1. Introduction
The enormous accumulation of non-biodegradable plastics in landfills and in the environment is one of the main problems of our century. Moreover, the depletion of fossil fuels has led to the need for new alternative bio-derived, sustainable polymers. Polyhydroxyalkanoates (PHAs) are fully biodegradable and biobased polyesters [
1] which can be considered a valuable alternative to conventional, non-biodegradable, fossil-based polyesters. PHAs are biosynthesized by a variety of bacterial genera [
2] in the form of intracellular granules as carbon and energy reserves under stress-feeding conditions, such as a carbon availability surplus or lack of macronutrients (phosphorous, nitrogen, and oxygen) [
3,
4]. In terms of chemical structure, PHAs are polyesters of hydroxy-alkanoic acids, which can be divided into short chain length (SCL; C3–C5), medium chain length (MCL; ≥C6) SCL-MCL copolymers, and long chain length LCL, depending on the number of carbon atoms per monomer [
1].
PHAs’ chemical structure and physicochemical properties mainly depend on the bacterial strain from which they are synthetized, the biosynthesis conditions, and the origin of the carbon source employed as a fermentation substrate [
5]. In fact, in accordance with specific growth conditions and the PHA producer strain used, PHAs can be both homopolymers and copolymers with different mechanical properties [
6]. Due to their biodegradability and biocompatibility, PHAs can be used in a wide range of applications such as packaging, agriculture (e.g., mulching and capsules for controlled pesticides release) [
7,
8], medicine, biomedical, drug encapsulation, and tissue engineering [
9,
10]. Nevertheless, the industrial large-scale production of PHAs is severely limited by the high production costs [
11]. In detail, their price is about six times higher than petrol-based plastics (1.18–6.12 vs. <1 €/Kg) [
12]; therefore, their market is reserved only for niche applications with high added value, such as biodegradable scaffolds for biomedical applications. Industrially, PHAs are mainly produced from single microbial cultures, using pure enzymatically derived sugars as a substrate. The cost of PHA production is mainly due to the sterilization of the production plant, the substrate, and the extraction of the polymer from cellular biomass [
13]. The cost of the substrate, consisting for example of glucose, fructose, or xylose, contributes to about 45% of the total cost of the process [
14], while the downstream processes account for about 50% [
15].
One of the strategies to lower substrate costs is to use organic waste raw materials such as industrial waste streams (e.g., sugarcane molasses from the sugar industry, cheese and milk whey from the dairy industry, effluents from olive, palm oil, and paper mills [
16]), the organic fraction of municipal solid waste (OFMSW) [
17,
18,
19], wastewater treatment-derived sludges [
2], volatile fatty acids (VFAs) from sludge [
20] and yeast industry wastewaters [
21], crude glycerol (a by-product of biodiesel production) [
22,
23]. Lignocellulosic biomass (LCB) has also been exploited as a low-cost substrate. Being a non-edible plant or plant-based material, they do not threaten the food availability for the growing world population. Examples of residual LCB used for PHA production are sugarcane bagasse [
24,
25], straw [
26,
27,
28], agro-industrial residues such as corncob [
29], corn stover [
30,
31], and peels [
32].
To obtain fermentable sugars from the cellulose and hemicellulose fractions of LCB, appropriate pretreatments must be performed to break the intramolecular bonds with the lignin structure (recalcitrant to further biocatalytic processes) and make the cellulose and hemicellulose more accessible for the enzymatic or chemical hydrolysis. Chemical alkaline or acid pretreatments are the most commonly used strategies to promote the degradation of the lignocellulosic matrix. However, they usually determine the release of phenolic compounds and furfural that act as inhibitors [
33]. To avoid the need for detoxification processes, milder conditions or cavitational treatments [
34] could replace the chemical ones to avoid the need for detoxification processes to remove inhibitors. Only a few biomasses such as oil palm trunk sap and mango peels were used as substrates without the need for pretreatments [
35,
36].
Enzymatic saccharification [
37,
38,
39] has been the most exploited process to enhance the production of reducing sugars from LCB [
40]. Alternatively, monosaccharides from pretreated biomasses can be recovered after chemical acid hydrolysis [
41,
42]. However, to avoid the acid catalyzed degradation of cellulose and hemicellulose into furan inhibitors (5-hydroxymethylfurfural and furfural), it is recommended to lower the operating temperatures or the reaction times. In this context, the use of alternative energy sources such as microwaves (MW), which transform electromagnetic energy into thermal energy, enable shorter conversion times than those provided by conventional heating. Furthermore, the effect of MW irradiation, in the case of lignocellulosic biomass, leads to a physical disruption of the internal composition of plant cells, thus enhancing the accessibility of biomass components to chemical treatments [
43].
Another strategy to lower the cost of PHA production relies on the use of mixed microbial cultures (MMC), open biological systems that do not require the sterilization of the productive plant. Furthermore, MMC can facilitate the fermentation of complex substrates such as LCB due to their robustness (i.e., the ability to preserve metabolic functions in the presence of internal or external perturbations) and community stability (i.e., the ability to create alternative metabolic pathways leading to the biosynthesis of the same product and excluding undesirable microorganisms within the consortium) [
44,
45]. Therefore, the use of MMC as biocatalysts for PHA production is gaining increasing attention at both the academic and industrial levels [
46]. In addition, the combined use of MMC and low-value feedstocks is currently under investigation in order to decrease operating costs. With this aim, hydrolysates from rubber wood waste [
47,
48] and poplar wood waste [
49] were used as carbon sources in combination with MMC to produce PHA.
This work describes the conversion of agricultural residues into monosaccharides with a negligible saline content with an MW-assisted acid hydrolysis and the intensification of the neutralization and desalination processes. The monosaccharides thus obtained were then used in combination with MMC recovered from a dairy waste active sludge to produce PHAs.
2. Materials and Methods
2.1. Residual Lignocellulosic Biomass
Corn straw (CS), a finely milled mixture of cuttings, leaves, and branches that remain at the end of the corn harvest period, was employed as a raw material. The CS moisture was calculated as the weight loss after overnight heating at 100 °C, while the organic content was determined as the weight loss after calcination of the sample at 650 °C for 4 h. The chemical characterization of CS was carried out using an analytical and standard process based on a Natural Renewable Energy Laboratory (NREL) procedure reported by Genevini et al. [
50], with some modifications. The results are listed in
Table 1.
2.2. MW-Assisted Acid Hydrolysis
The biomass conversion was carried out in a multimode MW reactor (SynthWave, Milestone Srl, Bergamo, Italy) designed to perform single or multiple reactions at temperatures up to 300 °C and a gas pressure up to 199 bars. Acid hydrolysis reactions were performed for 2 min at different combinations of HCl concentrations and temperatures with a N
2 pressure of 40 bar and a 1:10
w/
v solid/liquid ratio. The crude reaction was filtered under a vacuum, and the residual solid fraction was washed with deionized water. The collected liquid fractions were then freeze-dried. Biomass conversion and product yields were determined according to Equations (1) and (2), respectively:
where the sugar fraction accounts for the amount of cellulose and hemicellulose determined by NREL.
2.3. Concentration and Desalination with Membrane System
Concentration and desalination experiments of the liquid crude reaction were performed with a Hari PB-100 system (Hydro Air Research, Merlino (LO), Italy), equipped with a polyamide nanofiltration membrane (DKU-1812; 150–300 Da cut-off and 0.38 m2 filtering area). The liquid fractions recovered after filtrations (2.1 L) were collected and treated with KOH to pH 4.5 and then nano-filtered under a pressure of 5 bar. After 6 min of nanofiltration, the retentate volume was 1.2 L. To further reduce the saline concentration, an additional diafiltration step was conducted. For this purpose, 0.9 L of deionized water was added to the retentate obtained at the end of the nanofiltration. After 5 min of diafiltration, 1.2 L of retentate was collected.
2.4. Neutralization and Desalination with Ion-Exchange Resins
The liquid fraction recovered after filtration was firstly treated with the strong acid ion-exchange resin Amberlite™IR120H (Thermo Scientific™, Milan, Italy), which was dimensioned considering its total exchange capacity (1.8 eq/L). Subsequently, the eluate from the acid resin was treated with the weak basic ion-exchange resin Amberlite™IRA96 (total exchange capacity 1.25 eq/L). Both resin treatments were performed in a 10 L tank equipped with a vertical mixer (Stirrer LS, Velp Scientifica, Usmate (MB), Italy) using a liquid/solid ratio of 15:1 for the optimized protocol. The contact time was 5 min for the treatment with Amberlite™IR120H and 30 min for the Amberlite™IRA96. At the end of the whole process, the volume of the monosaccharide solution was reduced from 4 to 1 L using a 50 L rotary evaporator (Heidolph Instruments GmbH & Co. KG, D-91126 Schwabach ) at 0.09 MPa for the subsequent freeze-drying step conducted with a Lyotest (Telstar) freeze dryer at a temperature of −70 °C and a pressure of 0.55 bar.
