
A Preliminary Study for Identifying Genes Associated with Pellicle Development in Xinjiang Walnut (Juglans regia L.)

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Sample Collection
2.2. RNA Extraction, Library Preparation, RNA Sequencing
2.3. RNA-Seq Data Analysis
2.4. Functional Enrichment Analysis
2.5. Quantitative Real-Time PCR Validation
3. Results
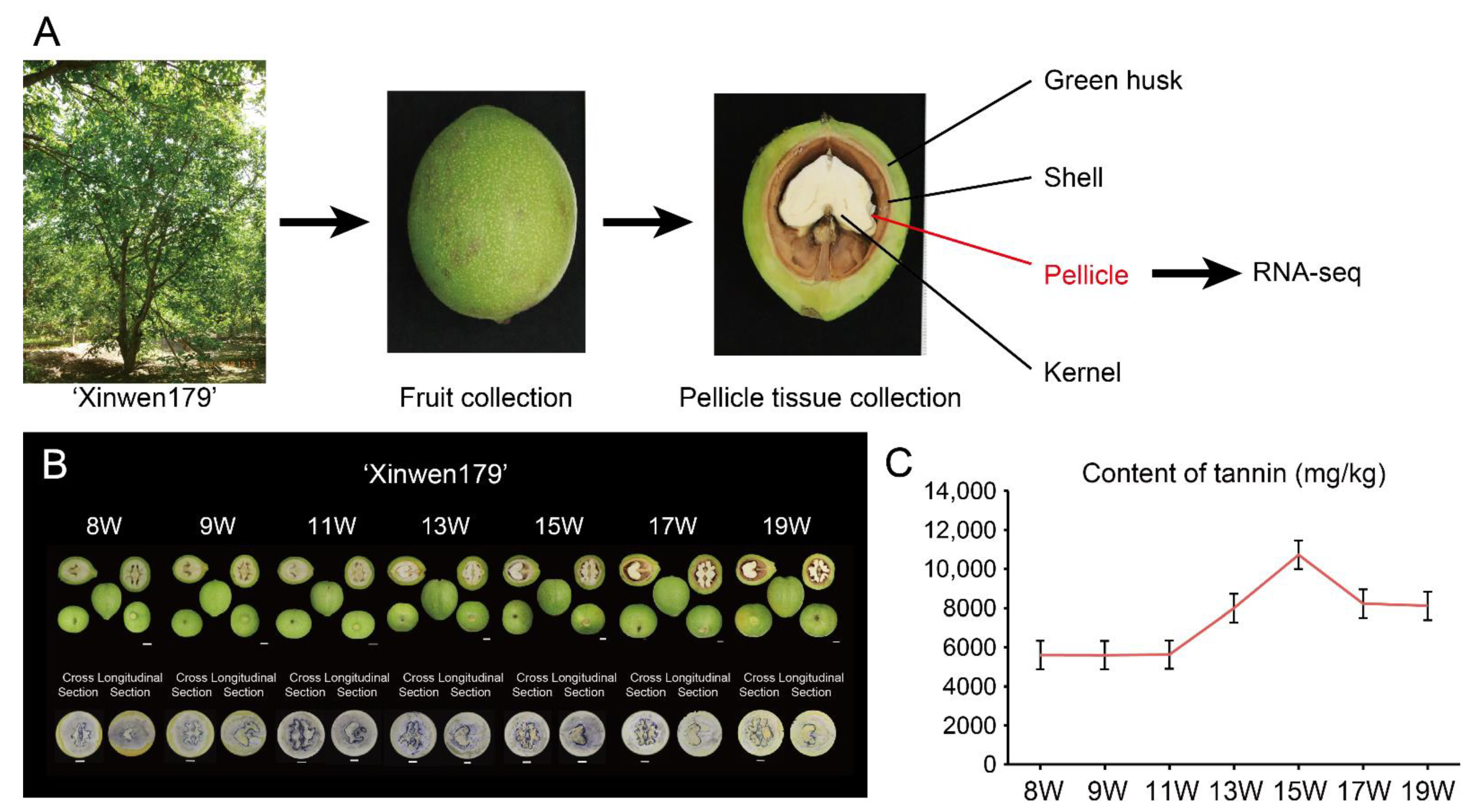
3.1. Fruit Phenotype and Dynamic Tannin Content in ‘Xinwen179’
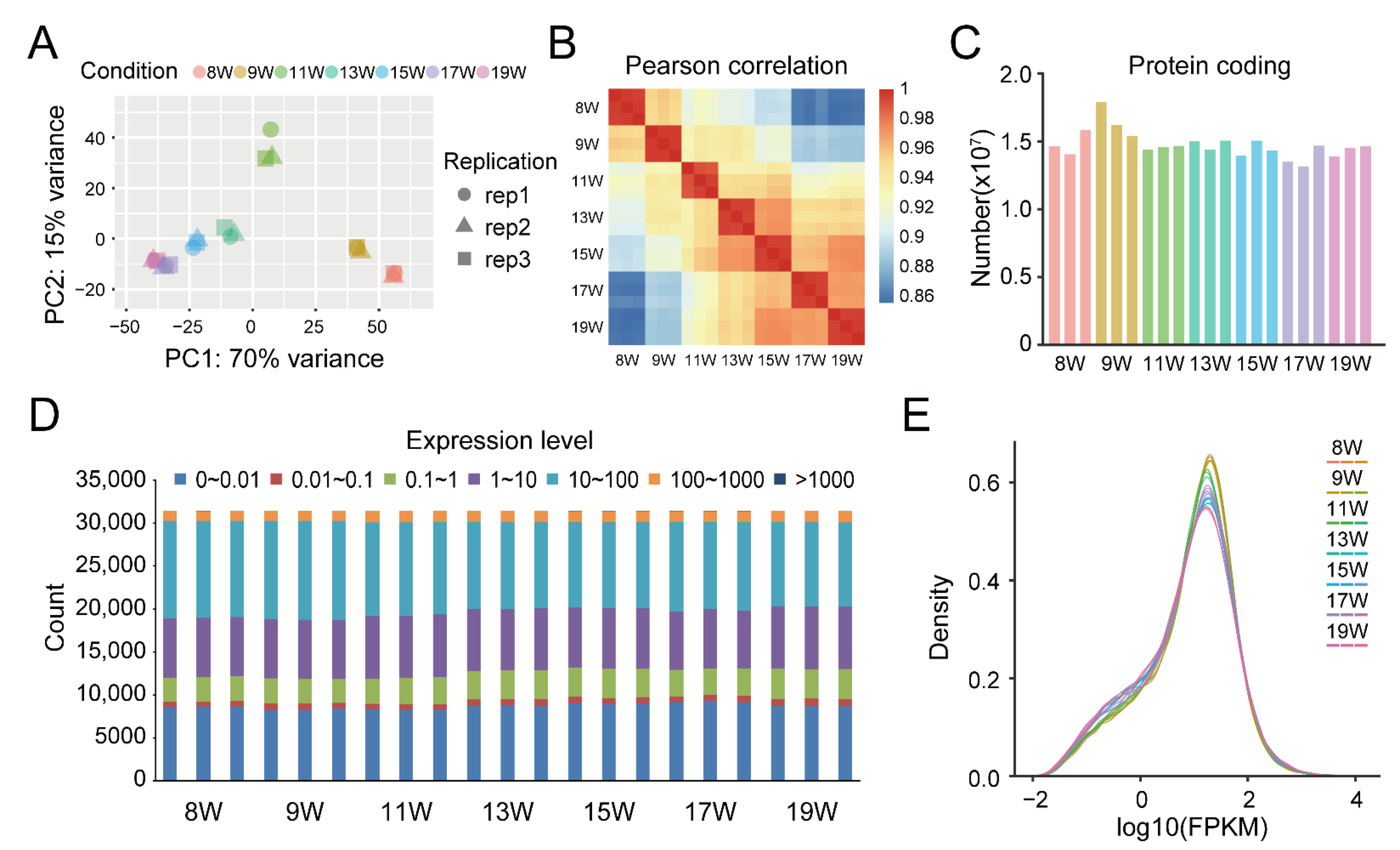
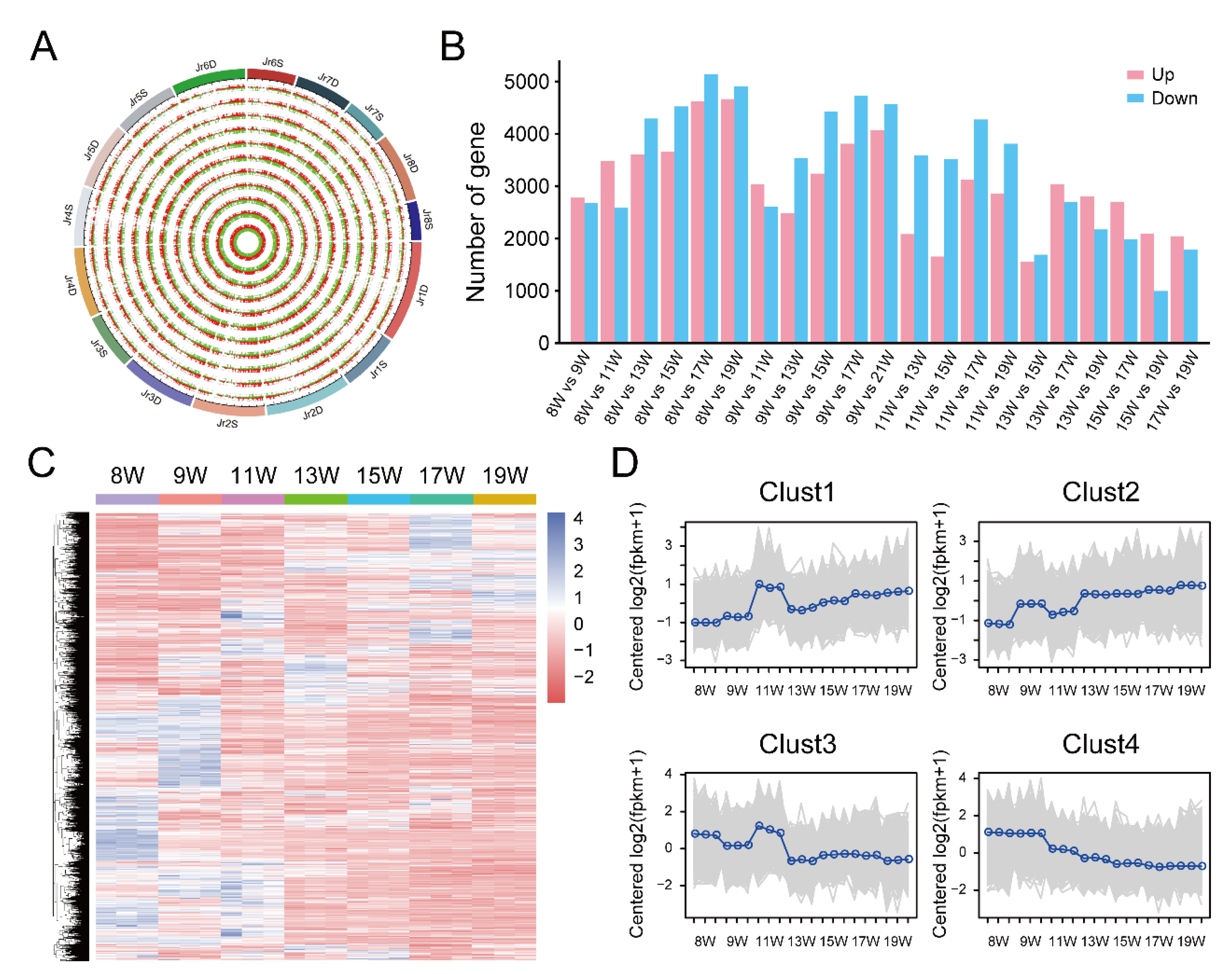
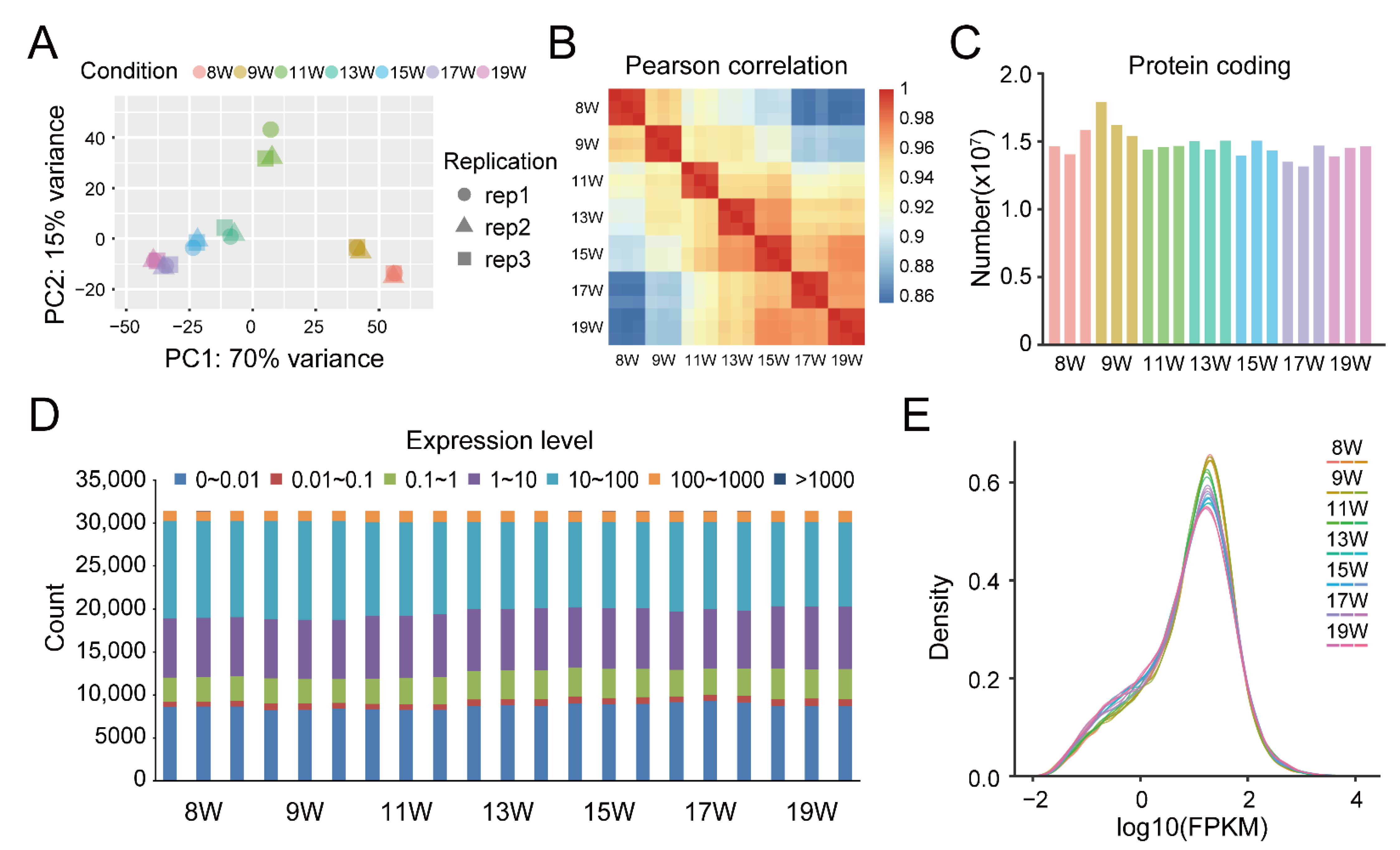
3.2. RNA-seq Results of Dynamic Development Stages of Walnut Pellicles
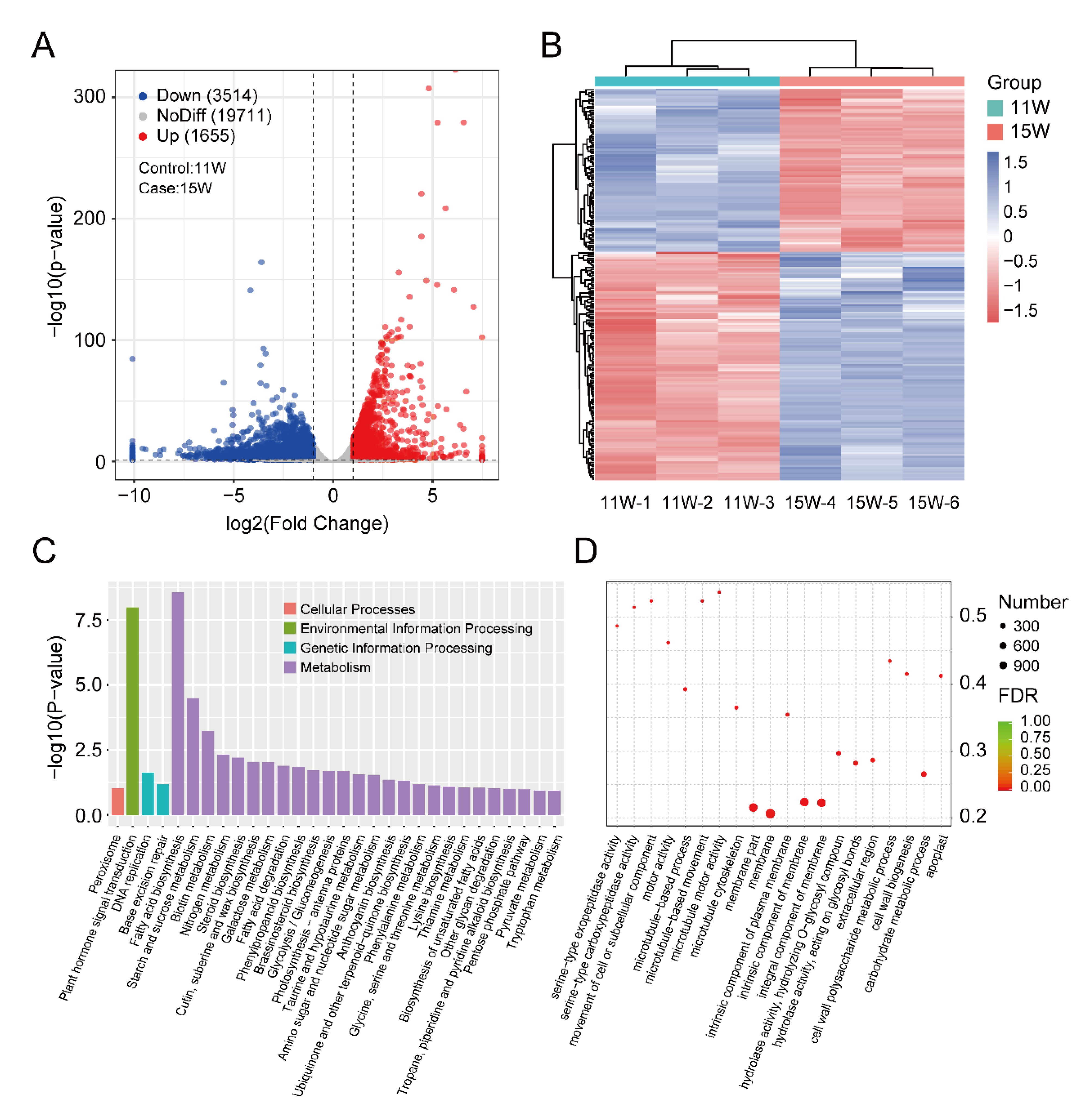
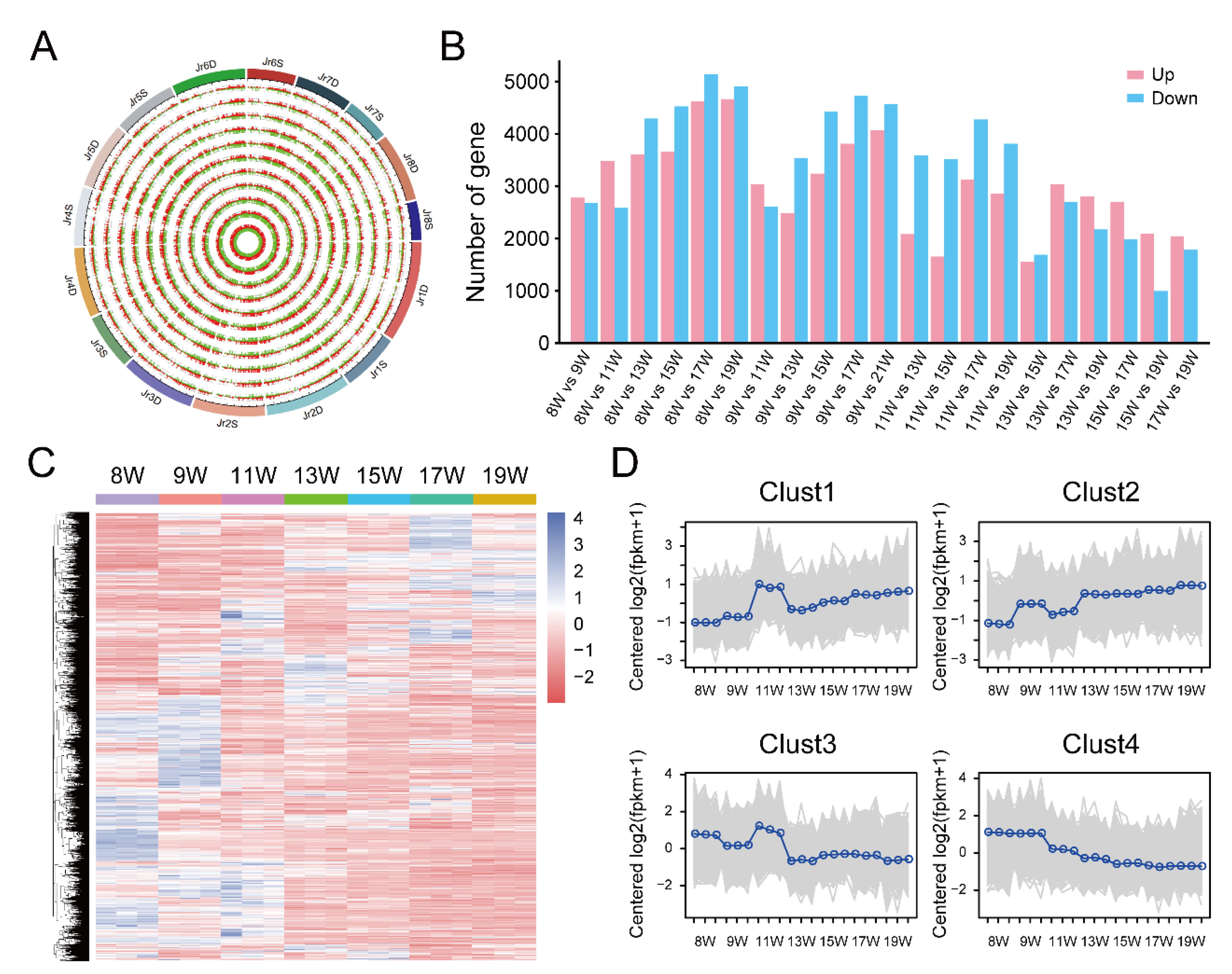
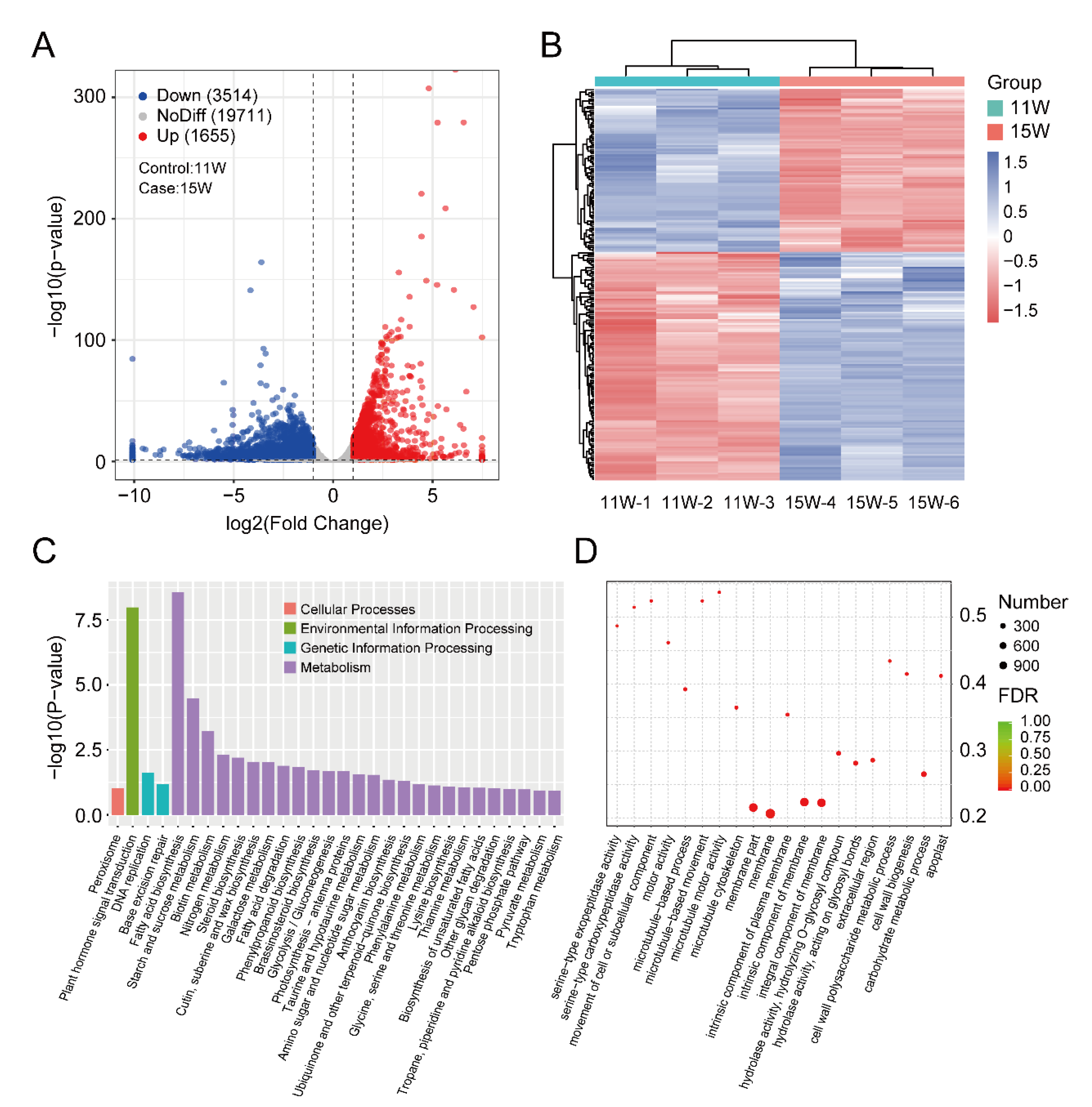
3.3. Identification and Enrichment Analysis of DEGs between 11 W and 15 W Sample
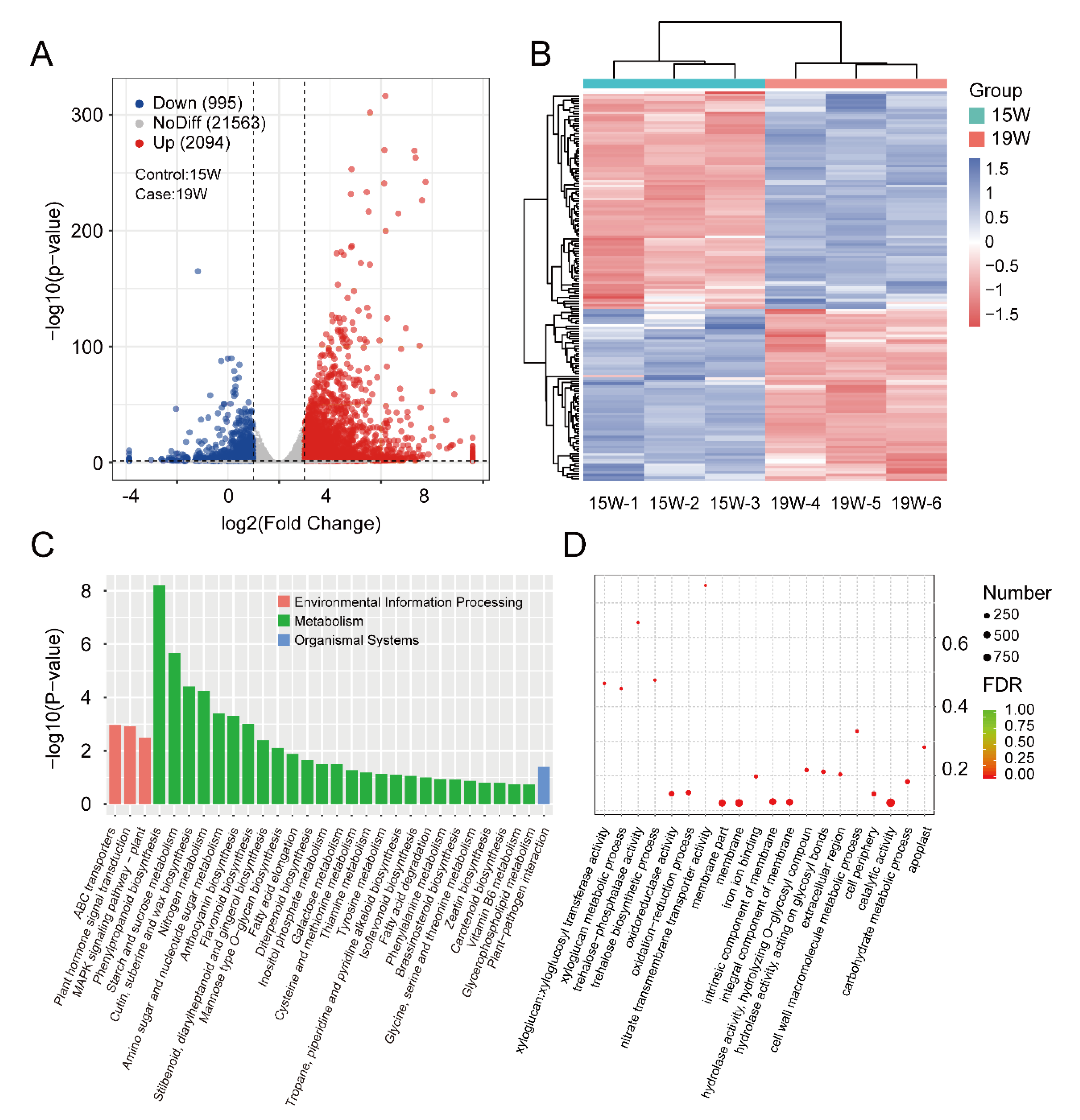
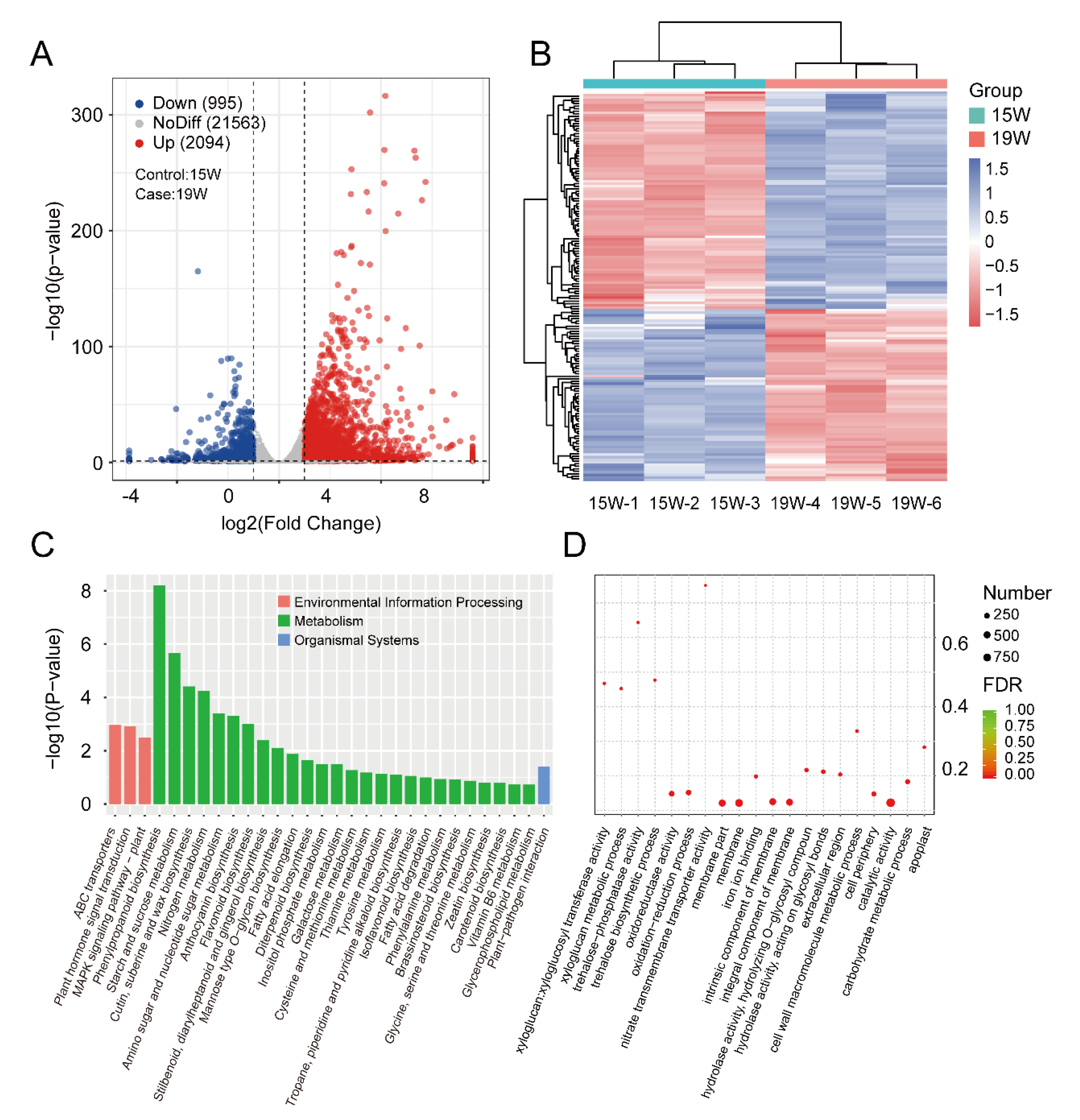
3.4. Identification and Enrichment Analysis of DEGs between the 15 W and 19 W Samples
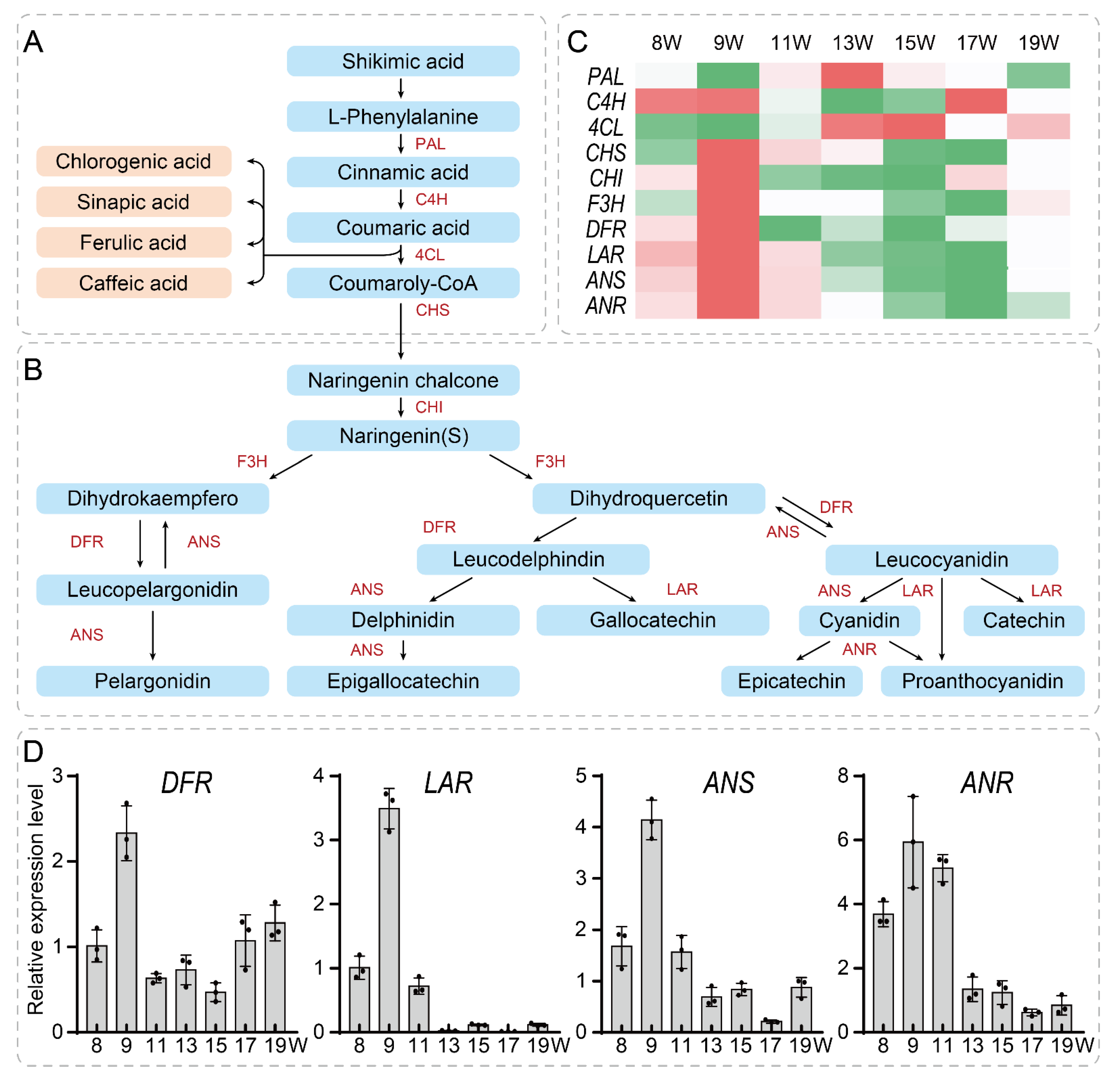
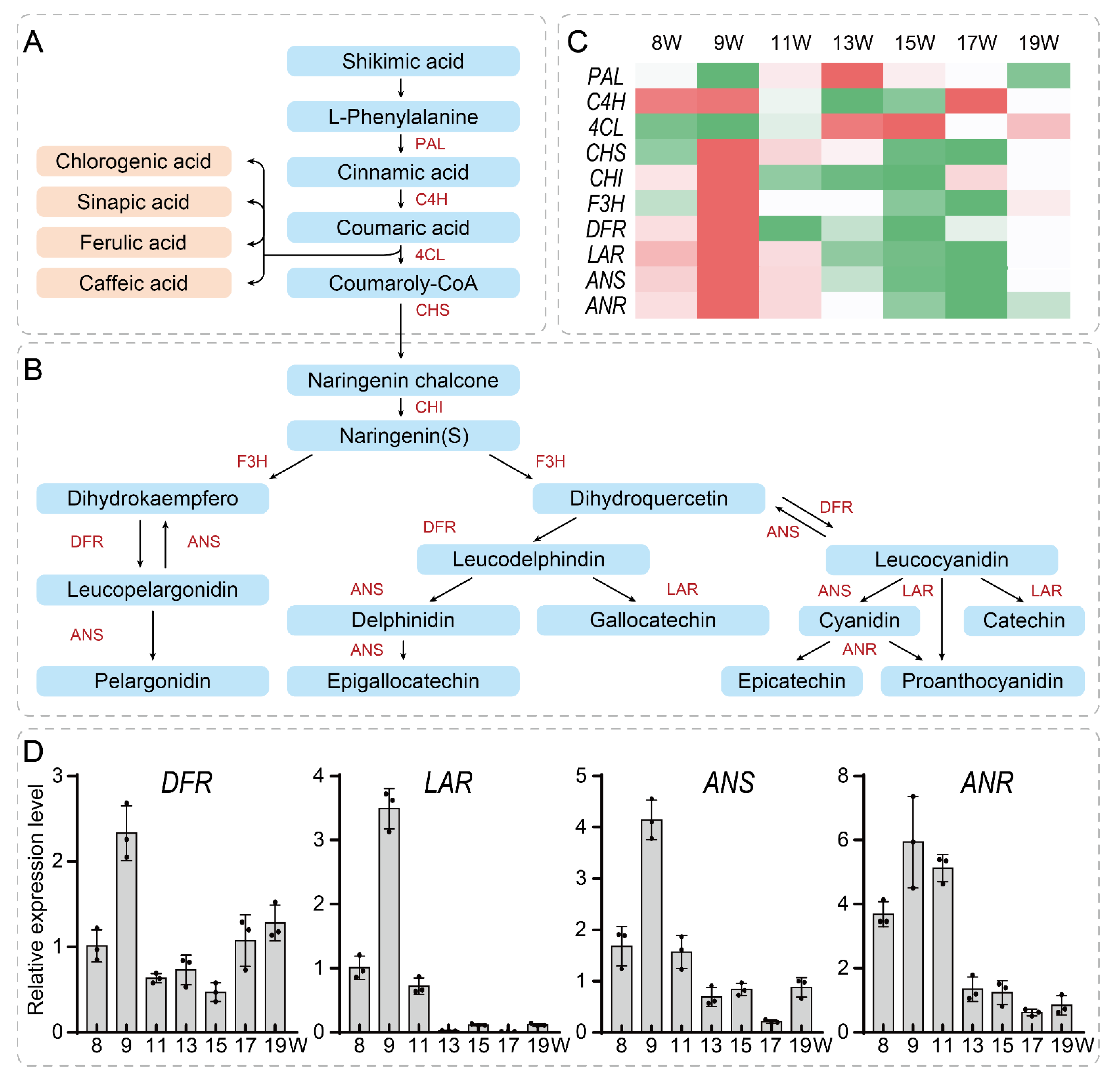
3.5. Network Construction and Validation of Key Genes Related to the Tannin Synthesis Pathway in the Walnut Pellicle
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bernard, A.; Lheureux, F.; Dirlewanger, E. Walnut: Past and future of genetic improvement. Tree Genet. Genomes 2018, 14, 1–28. [Google Scholar] [CrossRef]
