substitution at a flower colour locus produces a pollinator shift in monkeyflowers. Nature 2003, 426, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Schmitzer, V.; Veberic, R.; Osterc, G.; Stampar, F. Color and Phenolic Content Changes during Flower Development in Groundcover Rose. J. Am. Soc. Hortic. Sci. 2010, 135, 195–202. [Google Scholar] [CrossRef]
- Ye, L.-J.; Möller, M.; Luo, Y.-H.; Zou, J.-Y.; Zheng, W.; Wang, Y.-H.; Liu, J.; Zhu, A.-D.; Hu, J.-Y.; Li, D.-Z. Differential expressions of anthocyanin synthesis genes underlie flower color divergence in a sympatric Rhododendron sanguineum complex. BMC Plant Biol. 2021, 21, 204. [Google Scholar] [CrossRef] [PubMed]
- Mol, J.; Grotewold, E.; Koes, R. How genes paint flowers and seeds. Trends Plant Sci. 1998, 3, 212–217. [Google Scholar] [CrossRef]
- Chen, S.; Li, C.; Zhu, X.; Deng, Y.M.; Sun, W.; Wang, L.; Chen, F.; Zhang, Z. The identification of flavonoids and the expression of genes of anthocyanin biosynthesis in the chrysanthemum flowers. Biol. Plant. 2012, 56, 458–464. [Google Scholar] [CrossRef]
- Thill, J.; Miosic, S.; Ahmed, R.; Schlangen, K.; Muster, G.; Stich, K.; Halbwirth, H. ‘Le Rouge et le Noir’: A decline in flavone formation correlates with the rare color of black dahlia (Dahlia variabilis hort.) flowers. BMC Plant Biol. 2012, 12, 225. [Google Scholar] [CrossRef]
- Tatsuzawa, F.; Saito, N.; Toki, K.; Shinoda, K.; Honda, T. Flower Colors and their Anthocyanins in Matthiola incana Cultivars (Brassicaceae). J. Jpn. Soc. Hortic. Sci. 2012, 81, 91. [Google Scholar] [CrossRef]
- Zhao, D.; Tao, J.; Han, C.; Ge, J. Flower color diversity revealed by differential expression of flavonoid biosynthetic genes and flavonoid accumulation in herbaceous peony (Paeonia lactiflora Pall.). Mol. Biol. Rep. 2012, 39, 11263–11275. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef]
- Zhou, Z.; Gao, H.; Ming, J.; Ding, Z.; Lin, X.e.; Zhan, R. Combined Transcriptome and Metabolome analysis of Pitaya fruit unveiled the mechanisms underlying Peel and pulp color formation. BMC Genom. 2020, 21, 734. [Google Scholar] [CrossRef]
- Liu, W.; Feng, Y.; Yu, S.; Fan, Z.; Li, X.; Li, J.; Yin, H. The Flavonoid Biosynthesis Network in Plants. Int. J. Mol. Sci. 2021, 22, 12824. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, M.; Nakatsuka, T.; Yamamura, S. Flavonoid components and flower color change in transgenic tobacco plants by suppression of chalcone isomerase gene. FEBS Lett. 2005, 579, 6074–6078. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Li, M.; Li, Z.; Li, S.; Zhu, Y.; Ding, H.; Wang, Z. Transcriptome analysis of the molecular mechanism of Chrysanthemum flower color change under short-day photoperiods. Plant Physiol. Biochem. 2020, 146, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Malik, W.; Khan, A.; Masooma, H.; Aslam, U.; Iqbal, M.; Qayyum, A.; Yasmeen, A.; Bibi, N. Transcriptome Analysis of Pigment Related Genes in Colored Cotton. Int. J. Agric. Biol. 2015, 17, 1560–8530. [Google Scholar]
- Zheng, X.; Zhao, B.; Zeng, H.; Shen, H.; Xu, J. Comparative Analysis of Composition and Content of Pigments in Petals of Three Different Colors of Rhododendron calophytum in Qinling Mountains. J. Northwest For. Univ. 2017, 32, 62–68. (In Chinese) [Google Scholar]