2.5. Chemical Characterization of Monosaccharides Mixture
Qualitative characterization of monosaccharides was performed by GC-MS analysis with the Agilent 6890 (Agilent Technologies—Santa Clara, CA, USA) gas chromatograph equipped with an HP-5 capillary column (length: 30 m; internal diameter: 0.25 mm; film thickness: 0.25 μm) and connected to an Agilent Network 5973 mass detector. The sample was previously derivatized with BSTFA (N,O-Bis(trimethylsilyl)trifluoroacetamide) in the presence of 1% TMCS (trimethylchlorosilane). Next, 10 mg of the sample was completely dissolved into 1 mL of pyridine and 150 µL of BSTFA, stirred at 60 °C for 45 min, filtered with a 0.45 µm PTFE filter and then injected into the gas chromatograph [
51].
2.6. Enrichment of PHA-Producer Bacterial Strains
MMC inoculum was enriched from an activated sludge plant in a synthetic medium containing acetic acid (20 g/L) as carbon source (
Table S1) [
52]. The enrichment step was conducted in 500 mL Pyrex glass Erlenmeyer flasks equipped with baffles. The culture volume was 200 mL, of which 10%
v/
v was of standardized inoculum (active sludge) and 90%
v/
v was of Khardenavis fermentation medium. The flasks were incubated in a thermostatic incubator (Thermo Scientific MaxQ 6000, Thermo Scientific™, Milan, Italy) at a temperature of 30 °C and the agitation was ensured by a rotary shaker at 120 rpm. Optical density at 620 nm (OD
620) was used as a rapid method to monitor the cellular biomass growth and was measured with Perkin Elmer Lambda 465 spectrophotometer (PerkinElmer, Inc. Waltham, MA, USA), while the pH of the culture media was measured with WTW inoLab pH730 pH meter.
2.7. PHA Production
PHA was produced by batch fermentation using modified Khardenavis fermentation media. In detail, the acetic acid used as a carbon source for the enrichment step has been replaced by the biomass-derived monosaccharides at the same concentration (20 g/L). For flask fermentation, 10 flasks with a total volume of 200 mL (10% v/v of inoculum) were incubated in the thermostatic incubator at a temperature of 30°C, under agitation at 120 rpm, for a maximum time of 192 h. The inoculum collected from the enrichment step was properly diluted to have an optical density (OD620) value between 0.8 and 1 (standardized inoculum). The ammonium concentration was evaluated using the Boehringer Mannheim/R-Biopharm Enzymatic BioAnalysis/Food Analysis enzymatic kit (R-Biopharm AG, Darmstadt, Germany). The amount of sugars was monitored through the measurements of Brix degrees with a PAL-1 portable refractometer (Atago, Tokyo, Japan).
The PHA yield (Y
PHA/X) was calculated at different fermentation times (t), as the ratio of PHA dry weight (g) and biomass dry weight (g) (Equation (3)).
The productivity of PHA was calculated at different fermentation times (t) according to Equation (4), for the same sampling volume of 200 mL.
For the scale-up of the PHA production, the fermentation was performed in the R’ALF® (Bioengineering, Wald, Switzerland) fermenter characterized by a total volume of 6.7 L, equipped with an external jacket for the temperature control and a shaft on which two radial “Rushton” turbines with six blades were mounted. In addition, the presence of four baffles makes stirring more efficient. The protocol applied for the bioreactor fermentation was the same as described for flask fermentation, except for the total fermentation media volume (3 L), stirring speed (125 rpm), and maximum time (96 h). After 96 h, the residual fermentation media was transferred to two Erlenmeyer flasks equipped with baffles (200 mL for each flask). The pH of the fermentation media was reported to the initial value by adding NaOH (10 M) when necessary. Then, both flasks were placed for 72 h in a thermostatic incubator at 30 °C and stirred at 120 rpm. At the end of PHA production, the cellular biomass was recovered by centrifugation of the fermentation medium at 4100 rpm for 10 min. The pellet was washed twice with distilled water. The supernatant of the second wash was then recovered and centrifuged at 12,000 rpm for 5 min. The total pelleted biomass was dried in a laboratory oven at a temperature of 60 °C for about 72 h and its quantity was determined by gravimetric means.
2.8. PHA Extraction
The polymer was extracted from the dried cellular biomass (dried at 60°C for 72 h) using a 1:1 v/v solution of NaClO and CHCl3. The extraction was conducted in a 50 mL falcon tube, adding CHCl3 (10 mL) and NaClO (10 mL) to the microbial biomass. The solution was kept under vigorous stirring for 90 min. A centrifugation step at 4000 rpm for 10 min allowed the separation of the organic phase from the aqueous one. The organic phase was recovered and transferred inside a calibrated falcon tube. A second extraction was carried out by adding CHCl3 (10 mL) and NaClO (10 mL) and stirring for 30 min. The solvent was evaporated from the collected organic fractions and the recovered PHA was weighed and characterized.
2.9. Characterization of PHA
For the NMR analysis, 600 µL of CDCl3 was added to the samples, which was further filtered with a nylon syringe filter (Whatman® (0.45 μm)-Merck KGaA, Darmstadt, Germany). The sample was analyzed by 1H-NMR, with a Jeol JNM-ECZ600R spectrometer (Jeol, Tokyo, Japan) operating at a frequency of 600 MHz. FTIR analyses of PHAs were carried out with the Perkin Elmer BX FT-IR System spectrometer through the film casting method, dissolving PHA in CHCl3.
3. Results and Discussion
3.1. MW-Assisted Acid Hydrolysis
Acid hydrolysis is one of the most well-known methods to produce simple sugars. For this purpose, strong acids such as concentrated (10–30%) or diluted (2–5%) H
2SO
4 and HCl are commonly used [
53]. Generally, hydrolysis with concentrated acids requires more moderate temperatures and results in a high yield in glucose (90%) but involves corrosion and damage to the instruments, while hydrolysis with dilute acids requires high temperatures to achieve acceptable yields.
Based on an optimized protocol of a previous work [
54], the hydrolysis of CS was carried out in a multimode MW reactor designed to rapidly reach high temperatures, using HCl as an acid catalyst. A screening of HCl concentration and temperatures, with a fixed reaction time of 2 min and 3 g of biomass, was performed to select the best reaction conditions aiming to reduce the acid quantity maintaining reasonable biomass conversion and product yield. The screening data and results are listed in
Table 2.
Despite the highest overall conversion and yield being achieved at 200 °C with 0.5 M HCl (
Table 2, entry 2), levoglucosan was observed in the final product (
Figure S1), limiting monosaccharides selectivity. The presence of levoglucosan is due to high temperatures and a strong interaction of MW with cellulose through the activation of the CH
2OH pendant groups of the glucose monomeric units [
55]. Moreover, the GC-MS pattern showed a limited variety of monosaccharides, which could limit the MMC growth. The lowest biomass conversion was observed at 150 °C with HCl 0.1 M (entry 3); thus, no further yield and organic content evaluations were made, as the higher HCl concentration or higher temperature were necessary (entries 1 and 4, respectively,
Figures S2 and S3). As higher temperatures (200 °C) allowed us to reach a higher conversion with a lower amount of HCl (0.1 M), these were selected as reference conditions for further experiments. A scale-up was performed by hydrolyzing 30 g of CS obtaining the same biomass conversion (37.0%) and a slightly lower yield (50.1%).
3.2. Concentration and Desalination with Membrane System
To concentrate the filtered crude reaction obtained at the end of the acid hydrolysis step, a nanofiltration membrane system with a cut-off of 150–300 Da was used. The moderate acidity of the liquid fraction (pH = 3.5) allowed its direct concentration without damaging the membrane polymeric structure. After 6 min of nanofiltration with an applied pressure of 5 bars, the volume (2 L) was reduced by approximately 50%. The nanofiltration step was performed not only to concentrate the monosaccharide solution, but also as an alternative attempt for the desalination. For this purpose, an additional diafiltration step was conducted after nanofiltration using the same membrane system and adding deionized water (to reach the starting volume of 2 L). Unfortunately, only a slight decrease in the saline concentration was detected (1.2%). Therefore, no additional diafiltration steps were performed due to the low desalination efficiency observed.