- Jahanban-Esfahlan, A.; Ostadrahimi, A.; Tabibiazar, M.; Amarowicz, R. A comprehensive review on the chemical constituents and functional uses of walnut (Juglans spp.) husk. Int. J. Mol. Sci. 2019, 20, 3920. [Google Scholar] [CrossRef] [PubMed]
- Sheng, F.; Hu, B.; Jin, Q.; Wang, J.; Wu, C.; Luo, Z. The analysis of phenolic compounds in walnut husk and pellicle by UPLC-Q-Orbitrap HRMS and HPLC. Molecules 2021, 26, 3013. [Google Scholar] [CrossRef]
- Hardman, W.E. Walnuts have potential for cancer prevention and treatment in mice. J. Nutr. 2014, 144, 555S–560S. [Google Scholar] [CrossRef]
- Pandey, K.B.; Rizvi, S.I. Plant polyphenols as dietary antioxidants in human health and disease. Oxid. Med. Cell. Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Trandafir, I.; Cosmulescu, S.; Nour, V. Phenolic profile and antioxidant capacity of walnut extract as influenced by the extraction method and solvent. Int. J. Food Eng. 2017, 13, 1–8. [Google Scholar] [CrossRef]
- Martínez, M.L.; Labuckas, D.O.; Lamarque, A.L.; Maestri, D.M. Walnut (Juglans regia L.): Genetic resources, chemistry, by-products. J. Sci. Food Agric. 2010, 90, 1959–1967. [Google Scholar] [CrossRef]
- Jahanban-Esfahlan, A.; Amarowicz, R. Walnut (Juglans regia L.) shell pyroligneous acid: Chemical constituents and functional applications. RSC Adv. 2018, 8, 22376–22391. [Google Scholar] [CrossRef]
