
Dynamic Regulation Engineering of Plasmid Copy Number Based on CRISPRi in Saccharomyces cerevisiae

 , ,
, ,
Abstract
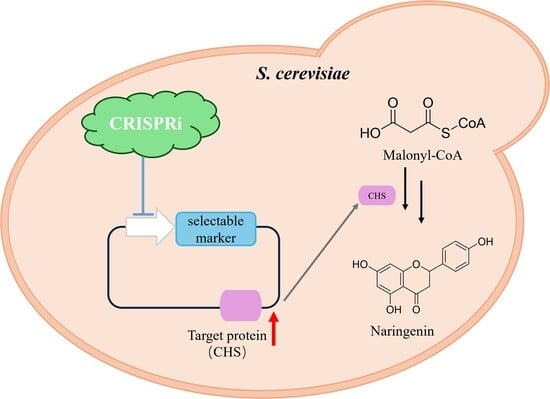
1. Introduction
2. Materials and Methods
2.1. Strains and Plasmids
2.2. Medium and Culture Conditions
2.3. CRISPR-Mediated Genomic Integration Method
- (1)
- Design and construction of gRNA plasmids
- (2)
- Design and construction of donor repair fragments
2.4. Measurement and Analysis of Fluorescence Data
2.5. Data Statistical Methods
2.6. HPLC Analysis of Naringenin Metabolites
3. Results
3.1. Construction of Plasmid Copy Number Regulation Tool Based on CRISPRi
3.2. Optimization of Plasmid Copy Number Regulation Tool
3.2.1. Screening of Targeted Sites for Inhibition
3.2.2. Optimization of the Inhibition Initiation Time
3.2.3. Optimization of Inducer Concentration
3.3. The Application of Plasmid Copy Number Regulation Tool
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PCN | Plasmid copy number |
| CRISPRi | CRISPR interference |
| Gal | Galactose |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Function |
|---|---|---|
| -ig-TEF1p-KanR-F | ATATGAAAGAAGAACCTCAGgttttagagctagaaatagcaagttaaaataag | For the construction of the PgKanR plasmid |
| -ig-TEFp-KanR-R | CTGAGGTTCTTCTTTCATATgatcatttatctttcactgcggag | |
| ADH1t: 0F | agcgacctcatgctatac | |
| ADHt-TEF1t: 0R | gtatagcatgaggtcgctatagcgccgatcaaagtatttg | |
| KaR2p-0R | tgagtcctctagtttttaccgc | |
| p41-T3-F | gcggtaaaaactagaggactcacccgggcagcttttgttc | |
| -igLEU2-315-F | AACATAACGAGAACACACAGgttttagagctagaaatagcaagttaaaataag | For the construction of the PgLEU2 plasmid |
| -igLEU2-315-R | CTGTGTGTTCTCGTTATGTTgatcatttatctttcactgcggag | |
| ADHt-CYC1t: 0R | ggtatagcatgaggtcgctgcaaattaaagccttcgagcg | |
| DC 16-T7p-F | taaaaactagaggactcaggccggtacccaattcg | For the construction of the PgKiURA3 plasmid |
| -ig-URA3-66-F | TGACGGGAGTGTATTGACGCgttttagagctagaaatagcaagttaaaataag | |
| -ig-URA3-66-R | GCGTCAATACACTCCCGTCAgatcatttatctttcactgcggag | |
| +ig-2uori-621-F | ctgcattatagagcgcacaagttttagagctagaaatagcaagttaaaataag | For the construction of the Pg2μ plasmid |
| +ig-2uori-621-R | ttgtgcgctctataatgcaggatcatttatctttcactgcggag | |
| +ig-CEN/ARS-374-R | cctagagtcttttacatcttgatcatttatctttcactgcggag | For the construction of the PgCEN/ARS plasmid |
| -ig-CEN/ARS-374-F | aagatgtaaaagactctagggttttagagctagaaatagcaagttaaaataag | |
| CHS-0R | ATGGTTACTGTTGAAGATGTTAGAAGAGC | For the construction of the Pg2μ-CHS plasmid |
| CHS-KaR2p-F | CATCTTCAACAGTAACCATatttgtaattaaaacatggtatgtttgatacgc | |
| TDH2t-0F | GCGAAAAGCCAATTAGTGTGATAC | |
| TDH2t-CYC1t-R | CACACTAATTGGCTTTTCGCgcaaattaaagccttcgagcg |
References
- Wu, Z.; Gao, J.; Gao, N.; Zhao, Y.; Zhou, Y.J. Engineering chronological lifespan toward a robust yeast cell factory. Proc. Natl. Acad. Sci. USA 2025, 122, e2515324122. [Google Scholar] [CrossRef]
- Miao, Z.; Ren, Y.; Tarabini, A.; Yang, L.; Li, H.; Ye, C.; Liti, G.; Fischer, G.; Li, J.; Yue, J. ScRAPdb: An integrated pan-omics database for the Saccharomyces cerevisiae reference assembly panel. Nucleic Acids Res. 2025, 53, D852–D863. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, T.; Zhang, Z.; Jiang, Q.; Yuwen, W.; Qu, L.; Fu, R.; Zhu, C.; Lei, H.; Fan, D. Engineering yeast expression platforms: Advanced strategies for heterologous protein production and glycosylation modification. Chem. Eng. J. 2025, 526, 171510. [Google Scholar] [CrossRef]