3.3. Neutralization and Desalination with Ion-Exchange Resins
Neutralization and desalination of the crude substrates is a crucial preliminary step allowing the use of biomass-derived monosaccharides as a carbon source for the fermentation process without altering the pH and the saline formulation of the liquid fermentation media. For this purpose, a strong acid ion-exchange resin (Amberlite™
IR120H) was firstly deployed for the crude desalination while a subsequent treatment with a weak basic ion-exchange resin (Amberlite™
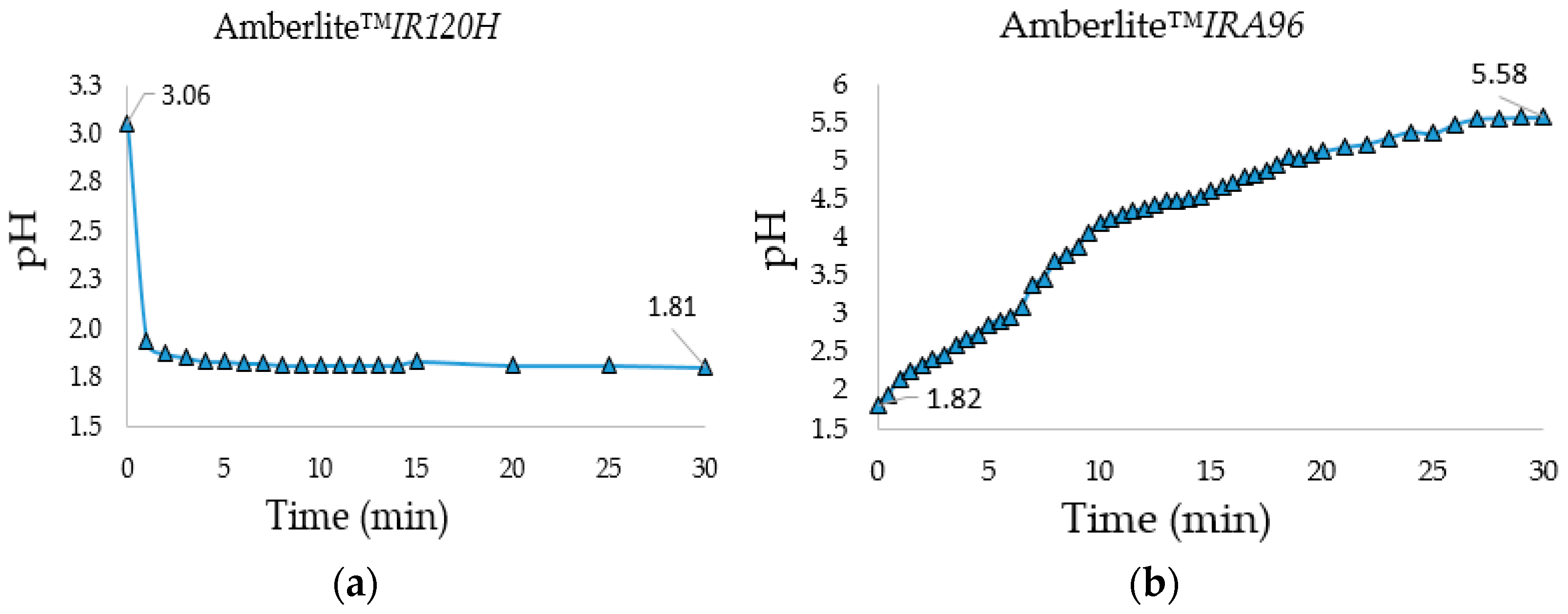
IRA96) allowed the neutralization of the solution. Both treatments were performed in a 10 L flask under stirring with the proper amount (bed volume) of resin calculated from the resins total exchange capacity and the amount of salts and HCl protons that had to be removed. Moreover, to establish the minimum treatment time with the two resins, the pH was monitored over time during desalination and neutralization steps (
Figure 1a,b).
During the desalination step, the pH became constant after only 5 min and its decrease indicated the effective exchange between the salts’ cations and resin protons. The neutralization process (to monosaccharides’ natural pH) was longer and required 30 min of stirring at 1000 rpm with a PTFE blade for stirrer shafts (blade diameter: 105 mm) to reach a pH = 5.58. The gravimetric analysis of the freeze-dried monosaccharides showed that the combined use of the two ion-exchange resins was efficient to neutralize the crude reaction and to reduce the starting saline content to a minimum value of 2.4% (
Table 3), an acceptable value to not alter excessively the saline formulation of the fermentation media, which allowed the concentration of the organic carbon source.
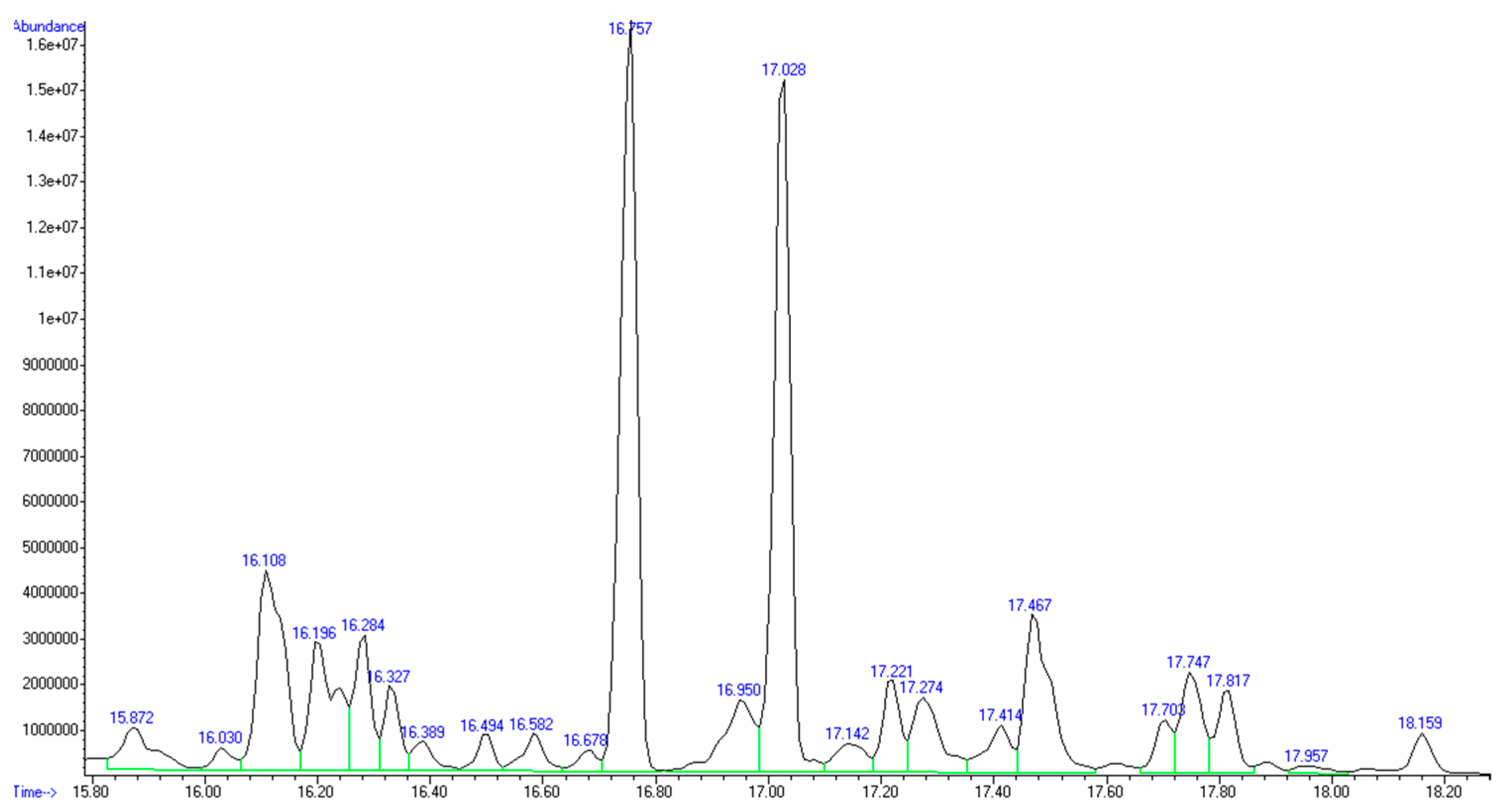
The GC-MS analysis confirmed the presence of mainly C5 and C6 monosaccharides inside the recovered product (
Figure 2 and
Table 4).