- Persic, M.; Mikulic-Petkovsek, M.; Slatnar, A.; Solar, A.; Veberic, R. Changes in phenolic profiles of red-colored pellicle walnut and hazelnut kernel during ripening. Food Chem. 2018, 252, 349–355. [Google Scholar] [CrossRef]
- Colaric, M.; Veberic, R.; Solar, A.; Hudina, M.; Stampar, F. Phenolic acids, syringaldehyde, and juglone in fruits of different cultivars of Juglans regia L. J. Agric. Food Chem. 2005, 53, 6390–6396. [Google Scholar] [CrossRef]
- Hu, B.; Sheng, F.; Jin, Q.; Wang, J.; Wu, C.; Chen, W.; Luo, Z. Characterization of the 5-enolpyruvylshikimate-3-phosphate synthase gene from walnut (Juglans regia L.). Hortic. J. 2022, 91, 176–185. [Google Scholar] [CrossRef]
- Quan, S.; Niu, J.; Zhou, L.; Xu, H.; Ma, L.; Qin, Y. Stages identifying and transcriptome profiling of the floral transition in Juglans regia. Sci. Rep. 2019, 9, 7092. [Google Scholar] [CrossRef] [PubMed]
- Hassankhah, A.; Rahemi, M.; Ramshini, H.; Sarikhani, S.; Vahdati, K. Flowering in Persian walnut: Patterns of gene expression during flower development. BMC Plant Biol. 2020, 20, 136. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhang, R.; Gao, S.; Pan, Z.; Guo, Z. Transcriptome analysis and phenotyping of walnut seedling roots under nitrogen stresses. Sci. Rep. 2022, 12, 12066. [Google Scholar] [CrossRef]