- Du, H.; Lai, L.; Wang, F.; Sun, W.; Zhang, L.; Li, X.; Wang, L.; Jiang, L.; Zheng, Y. Characterisation of flower colouration in 30 Rhododendron species via anthocyanin and flavonol identification and quantitative traits. Plant Biol. 2018, 20, 121–129. [Google Scholar] [CrossRef]
- Jaakola, L.; Määttä, K.; Pirttilä, A.M.; Törrönen, R.; Kärenlampi, S.; Hohtola, A. Expression of genes involved in anthocyanin biosynthesis in relation to anthocyanin, proanthocyanidin, and flavonol levels during bilberry fruit development. Plant Physiol. 2002, 130, 729–739. [Google Scholar] [CrossRef]
- Zipor, G.; Duarte, P.; Carqueijeiro, I.; Shahar, L.; Ovadia, R.; Teper-Bamnolker, P.; Eshel, D.; Levin, Y.; Doron-Faigenboim, A.; Sottomayor, M.; et al. In planta anthocyanin degradation by a vacuolar class III peroxidase in Brunfelsia calycina flowers. New Phytol. 2015, 205, 653–665. [Google Scholar] [CrossRef]
- Liu, Y.; Tikunov, Y.; Schouten, R.E.; Marcelis, L.F.M.; Visser, R.G.F.; Bovy, A. Anthocyanin Biosynthesis and Degradation Mechanisms in Solanaceous Vegetables: A Review. Front. Chem. 2018, 6, 52. [Google Scholar] [CrossRef]
- Mizuta, D.; Ban, T.; Miyajima, I.; Nakatsuka, A.; Kobayashi, N. Comparison of flower color with anthocyanin composition patterns in evergreen azalea. Sci. Hortic. 2009, 122, 594–602. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Y.; Zou, Q.; Shan, W.; Li, X.; Zhang, L. Effects of IBA concentration and cutting time on rooting of Rhododendron kiangsiense and Rh. liliiflorum cuttings. Guihaia 2016, 36, 1468–1475. (In Chinese) [Google Scholar]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformation 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef]
- Deng, X.; Bashandy, H.; Ainasoja, M.; Kontturi, J.; Pietiäinen, M.; Laitinen, R.A.E.; Albert, V.A.; Valkonen, J.P.T.; Elomaa, P.; Teeri, T.H. Functional diversification of duplicated chalcone synthase genes in anthocyanin biosynthesis of Gerbera hybrida. New Phytol. 2014, 201, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Schijlen, E.; De Vos, R.; Martens, S.; Jonker, H.; Rosin, F.; Molthoff, J.; Tikunov, Y.; Angenent, G.; van tunen, A.; Bovy, A. RNA Interference Silencing of Chalcone Synthase, the First Step in the Flavonoid Biosynthesis Pathway, Leads to Parthenocarpic Tomato Fruits. Plant Physiol. 2007, 144, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Dubouzet, J.G.; Matsumura, H.; Yasuda, K.; Iwashina, T. A new allele of flower color gene W1 encoding flavonoid 3’5’-hydroxylase is responsible for light purple flowers in wild soybean Glycine soja. BMC Plant Biol. 2010, 10, 155. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Shi, Y.; Fu, Z.; Li, W.-W.; Lai, S.; Wu, Y.; Wang, Y.; Liu, Y.; Gao, L.; Xia, T. Functional characterization of three flavonol synthase genes from Camellia sinensis: Roles in flavonol accumulation. Plant Sci. 2020, 300, 110632. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, D.-H.; Yang, J.-H.; Lee, J.-Y.; Lim, S. Increased Flavonol Levels in Tobacco Expressing AcFLS Affect Flower Color and Root Growth. Int. J. Mol. Sci. 2020, 21, 1011. [Google Scholar] [CrossRef]
- Veremeichik, G.N.; Grigorchuk, V.P.; Butovets, E.S.; Lukyanchuk, L.M.; Brodovskaya, E.V.; Bulgakov, D.V.; Bulgakov, V.P. Isoflavonoid biosynthesis in cultivated and wild soybeans grown in the field under adverse climate conditions. Food Chem. 2021, 342, 128292. [Google Scholar] [CrossRef]