- Yang, S.; Song, L.; Wang, J.; Zhao, J.; Tang, H.; Bao, X. Engineering Saccharomyces cerevisiae for efficient production of recombinant proteins. Eng. Microbiol. 2024, 4, 100122. [Google Scholar] [CrossRef]
- Yu, A.; Mao, J.; Xu, N. Metabolic Engineering of Yeasts: A Key Cell Factory Platform for Advanced Biomanufacturing. J. Fungi 2025, 11, 863. [Google Scholar] [CrossRef]
- Zhao, M.; Ma, J.; Zhang, L.; Qi, H. Engineering strategies for enhanced heterologous protein production by Saccharomyces cerevisiae. Microb. Cell Factories 2024, 23, 32. [Google Scholar] [CrossRef] [PubMed]
- Rouches, M.V.; Xu, Y.; Cortes, L.B.G.; Lambert, G. A plasmid system with tunable copy number. Nat. Commun. 2022, 13, 3908. [Google Scholar] [CrossRef]
- Ramiro-Martínez, P.; de Quinto, I.; Lanza, V.F.; Gama, J.A.; Rodríguez-Beltrán, J. Universal rules govern plasmid copy number. Nat. Commun. 2025, 16, 6022. [Google Scholar] [CrossRef]
- Son, Y.J.; Ryu, A.J.; Li, L.; Han, N.S.; Jeong, K.J. Development of a high-copy plasmid for enhanced production of recombinant proteins in Leuconostoc citreum. Microb. Cell Factories 2016, 15, 12. [Google Scholar] [CrossRef]
- Chen, Y.; Partow, S.; Scalcinati, G.; Siewers, V.; Nielsen, J. Enhancing the copy number of episomal plasmids in Saccharomyces cerevisiae for improved protein production. FEMS Yeast Res. 2012, 12, 598–607. [Google Scholar] [CrossRef]
- Wang, G.; Wang, Q.; Qi, Q.; Wang, Q. Dynamic plasmid copy number control for synthetic biology. Trends Biotechnol. 2024, 42, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Roux, I.; Woodcraft, C.; Sbaraini, N.; Pepper, A.; Wong, E.; Bracegirdle, J.; Chooi, Y. Next-generation AMA1-based plasmids for enhanced heterologous expression in filamentous fungi. Microb. Biotechnol. 2024, 17, e70010. [Google Scholar] [CrossRef] [PubMed]
- Park, G.; Yang, J.; Seo, S.W. Dynamic control of the plasmid copy number maintained without antibiotics in Escherichia coli. J. Biol. Eng. 2024, 18, 71. [Google Scholar] [CrossRef] [PubMed]
- Weiss, F.; Requena-Moreno, G.; Pichler, C.; Valero, F.; Glieder, A.; Garcia-Ortega, X. Scalable protein production by Komagataella phaffii enabled by ARS plasmids and carbon source-based selection. Microb. Cell Factories 2024, 23, 116. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- McQuaid, M.E.; Pinder, J.B.; Arumuggam, N.; Lacoste, J.S.C.; Chew, J.S.K.; Dobson, M.J. The yeast 2-μm plasmid Raf protein contributes to plasmid inheritance by stabilizing the Rep1 and Rep2 partitioning proteins. Nucleic Acids Res. 2017, 45, 10518–10533. [Google Scholar] [CrossRef]
- Mereshchuk, A.; Johnstone, P.S.; Chew, J.S.K.; Dobson, M.J. The yeast 2-micron plasmid Rep2 protein has Rep1-independent partitioning function. Nucleic Acids Res. 2022, 50, 10571–10585. [Google Scholar] [CrossRef]
- Mereshchuk, A.; Chew, J.S.K.; Dobson, M.J. Use of Yeast Plasmids: Transformation and Inheritance Assays. Methods Mol. Biol. 2021, 2196, 1–13. [Google Scholar]
- Lian, J.; Jin, R.; Zhao, H. Construction of plasmids with tunable copy numbers in Saccharomyces cerevisiae and their applications in pathway optimization and multiplex genome integration. Biotechnol. Bioeng. 2016, 113, 2462–2473. [Google Scholar] [CrossRef]