- Marrano, A.; Britton, M.; Zaini, P.A.; Zimin, A.V.; Workman, R.E.; Puiu, D.; Bianco, L.; Di Pierro, E.A.; Allen, B.J.; Chakraborty, S.; et al. High-quality chromosome-scale assembly of the walnut (Juglans regia L.) reference genome. Gigascience 2020, 9, giaa050. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, W.; Ji, F.; Qiu, J.; Song, X.; Bu, D.; Pan, G.; Ma, Q.; Chen, J.; Huang, R.; et al. A high-quality walnut genome assembly reveals extensive gene expression divergences after whole-genome duplication. Plant Biotechnol. J. 2020, 18, 1848–1850. [Google Scholar] [CrossRef]
- Zhu, T.; Wang, L.; You, F.M.; Rodriguez, J.C.; Deal, K.R.; Chen, L.; Li, J.; Chakraborty, S.; Balan, B.; Jiang, C.Z.; et al. Sequencing a Juglans regia × J. microcarpa hybrid yields high-quality genome assemblies of parental species. Hortic. Res. 2019, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Mo, R.; Chen, W.; Zhang, Q.; Sheng, F.; Wu, C.; Zhang, R.; Luo, Z. Identification and comparative analysis of genes and microRNAs involved in the floral transition of the Xinjiang early-flowering walnut (Juglans regia L.). Horticulturae 2022, 8, 136. [Google Scholar] [CrossRef]
- Wang, W.; Wen, H.; Jin, Q.; Yu, W.; Li, G.; Wu, M.; Bai, H.; Shen, L.; Wu, C. Comparative transcriptome analysis on candidate genes involved in lipid biosynthesis of developing kernels for three walnut cultivars in Xinjiang. Food Sci. Hum. Wellness 2022, 11, 1201–1214. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, X.; Su, Y.; Chen, Y.; Liu, Y.; Sun, M.; Qi, G. Transcriptome analysis reveals dynamic fat accumulation in the walnut kernel. Int. J. Genom. 2018, 2018, 8931651. [Google Scholar] [CrossRef]
- Gou, Y.; Cheng, S.; Ye, J.; Zhou, X.; Xu, F.; Chen, Z.; Zhang, W.; Liao, Y. Comparative transcriptome analysis reveals the potential molecular mechanism involved in fatty acids biosynthesis of Juglans regia. Sci. Hortic. 2020, 269, 109388. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. AgriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Zhang, P.; Li, X.; Wang, Y.; Guo, W.; Chachar, S.; Riaz, A.; Geng, Y.; Gu, X.; Yang, L. PRMT6 physically associates with nuclear factor Y to regulate photoperiodic flowering in Arabidopsis. aBIOTECH 2021, 2, 403–414. [Google Scholar] [CrossRef]
- Zhu, Z.; Wang, H.; Wang, Y.; Guan, S.; Wang, F.; Tang, J.; Zhang, R.; Xie, L.; Lu, Y. Characterization of the cis elements in the proximal promoter regions of the anthocyanin pathway genes reveals a common regulatory logic that governs pathway regulation. J. Exp. Bot. 2015, 66, 3775–3789. [Google Scholar] [CrossRef]
- Jaakola, L. New insights into the regulation of anthocyanin biosynthesis in fruits. Trends Plant Sci. 2013, 18, 477–483. [Google Scholar] [CrossRef]