- Uchida, K.; Akashi, T.; Aoki, T. The Missing Link in Leguminous Pterocarpan Biosynthesis is a Dirigent Domain-Containing Protein with Isoflavanol Dehydratase Activity. Plant Cell Physiol. 2017, 58, 398–408. [Google Scholar] [CrossRef]
- Yao, S.; Lan, Z.; Huang, R.; Tan, Y.; Huang, D.; Gu, J.; Pan, C. Hormonal and transcriptional analyses provides new insights into the molecular mechanisms underlying root thickening and isoflavonoid biosynthesis in Callerya speciosa (Champ. ex Benth.) Schot. Sci. Rep. 2021, 11, 9. [Google Scholar] [CrossRef]
- Sun, w.; Zhou, N.; Feng, C.; Sun, S.; Tang, M.; Tang, X.; Ju, Z.; Yi, Y. Functional analysis of a dihydroflavonol 4-reductase gene in Ophiorrhiza japonica (OjDFR1) reveals its role in the regulation of anthocyanin. PeerJ 2021, 9, e12323. [Google Scholar] [CrossRef]
- Halbwirth, H.; Muster, G.; Stich, K. Unraveling the Biochemical Base of Dahlia Flower Coloration. Nat. Prod. Commun. 2008, 3, 1934578X0800300807. [Google Scholar] [CrossRef]
- Li, H.; Tian, J.; Yao, Y.-y.; Zhang, J.; Song, T.-t.; Li, K.-t.; Yao, Y.-c. Identification of leucoanthocyanidin reductase and anthocyanidin reductase genes involved in proanthocyanidin biosynthesis in Malus crabapple plants. Plant Physiol. Biochem. 2019, 139, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, X.; Shulaev, V.; Dixon, R.A. A role for leucoanthocyanidin reductase in the extension of proanthocyanidins. Nat. Plants 2016, 2, 16182. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yang, B.; Wu, J.; Luo, P.; Anwar, M.; Allan, A.C.; Lin-Wang, K.; Espley, R.V.; Zeng, L. Identification of Genes Involved in Flavonoid Biosynthesis of Chinese Narcissus (Narcissus tazetta L. var. chinensis). Plant Mol. Biol. Report. 2018, 36, 812–821. [Google Scholar] [CrossRef]
- Zou, Q.; Wang, T.; Guo, Q.; Yang, F.; Chen, J.; Zhang, W. Combined metabolomic and transcriptomic analysis reveals redirection of the phenylpropanoid metabolic flux in different colored medicinal Chrysanthemum morifolium. Ind. Crops Prod. 2021, 164, 113343. [Google Scholar] [CrossRef]
- Zhu, J.; Guo, X.; Li, X.; Tang, D. Composition of Flavonoids in the Petals of Freesia and Prediction of Four Novel Transcription Factors Involving in Freesia Flavonoid Pathway. Front. Plant Sci. 2021, 12, 756300. [Google Scholar] [CrossRef]
- Wu, Y.-q.; Wei, M.-r.; Zhao, D.-q.; Tao, J. Flavonoid content and expression analysis of flavonoid biosynthetic genes in herbaceous peony (Paeonia lactiflora Pall.) with double colors. J. Integr. Agric. 2016, 15, 2023–2031. [Google Scholar] [CrossRef]
- Jin, X.; Huang, H.; Wang, L.; Sun, Y.; Dai, S. Transcriptomics and Metabolite Analysis Reveals the Molecular Mechanism of Anthocyanin Biosynthesis Branch Pathway in Different Senecio cruentus Cultivars. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef]
- Xing, A.S.; Wang, X.Y.; Nazir, M.F.; Zhang, X.M.; Wang, X.X.; Yang, R.; Chen, B.J.; Fu, G.Y.; Wang, J.J.; Ge, H.; et al. Transcriptomic and metabolomic profiling of flavonoid biosynthesis provides novel insights into petals coloration in Asian cotton (Gossypium arboreum L.). BMC Plant Biol. 2022, 22, 416. [Google Scholar] [CrossRef]