- Herman-Antosiewicz, A.; Obuchowski, M.; Węgrzyn, G. A plasmid cloning vector with precisely regulatable copy number in Escherichia coli. Mol. Biotechnol. 2001, 17, 193–199. [Google Scholar] [CrossRef]
- Hohnholz, R.; Pohlmann, K.J.; Achstetter, T. Impact of plasmid architecture on stability and yEGFP3 reporter gene expression in a set of isomeric multicopy vectors in yeast. Appl. Microbiol. Biotechnol. 2017, 101, 8455–8463. [Google Scholar] [CrossRef]
- Segall-Shapiro, T.H.; Sontag, E.D.; Voigt, C.A. Engineered promoters enable constant gene expression at any copy number in bacteria. Nat. Biotechnol. 2018, 36, 352–358. [Google Scholar] [CrossRef]
- Byun, G.; Yang, J.; Seo, S.W. CRISPRi-mediated tunable control of gene expression level with engineered single-guide RNA in Escherichia coli. Nucleic Acids Res. 2023, 51, 4650–4659. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jeon, H.H.; Seo, E.; Park, S.; Choe, D.; Cho, B.-K.; Lee, J.W. Direct mRNA-to-sgRNA conversion generates design-free ultra-dense CRISPRi libraries for systematic phenotypic screening. Metab. Eng. 2025, 89, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; HamediRad, M.; Hu, S.; Zhao, H. Combinatorial metabolic engineering using an orthogonal tri-functional CRISPR system. Nat. Commun. 2017, 8, 1688. [Google Scholar] [CrossRef] [PubMed]
- Shaw, W.M.; Studená, L.; Roy, K.; Hapeta, P.; McCarty, N.S.; Graham, A.E.; Ellis, T.; Ledesma-Amaro, R. Inducible expression of large gRNA arrays for multiplexed CRISPRai applications. Nat. Commun. 2022, 13, 4984. [Google Scholar] [CrossRef]
- Chen, Y.; Duan, Q.; Wang, L.; He, Q.; Fang, L.; Xiao, L.; Song, H.; Cao, Y. Genome-scale CRISPRi and base-editing libraries for genetic decoding and strain engineering in Shewanella. Trends Biotechnol. 2026, 44, 496–520. [Google Scholar] [CrossRef]
- Mans, R.; van Rossum, H.M.; Wijsman, M.; Backx, A.; Kuijpers, N.G.A.; van den Broek, M.; Daran-Lapujade, P.; Pronk, J.T.; van Maris, A.J.A.; Daran, J.-M.G. CRISPR/Cas9: A molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res. 2015, 15, fov004. [Google Scholar] [CrossRef]
- Stovicek, V.; Borja, G.M.; Forster, J.; Borodina, I. EasyClone 2.0: Expanded toolkit of integrative vectors for stable gene expression in industrial Saccharomyces cerevisiae strains. J. Ind. Microbiol. Biotechnol. 2015, 42, 1519–1531. [Google Scholar] [CrossRef]
- Jessop-Fabre, M.M.; Jakočiūnas, T.; Stovicek, V.; Dai, Z.; Jensen, M.K.; Keasling, J.D.; Borodina, I. EasyClone-MarkerFree: A vector toolkit for marker-less integration of genes into Saccharomyces cerevisiae via CRISPR-Cas9. Biotechnology 2016, 11, 1110–1117. [Google Scholar] [CrossRef]
- Lee, J.; Lim, K.; Kim, A.; Mok, Y.G.; Chung, E.; Cho, S.-I.; Lee, J.M.; Kim, J.-S. Prime editing with genuine Cas9 nickases minimizes unwanted indels. Nat. Commun. 2023, 14, 1786. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Márquez, D.L.; García, L.M. Evaluation of Plasmid Stability by Negative Selection in Gram-negative Bacteria. BioProtocol 2017, 7, e2261. [Google Scholar]
- Cai, Q.A.-O.; Wang, M.; Zhu, J.; Ye, B.; Chu, X.; Wu, J. Modular RNAi Pathway Engineering Enhances Plasmid Copy Number Control in Yeast Bioproduction System. Biotechnol. Bioeng. 2025, 122, 2888–2898. [Google Scholar] [CrossRef]