- Chen, W.; Zheng, Q.; Li, J.; Liu, Y.; Xu, L.; Zhang, Q.; Luo, Z. DkMYB14 is a bifunctional transcription factor that regulates the accumulation of proanthocyanidin in persimmon fruit. Plant J. 2021, 106, 1708–1727. [Google Scholar] [CrossRef]
- Shaikh, A.A.; Chachar, S.; Chachar, M.; Ahmed, N.; Guan, C.; Zhang, P. Recent advances in DNA methylation and their potential breeding applications in plants. Horticulturae 2022, 8, 562. [Google Scholar] [CrossRef]
- Chachar, S.; Chachar, M.; Riaz, A.; Shaikh, A.A.; Li, X.; Li, X.; Guan, C.; Zhang, P. Epigenetic modification for horticultural plant improvement comes of age. Sci. Hortic. 2022, 292, 110633. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total_Reads n | Clean_Reads n | Total_Mapped_Reads n | Mapped_to_Gene n | Clean_Reads % | Total_Mapped Reads % | Mapped_to_Gene % | Q20 (%) | Q30 (%) |
|---|---|---|---|---|---|---|---|---|---|
| 8 W-rep1 | 42,511,740 | 39,159,426 | 37,524,508 | 31,708,623 | 92.11 | 95.82 | 86.31 | 97.79 | 93.94 |
| 8 W-rep2 | 40,938,144 | 37,770,668 | 36,109,590 | 30,471,198 | 92.26 | 95.60 | 86.23 | 97.62 | 93.58 |
| 8 W-rep3 | 46,185,768 | 42,363,092 | 40,505,002 | 34,203,893 | 91.72 | 95.61 | 86.29 | 97.85 | 94.14 |
| 9 W-rep1 | 51,196,102 | 47,305,832 | 45,316,194 | 38,411,566 | 92.40 | 95.79 | 86.61 | 97.74 | 93.84 |
| 9 W-rep2 | 46,607,498 | 43,072,262 | 41,186,929 | 34,938,139 | 92.41 | 95.62 | 86.67 | 97.47 | 93.21 |
| 9 W-rep3 | 44,124,428 | 40,700,108 | 39,029,827 | 33,085,504 | 92.23 | 95.90 | 86.63 | 97.82 | 94.02 |