3.4. Enrichment of PHA-Producer Bacterial Strains
The presence of PHA-producing strains in MMC from activated sludge was confirmed in a previous work, leading to the satisfactory identification of two bacterial strains,
Citrobacter freundii and
Leuconostoc spp., whose ability to accumulate PHAs in synthetic media was confirmed [
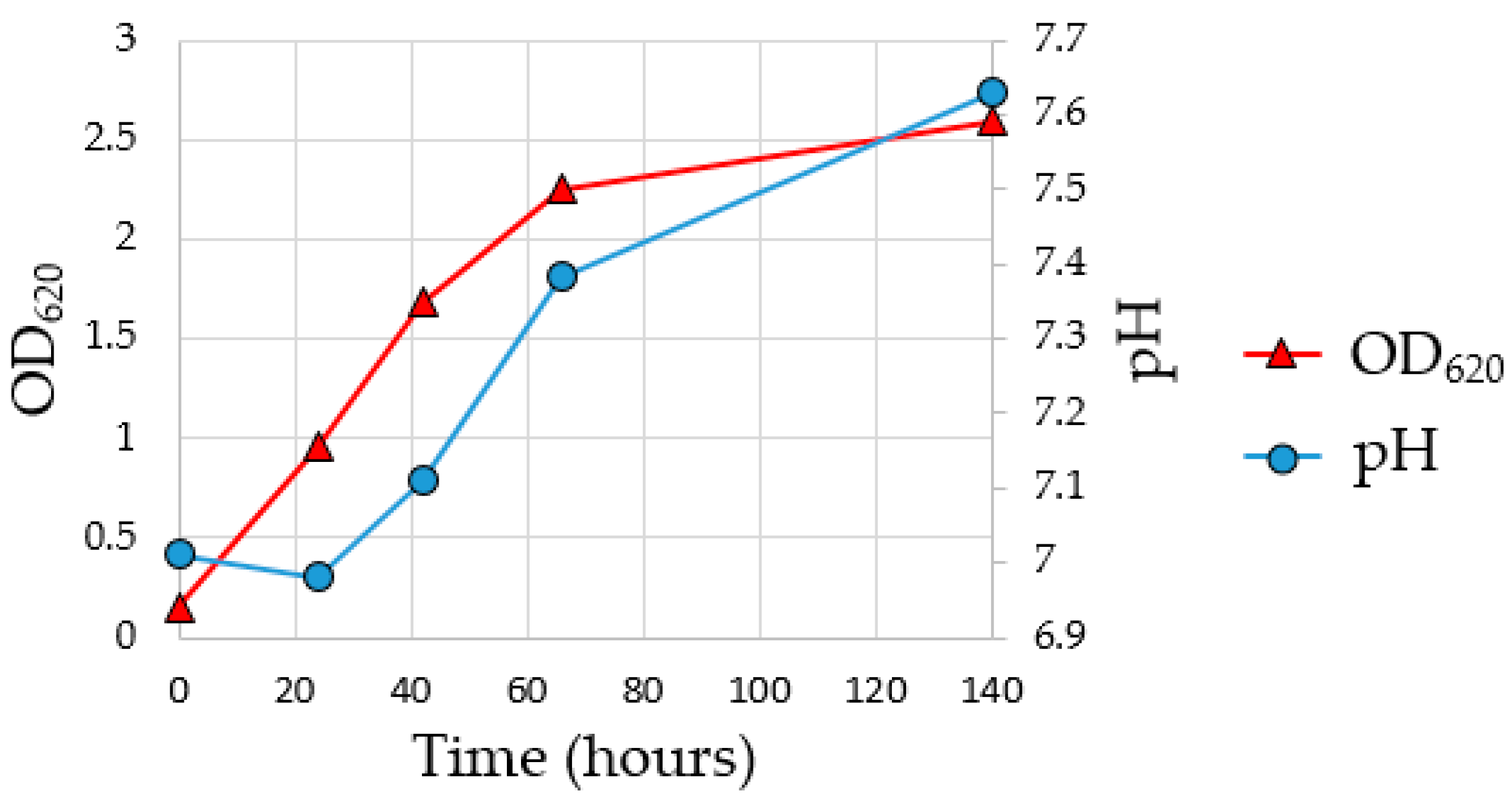
56]. To assess the growth of the MMC and the achievement of the exponential growth phase during the enrichment fermentation, a pre-culture test was carried out for 140 h. As indicated in
Figure 3, the pH of the fermentation media increased from 7.0 to 7.6 due to the consumption of acetic acid from MMC, while the OD
620 indicates that the stationary phase was reached at about 140 h of enrichment fermentation.
To collect the inoculum in the exponential growth phase for the subsequent flask fermentation, it was decided to conduct the subsequent enrichment fermentation for a total time of 69 h reaching an OD620 value of 2.01693.
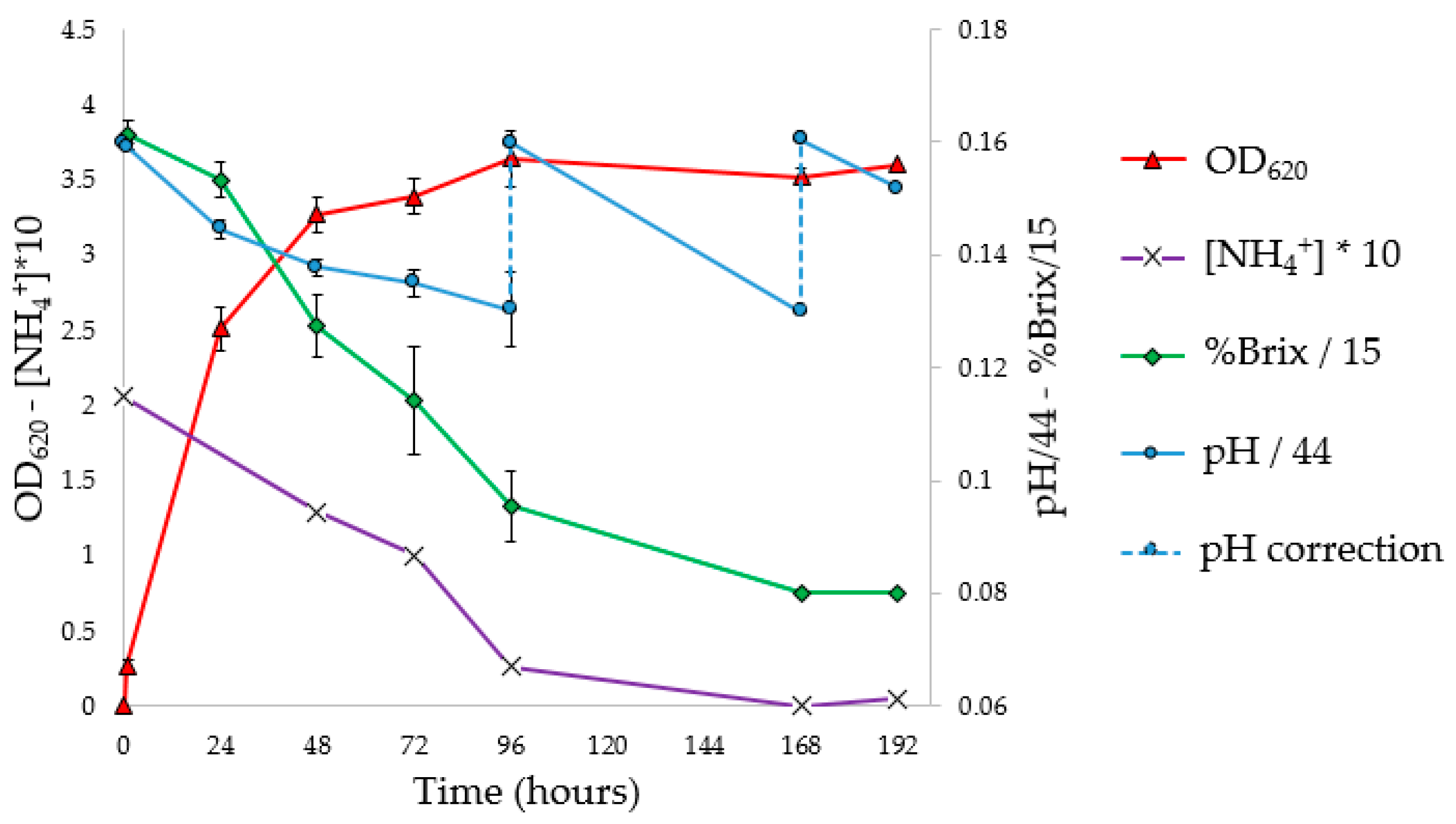
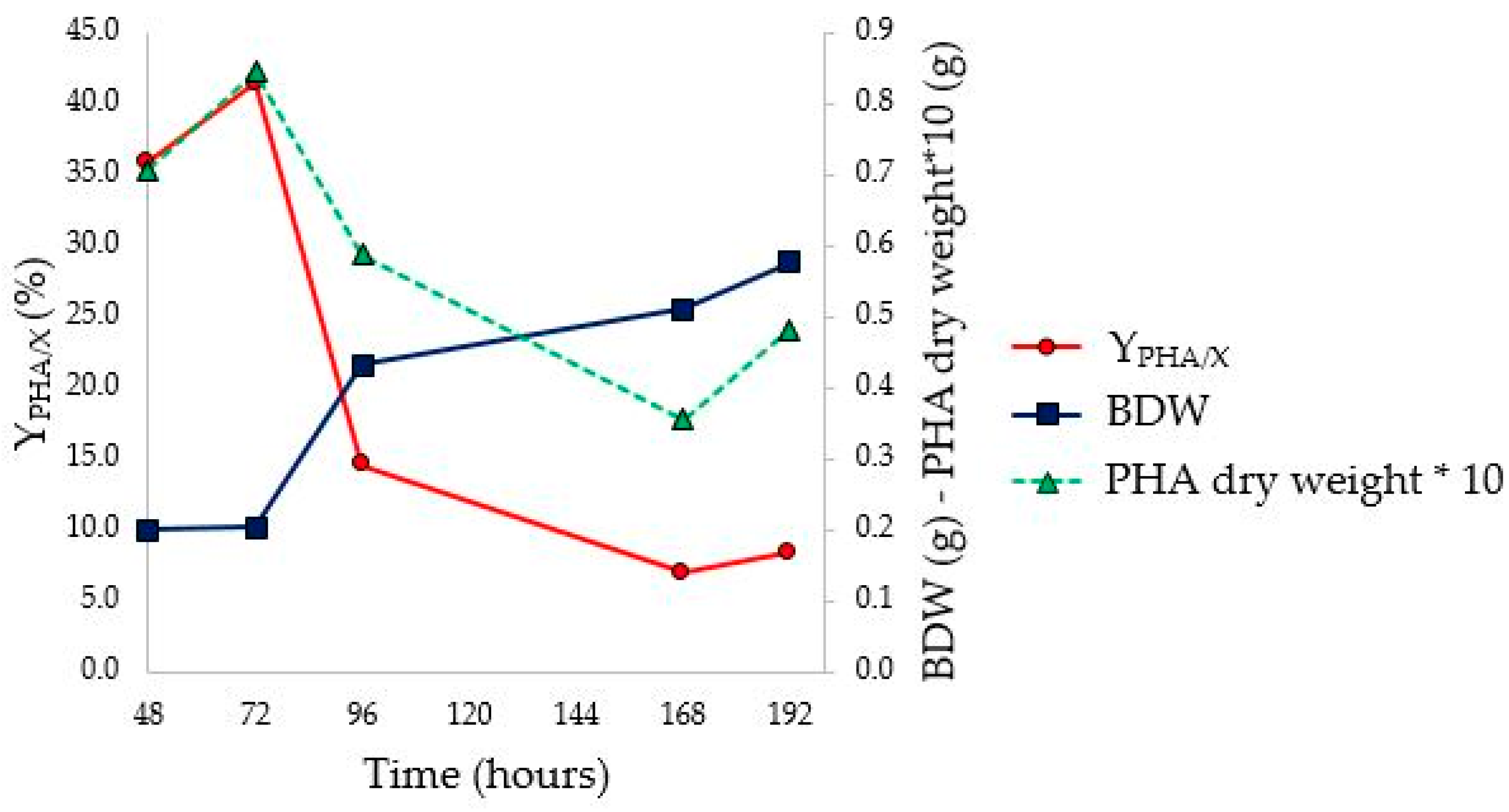
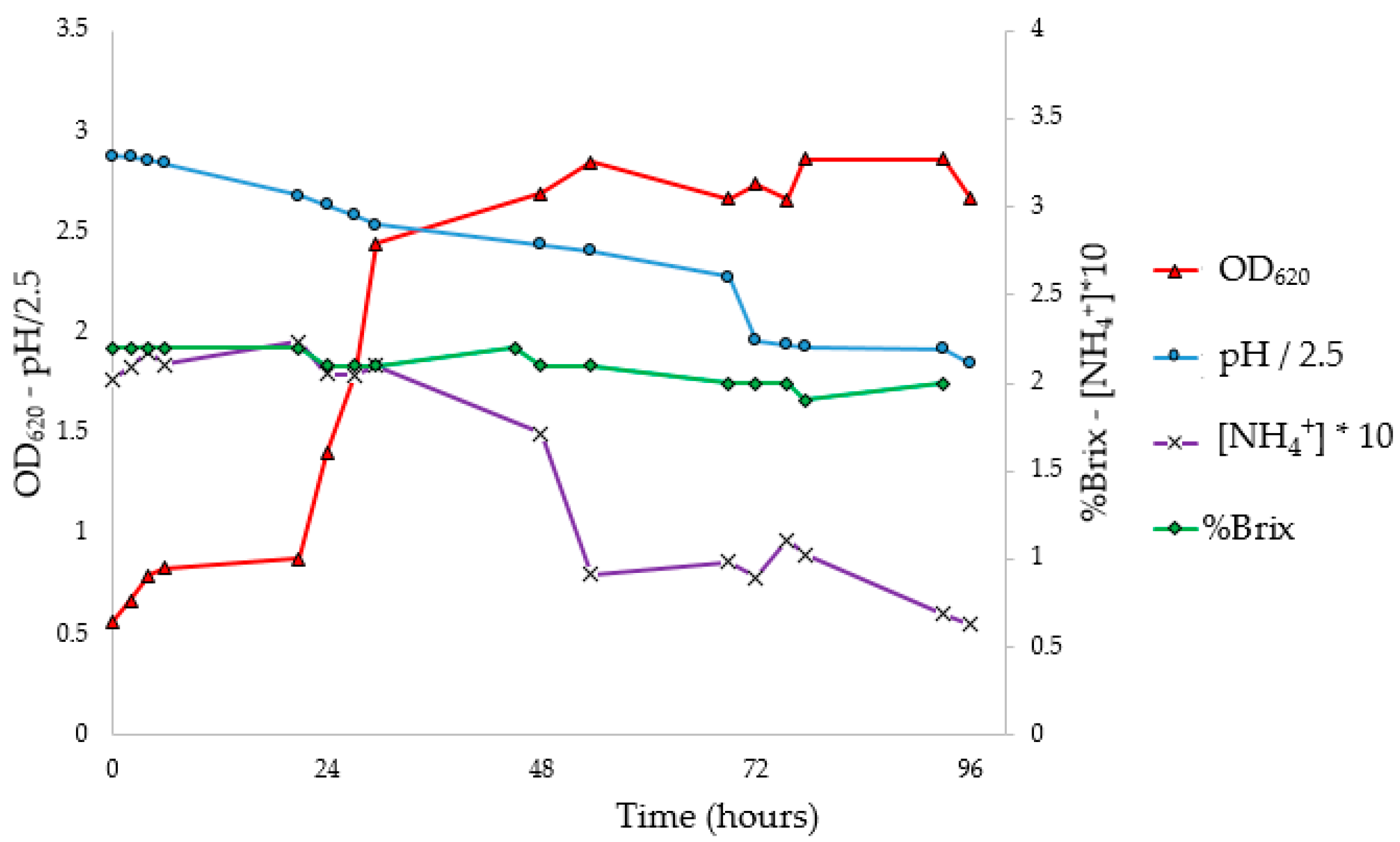
3.5. Flask PHA Production in Shaking Flasks
To investigate the progress of fermentation, the experiments were performed in 10 flasks, analyzing optical density, pH, NH
4+ concentration (g/L), Brix degrees (°Br) expressed as %Brix (
Figure 4), and Y
PHA/X (
Figure 5). To determine Y