- Wang, R.; Cress, B.F.; Yang, Z.; Hordines, J.C., III; Zhao, S.; Jung, G.Y.; Wang, Z.; Koffas, M.A.G. Design and Characterization of Biosensors for the Screening of Modular Assembled naringenin Biosynthetic Library in Saccharomyces cerevisiae. ACS Synth. Biol. 2019, 8, 2121–2130. [Google Scholar] [CrossRef]
- Tong, Y.; Li, N.; Zhou, S.; Zhang, L.; Xu, S.; Zhou, J. Improvement of Chalcone Synthase Activity and High-Efficiency Fermentative Production of (2S)-naringenin via In Vivo Biosensor-Guided Directed Evolution. ACS Synth. Biol. 2024, 13, 1454–1466. [Google Scholar] [CrossRef]








| Name | Description or Relevant Genotype | Source |
|---|---|---|
| IMX581 | parent strain | Purchased from Euroscarf (Scientific Research and Development GmbH), Frankfurt, Hesse, Germany; accno = Y40593 |
| KD01 | IMX581-derived naringenin-producing strain | This study |
| XY002 | KD01 Δleu2 | This study |
| XY010 | KD01, SpCas9::CRISPRi | This study |
| XY011 | XY002, SpCas9::CRISPRi | This study |
| XY012 | XY003 inserted Leu2p-sfGFP | This study |
| XY013 | XY012, SpCas9::CRISPRi | This study |
| pXY016 | 2μ-based plasmid, KiURA3 selectable marker | Lab stock |
| pXY018 | CEN/ARS -based plasmid, LEU2 selectable marker | Lab stock |
| pXY041 | CEN/ARS -based plasmid, KanR selectable marker | Lab stock |
| pXY016-2 | Constructed by integrating the KAR2p-yeGFP expression cassette into pXY016 | This study |
| pXY018-2 | Constructed by integrating the KAR2p-yeGFP expression cassette into pXY018 | This study |
| pXY041-2 | Constructed by integrating the KAR2p-yeGFP expression cassette into pXY041 | This study |
| PgKanR | pXY041-2-derived plasmid with an extra gRNA cassette targeting position 265–284 nt of the TEF1p (KanR promoter) | This study |
| PgLEU2 | pXY018-2-derived plasmid with an extra gRNA cassette targeting position 72–91 nt of the LEU2p | This study |
| PgKiURA3 | pXY016-2-derived plasmid with an extra gRNA cassette targeting position 69–88 nt of the KiURA3 promoter | This study |
| Pg2μ | pXY016-2-derived plasmid with an extra gRNA cassette targeting position 621–640 nt of the 2μ origin of replication | This study |
| PgCEN/ARS | pXY018-2-derived plasmid with an extra gRNA cassette targeting position 72–91 nt of CEN/ARS | This study |
| Pg2μ-CHS | Pg2μ-derived plasmid with yeGFP replaced by CHS | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Xu, Y.; Xu, T.; Jiang, T.; Wang, X.; Zhao, P.; Xu, K.; Xia, X.; Zhang, L. Dynamic Regulation Engineering of Plasmid Copy Number Based on CRISPRi in Saccharomyces cerevisiae. Fermentation 2026, 12, 177. https://doi.org/10.3390/fermentation12040177
Xu Y, Xu T, Jiang T, Wang X, Zhao P, Xu K, Xia X, Zhang L. Dynamic Regulation Engineering of Plasmid Copy Number Based on CRISPRi in Saccharomyces cerevisiae. Fermentation. 2026; 12(4):177. https://doi.org/10.3390/fermentation12040177
Chicago/Turabian StyleXu, Ying, Tingting Xu, Tao Jiang, Xiaoyi Wang, Peipei Zhao, Kuidong Xu, Xuekui Xia, and Lixin Zhang. 2026. "Dynamic Regulation Engineering of Plasmid Copy Number Based on CRISPRi in Saccharomyces cerevisiae" Fermentation 12, no. 4: 177. https://doi.org/10.3390/fermentation12040177
APA StyleXu, Y., Xu, T., Jiang, T., Wang, X., Zhao, P., Xu, K., Xia, X., & Zhang, L. (2026). Dynamic Regulation Engineering of Plasmid Copy Number Based on CRISPRi in Saccharomyces cerevisiae. Fermentation, 12(4), 177. https://doi.org/10.3390/fermentation12040177