- Lang, X.; Li, N.; Li, L.; Zhang, S. Integrated Metabolome and Transcriptome Analysis Uncovers the Role of Anthocyanin Metabolism in Michelia maudiae. Int. J. Genom. 2019, 2019, 4393905. [Google Scholar] [CrossRef]
- Wang, R.; Ren, C.; Dong, S.; Chen, C.; Xian, B.; Wu, Q.; Wang, J.; Pei, J.; Chen, J. Integrated Metabolomics and Transcriptome Analysis of Flavonoid Biosynthesis in Safflower (Carthamus tinctorius L.) with Different Colors. Front. Plant Sci. 2021, 12. [Google Scholar] [CrossRef]
- Bao, M.; Liu, M.; Zhang, Q.; Wang, T.; Sun, X.; Xu, J. Factors Affecting the Color of Herbaceous Peony. J. Am. Soc. Hortic. Sci. J. Amer. Soc. Hort. Sci. 2020, 145, 257–266. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Clean Bases (G) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|
| W1 | 60,163,298 | 58,377,468 | 8.76 | 98.02 | 94.29 | 46.69 |
| W2 | 63,100,240 | 61,086,422 | 9.16 | 98.05 | 94.35 | 46.59 |
| W3 | 58,213,406 | 56,532,014 | 8.48 | 97.91 | 94.03 | 46.76 |
| Y1 | 51,367,344 | 49,409,492 | 7.41 | 98.11 | 94.51 | 46.36 |
| Y2 | 48,489,068 | 46,025,466 | 6.9 | 98.11 | 94.53 | 46.14 |
| Y3 | 54,472,478 | 52,095,038 | 7.81 | 98.13 | 94.55 | 46.16 |
| Item | Total Sequence Number | Average Length (bp) | N50 Length (bp) | N90 Length (bp) |
|---|---|---|---|---|
| Transcripts | 219,221 | 1135 | 1928 | 470 |
| Unigenes | 180,677 | 1317 | 2016 | 590 |
| Sequence Number | 200–500 bp | 500–1000 bp | 1000–2000 bp | ≥2000 bp |
|---|---|---|---|---|
| Transcripts | 85,520 | 50,964 | 45,446 | 37,291 |
| Unigenes | 47,886 | 50,072 | 45,429 | 37,290 |
| Database | Number of Genes | Percentage (%) |
|---|---|---|
| KEGG | 81,784 | 47.56% |
| Nr | 108,753 | 63.25% |
| Swiss-Prot | 78,209 | 45.48% |
| Trembl | 108,159 | 62.90% |
| KOG | 66,156 | 38.47% |
| GO | 90,560 | 52.67% |
| Pfam | 81,639 | 47.48% |
| Gene Name | EC No. | Number of DEGs |
|---|---|---|
| CYP73A | E1.14.13.11 | 1 |
| 4CL | E6.2.1.12 | 3 |
| CHS | E2.3.1.74 | 2 |
| DFR | E1.1.12.34 | 2 |
| F3′5′H | E1.14.14.81 | 1 |
| FLS | E1.14.11.23 | 1 |
| LAR | E1.14.11.19 | 2 |
| CCoAOMT | E2.1.1.104 | 2 |
| HCT | E2.3.1.133 | 16 |
| I2′H | E1.14.13.89 | 2 |
| HID | E4.2.1.105 | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Chen, M.; Wang, X.; Dai, J.; Zhang, X.; Zhang, Z.; Zhang, X.; Tang, M.; Tang, J.; Gong, J.; et al. Transcriptome Analysis of Rhododendron liliiflorum H. Lév. Flower Colour Differences. Horticulturae 2023, 9, 82. https://doi.org/10.3390/horticulturae9010082
Zhang H, Chen M, Wang X, Dai J, Zhang X, Zhang Z, Zhang X, Tang M, Tang J, Gong J, et al. Transcriptome Analysis of Rhododendron liliiflorum H. Lév. Flower Colour Differences. Horticulturae. 2023; 9(1):82. https://doi.org/10.3390/horticulturae9010082
Chicago/Turabian StyleZhang, Hang, Meifeng Chen, Xinglin Wang, Jin Dai, Xu Zhang, Zhengdong Zhang, Ximin Zhang, Ming Tang, Jing Tang, Jiyi Gong, and et al. 2023. "Transcriptome Analysis of Rhododendron liliiflorum H. Lév. Flower Colour Differences" Horticulturae 9, no. 1: 82. https://doi.org/10.3390/horticulturae9010082
APA StyleZhang, H., Chen, M., Wang, X., Dai, J., Zhang, X., Zhang, Z., Zhang, X., Tang, M., Tang, J., Gong, J., Liu, L., & Yi, Y. (2023). Transcriptome Analysis of Rhododendron liliiflorum H. Lév. Flower Colour Differences. Horticulturae, 9(1), 82. https://doi.org/10.3390/horticulturae9010082