PHA/X, it was necessary to separate the cellular biomass from the growth medium and then carry out the subsequent PHA extractions. Two flasks were therefore sacrificed every 24 h from the beginning of fermentation, starting after 48 h from the set-up of the fermentation line. Finally, for the last two remaining flasks, the fermentation was prolonged for a further 72 and 96 h, correcting the pH and sacrificing them, respectively, at a total fermentation time of 168 and 192 h. The recovery of the cellular biomass was conducted as indicated in
Section 2.7. Subsequently, PHAs were extracted with the conventional procedure described in
Section 2.8.
From the OD
620 curve of the flask fermentation (
Figure 4) and the evaluation of Y
PHA/X (
Figure 5 and
Table 5), it is possible to assess that the stationary phase of growth was reached between 72 and 96 h, with a maximum Y
PHA/X (41.4%) at 72 h. The pH corrections (96 and 168 h) had no impact on microbial growth. In fact, it was observed that the pH tends to return to the pre-correction values, without however causing an increase in the concentration of biomass or the Y
PHA/X; probably the pH does not have an inhibiting effect, at least under these conditions. At the stationary phase, constant values of ammonium concentration and Brix degrees were reached; moreover, a decrease in the weight of extracted PHA was observed. Indeed, with the decrease in the C/N ratio, concentrations of nutrients unfavorable for the accumulation of PHA were reached. Under these conditions, microorganisms, therefore, use intracellular granules as a source of carbon, resulting in a decrease in the concentration of polymer in the cytoplasm [
57].