| 11 W-rep1 | 41,750,830 | 38,408,438 | 36,772,379 | 30,863,355 | 91.99 | 95.74 | 85.98 | 97.57 | 93.47 |
| 11 W-rep2 | 42,265,150 | 38,826,206 | 37,207,857 | 31,305,853 | 91.86 | 95.83 | 86.20 | 97.75 | 93.87 |
| 11 W-rep3 | 42,840,582 | 39,402,138 | 37,728,901 | 31,571,366 | 91.97 | 95.75 | 85.80 | 97.77 | 93.90 |
| 13 W-rep1 | 43,713,166 | 40,264,612 | 38,592,256 | 32,383,080 | 92.11 | 95.85 | 86.03 | 97.81 | 93.96 |
| 13 W-rep2 | 41,997,990 | 38,622,044 | 37,020,229 | 31,078,115 | 91.96 | 95.85 | 86.09 | 97.72 | 93.77 |
| 13 W-rep3 | 43,851,814 | 40,468,318 | 38,805,463 | 32,511,695 | 92.28 | 95.89 | 85.82 | 97.77 | 93.88 |
| 15 W-rep1 | 40,476,880 | 37,368,500 | 35,870,261 | 29,940,084 | 92.32 | 95.99 | 85.74 | 97.82 | 93.98 |
| 15 W-rep2 | 43,758,020 | 40,116,420 | 38,471,384 | 32,069,305 | 91.67 | 95.90 | 85.60 | 97.77 | 93.96 |
| 15 W-rep3 | 41,861,892 | 38,498,628 | 36,864,470 | 30,703,181 | 91.96 | 95.76 | 85.60 | 97.53 | 93.40 |
| 17 W-rep1 | 40,003,876 | 36,753,148 | 35,241,476 | 28,758,694 | 91.87 | 95.89 | 83.77 | 97.68 | 93.71 |
| 17 W-rep2 | 38,970,490 | 35,921,270 | 34,387,512 | 28,016,518 | 92.17 | 95.73 | 83.62 | 97.54 | 93.45 |
| 17 W-rep3 | 43,878,416 | 40,368,632 | 38,648,771 | 31,730,672 | 92.00 | 95.74 | 84.21 | 97.63 | 93.61 |
| 19 W-rep1 | 40,618,986 | 37,310,474 | 35,784,315 | 29,355,853 | 91.85 | 95.91 | 84.38 | 97.74 | 93.87 |