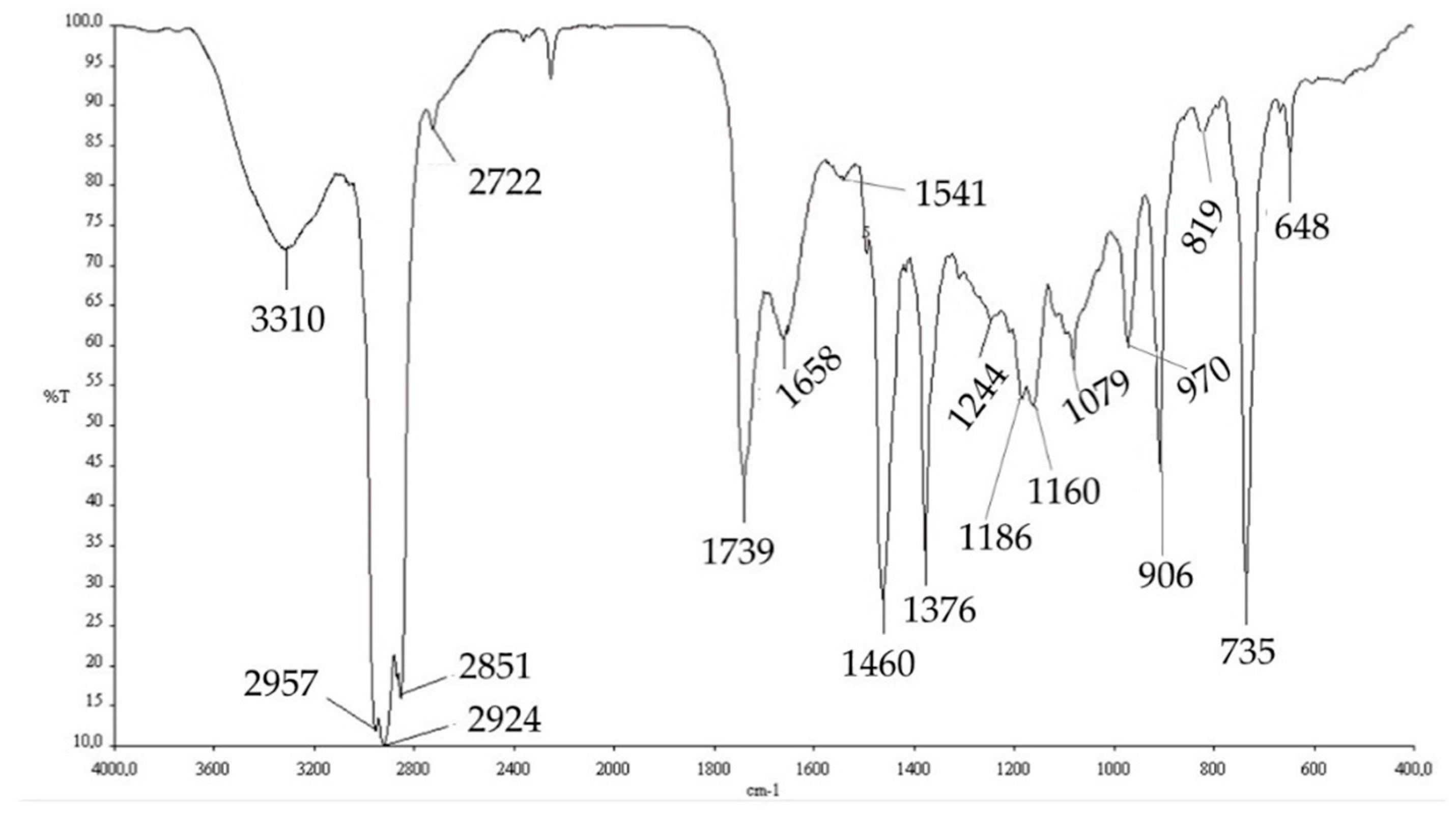
The extracted PHAs were then characterized by FT-IR analysis and NMR analysis. The comparison of the FT-IR spectra obtained experimentally (
Figure 6) with those reported in the literature, obtained from the strain
Pseudomonas putida [
58], and with a commercial standard, allowed us to hypothesize that the PHA produced by this mixed microbial culture is poly-(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV copolymer).
However, the extracted polymer contained some impurities, including, probably, cellular protein residues and DNA residues (1658.74, 1541.25, and 1244.75 cm
−1). Absorption band assignment is reported in
Table 6.
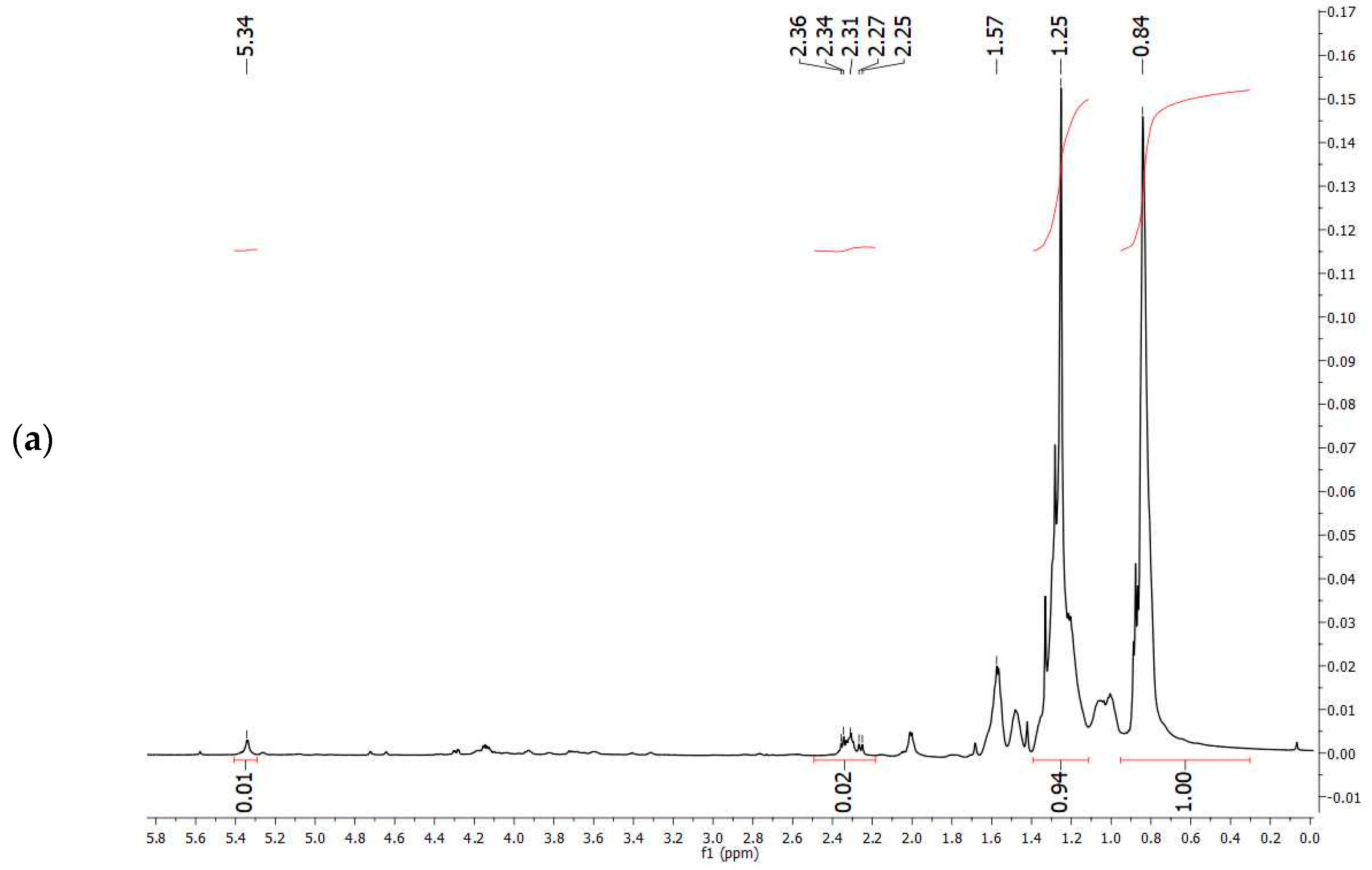
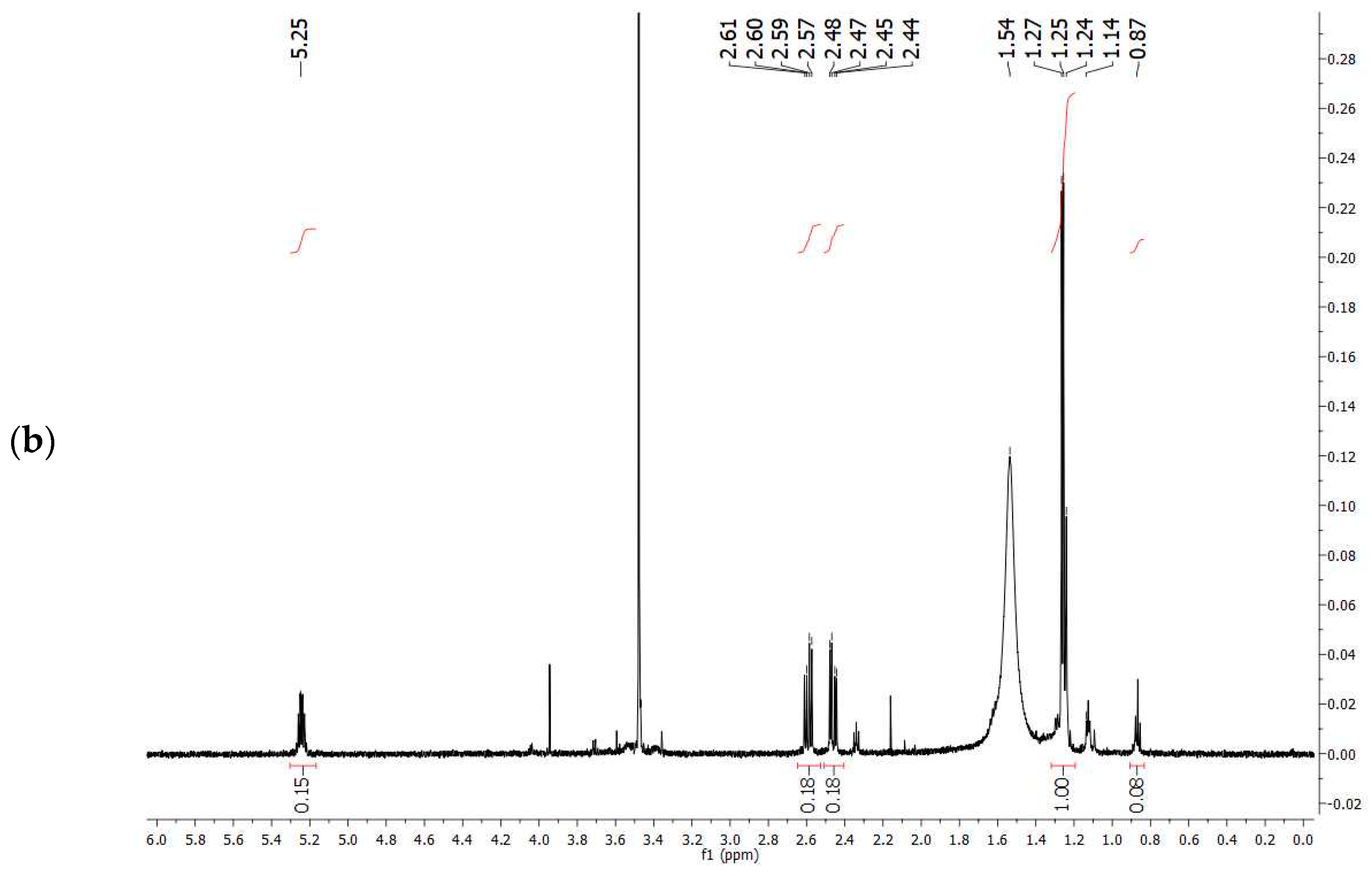
The comparison of
1H-NMR spectrum obtained experimentally (
Figure 7a) with that reported in the literature related to PHBV extracted from
Halomonas campisalis MCM B-1027 [
59], and with a commercial standard (
Figure 7b) supports the hypothesis that the PHA obtained from mixed microbial culture is PHBV with the presence of impurities. The composition of PHB and PHV in the copolymer was calculated from the area ratio of absorption peaks in methyl groups corresponding to the HB and HV groups (1.28 and 0.84 ppm, respectively) according to the method described by Kulkarni et al. [
59] (
Table 7). The molar percentage of the HB unit in the copolymer is 47% and the HV unit in the copolymer accounts for 53%.
3.6. PHA Production in Bioreactor Test
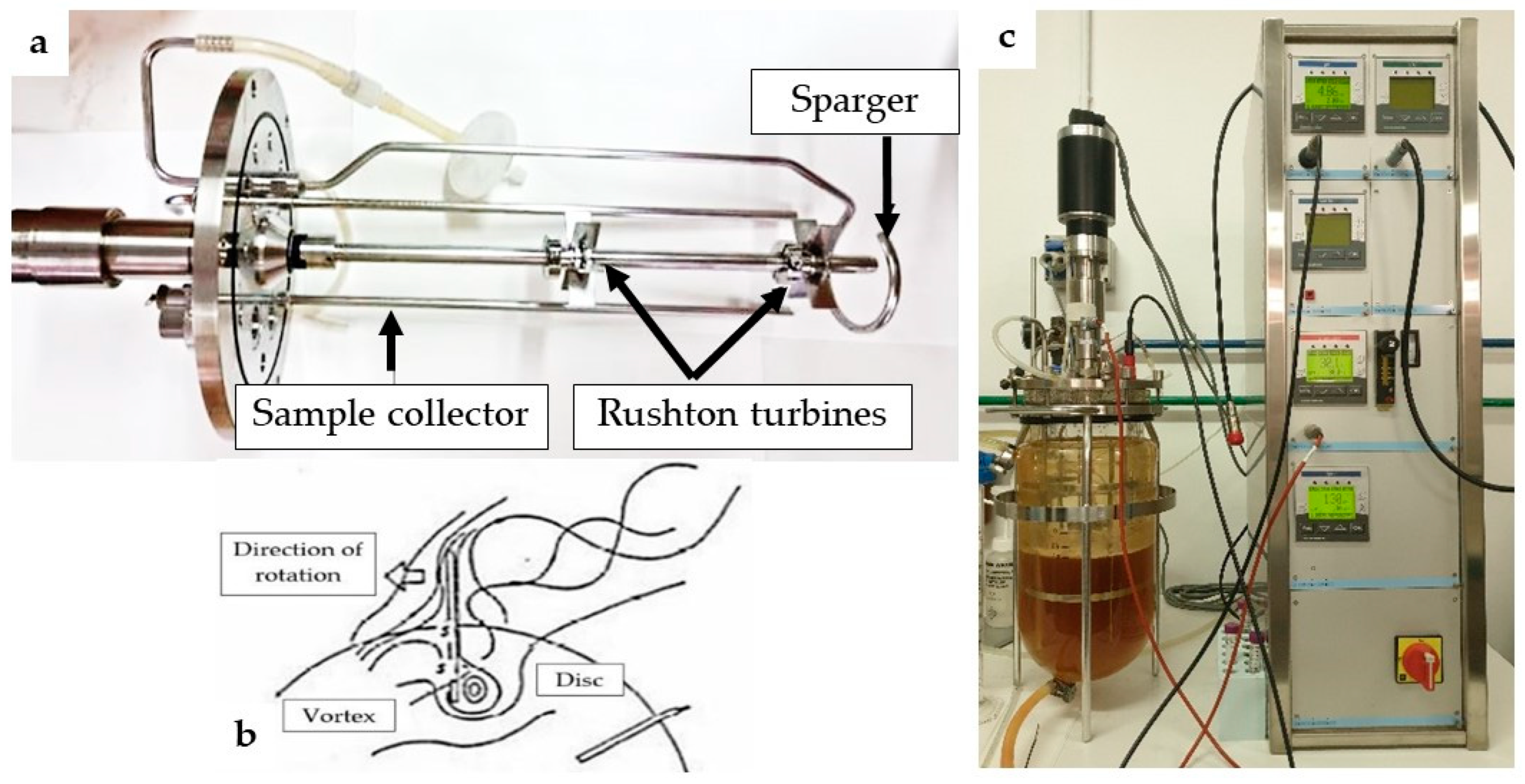
To scale-up the fermentation process, a stirred tank reactor equipped with two radial turbines of the “Rushton” type with six blades and the presence of four baffles (
Figure 8a) was used. This system is more efficient and ensures better turbulence and aeration, compared to the stirred flask, due to the formation of pairs of counter-rotating vortices, one above and the other under the impeller disc, on the back of each blade (
Figure 8b).
The fermentation was performed with 3 L of the medium inoculated following the same procedure described in the previous paragraph, at 30 °C, with an agitation speed of 125 rpm, for a total time of 68 h. For the monitoring of fermentation parameters, aliquots of samples (10 mL) were taken at regular intervals for the measurement of optical density, ammonium concentration, and Brix degrees, while a larger volume (200 mL) was taken every 24 h for PHA extraction.