| 19 W-rep2 | 42,697,630 | 39,273,190 | 37,627,973 | 30,838,835 | 91.97 | 95.81 | 84.29 | 97.65 | 93.67 |
| 19 W-rep3 | 43,254,244 | 39,778,674 | 38,088,212 | 31,127,183 | 91.96 | 95.75 | 84.11 | 97.62 | 93.59 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Q.; Gao, S.; Mo, R.; Sheng, F.; Zhang, Q.; Wu, C.; Zhang, R.; Luo, Z. A Preliminary Study for Identifying Genes Associated with Pellicle Development in Xinjiang Walnut (Juglans regia L.). Horticulturae 2022, 8, 784. https://doi.org/10.3390/horticulturae8090784
Jin Q, Gao S, Mo R, Sheng F, Zhang Q, Wu C, Zhang R, Luo Z. A Preliminary Study for Identifying Genes Associated with Pellicle Development in Xinjiang Walnut (Juglans regia L.). Horticulturae. 2022; 8(9):784. https://doi.org/10.3390/horticulturae8090784
Chicago/Turabian StyleJin, Qiang, Shan Gao, Rongli Mo, Fang Sheng, Qinglin Zhang, Cuiyun Wu, Rui Zhang, and Zhengrong Luo. 2022. "A Preliminary Study for Identifying Genes Associated with Pellicle Development in Xinjiang Walnut (Juglans regia L.)" Horticulturae 8, no. 9: 784. https://doi.org/10.3390/horticulturae8090784
APA StyleJin, Q., Gao, S., Mo, R., Sheng, F., Zhang, Q., Wu, C., Zhang, R., & Luo, Z. (2022). A Preliminary Study for Identifying Genes Associated with Pellicle Development in Xinjiang Walnut (Juglans regia L.). Horticulturae, 8(9), 784. https://doi.org/10.3390/horticulturae8090784