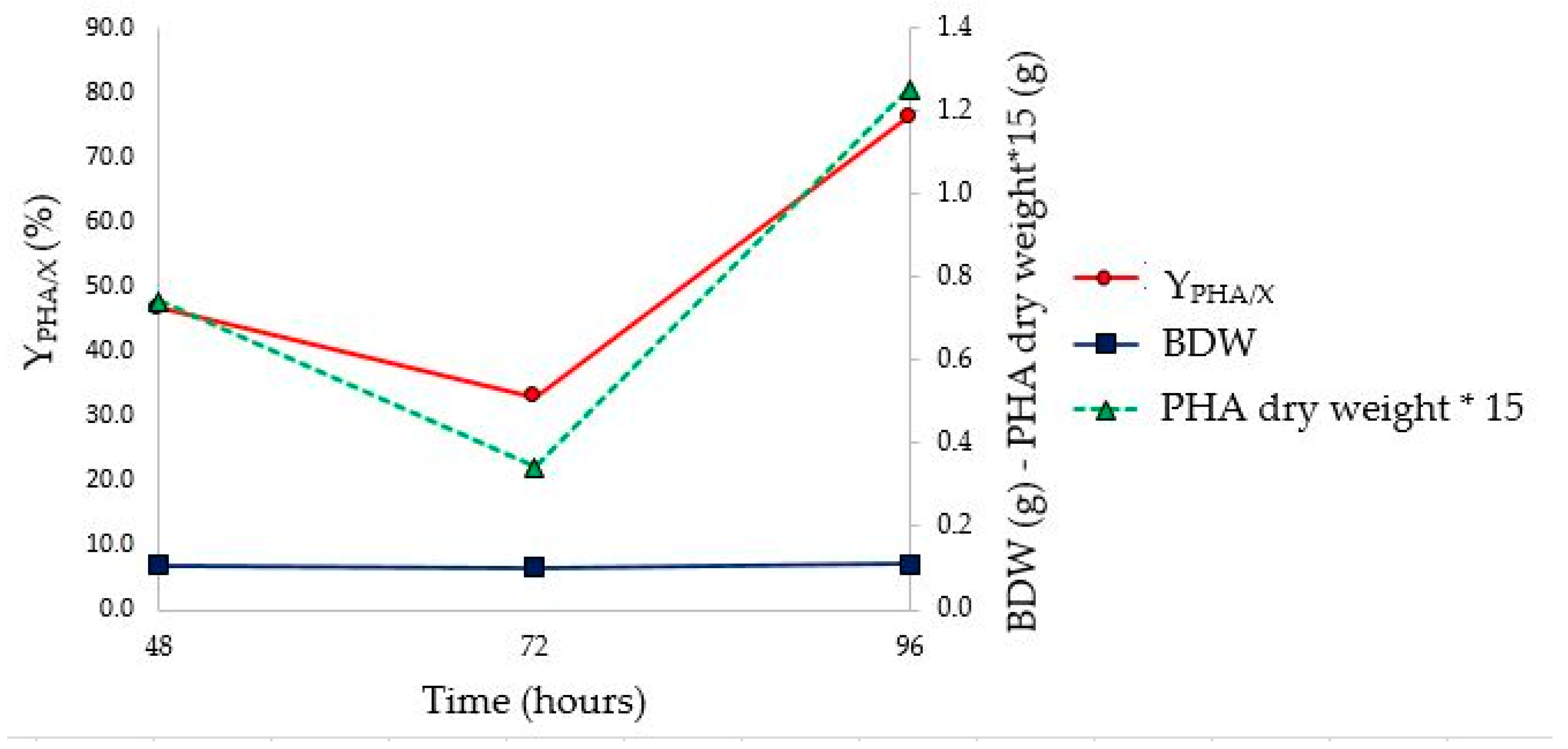
As shown in
Figure 9, the stationary phase of growth was reached between 48 and 72 h, earlier than what was observed in the tests in flask (between 72 and 96 h). However, the highest Y
PHA/X (76.3%) was observed at 96 h (
Figure 10 and
Table 8). Furthermore, no change in the value of Brix degrees was observed throughout fermentation. This phenomenon could be attributed to the fluid dynamics of the bioreactor, which would allow the continuous solubilization of solid monosaccharides in suspension, as well as influencing the pH on the solubility of the substrate. In fact, assuming that the driving force of the solubilization process is the same both in the flask and bioreactor, the different stirring efficiency could influence this parameter. A minimum pH value lower than that observed in flask fermentation (4.65 vs 5.75) was achieved. This, probably, could be ascribed to the fact that, due to the greater efficiency of agitation, a greater amount of monosaccharides was present in the culture medium, which were converted into organic acids by microorganisms. The characterization of PHA extracted at the end of the bioreactor fermentation confirms the presence of PHBV (FT-IR and
1H-NMR analysis results are reported in
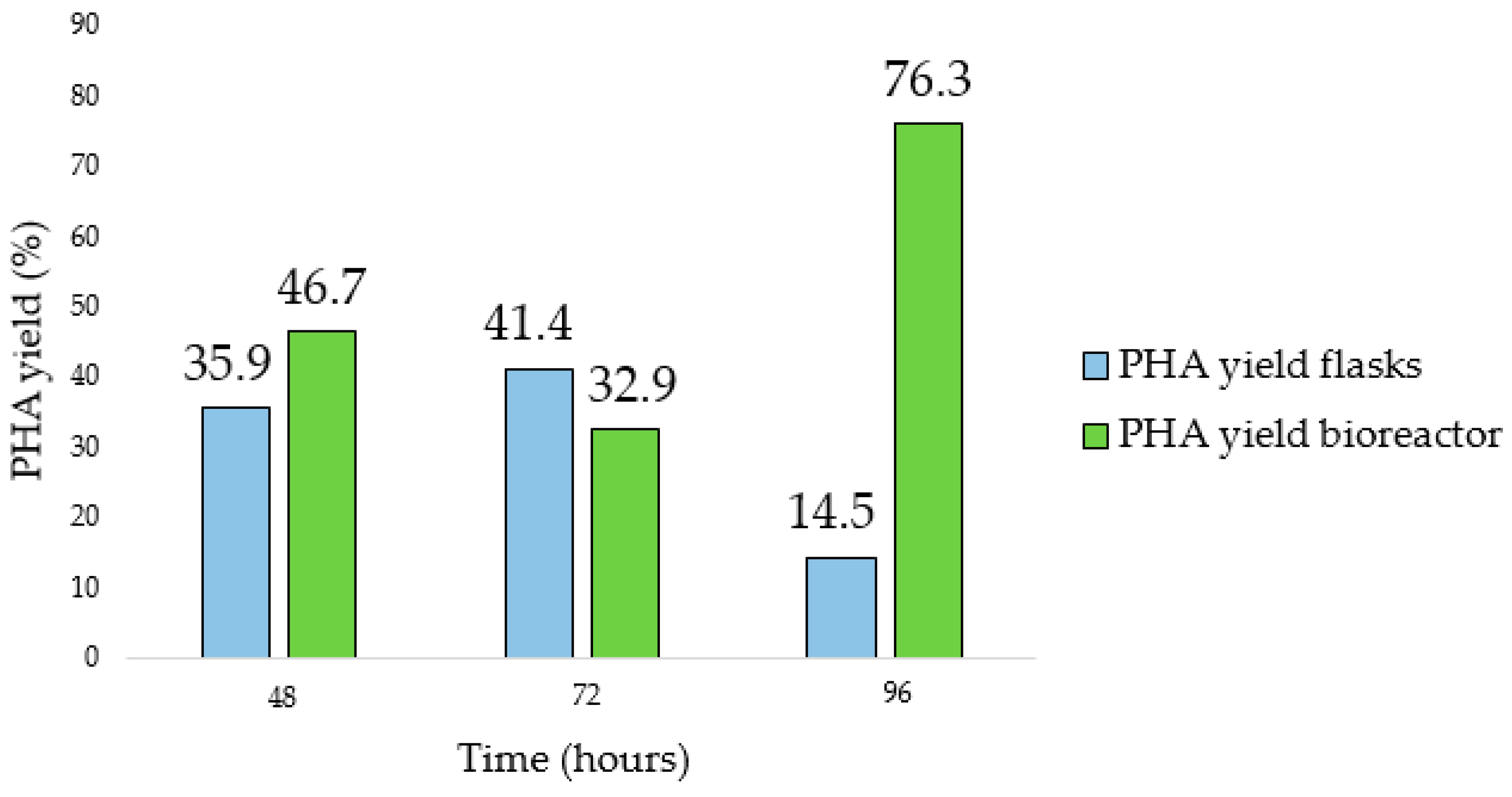
Figures S4 and S5). Comparing the yields obtained during the flask and the bioreactor tests (
Figure 11), the last one allowed us to obtain a higher yield (41.4% and 76.3%, respectively). In addition, the maximum Y
PHA/X was observed at a time postponed by 24 h compared to what was observed in the flask.
The maximum productivity of PHA (P
PHA) (Equation (4)) in the bioreactor test was 0.0052 g*L
−1*h
−1 while in flask fermentation it was 0.0074 g*L
−1*h
−1. For both tests, the highest productivity of PHA was obtained after 48 h of fermentation. These results are in accordance with those previously reported using the same MMC in bioreactor fermentation with Scotta whey cheese as a carbon source (52% PHA yield, [
56]), even if the productivity was lower, thus requiring us to replicate the experiments and undergo further investigation.
After 96 h of fermentation in the bioreactor, a part of the fermentation media was transferred to two flasks (200 mL each) and subsequently incubated for a further 72 h (for a total time of 168 h from the beginning of bioreactor fermentation) at 30 ° C in the thermostatic incubator. On one of the two flasks (FlaskA), a pH correction was made, bringing it to neutrality, while the other (FlaskB) was incubated as it is. The biomass from the two flasks was extracted with the conventional protocol. The results of this fermentation phase are shown in
Table 9.
Despite the pH correction, the fermentation broth tends to return to the pH value before correction, probably due to the acids produced by the microbial consortium. Although the pH variation did not seem to influence microbial growth, it could affect the accumulation of PHA. It is possible that, for the same weight of biomass, the flask where the pH was adjusted contained a greater number of PHA producers than the flask without correction. The decrease in Brix degrees observed in the flask, compared to the bioreactor, could confirm the hypothesis of the effect of stirring efficiency on the solubilization of suspended solids and pH variation. However, these preliminary results would need further confirmation by replicates.


 ,
, 
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
