1. Introduction
Multicopper oxidase (MCO) enzymes, called laccases [
1,
2], belong to the group EC 1.10.3.2 and are capable of oxidizing various compounds like phenolic compounds (diphenols, polyphenols, and aminophenols), amines and polyamines [
3], and even lignin. These enzymes utilize molecular oxygen as the final acceptor of electrons.
Laccases are widely distributed in animals, plants, fungi, and bacteria. To date, most of the isolated and characterized laccases have been produced by fungi, such as
Trametes versicolor and
Botrytis cinerea, as well as species from
Aspergillus and
Penicillium. Additionally, certain bacteria, including
Azospirillum lipoferum,
Bacillus subtilis [
4], and
E. coli, as well as other bacteria from various genera including
Streptomyces, Staphylococcus, Pseudomonas, Enterobacter, and
Alteromonas, have been found to produce laccases.
Fungal and bacterial laccases differ in several ways, including cellular localization, redox potential, performance conditions, and production methods. Fungal laccases are extracellular, have a high redox potential, work in acidic conditions and mesophilic temperatures, and have been extensively examined [
5,
6,
7,
8,
9]. However, they typically do not function in harsh industrial environments, and their heterologous production is complex [
10]. In contrast, bacterial laccases have low redox potential and are intracellular enzymes [
11]. They offer several advantages over fungal laccases, including a broader range of pH in which they can operate, greater thermal stability, easier heterologous expression, increased tolerance to common laccase inhibitors [
3,
12,
13], and greater resistance to copper and chlorine [
4].
Laccases have been detected in a specific group of bacteria known as lactic acid bacteria (LAB). These laccases demonstrate the ability to degrade biogenic amines (BAs), which are toxic to humans and were discovered some years ago [
13,
14,
15,
16].
Biogenic amines result from bacterial amino acid decarboxylation or amination and transamination of aldehydes and ketones [
17]. The EFSA has reported that the presence of BAs in food and wine is influenced by the existence of microorganisms that produce amino acid decarboxylase enzymes as well as conditions that allow their growth, particularly temperature and pH levels, and the availability of free amino acids. Such conditions promote intensive microbial activity and BA production (EFSA Panel on Biological Hazards) [
18]. Some chemical and biochemical processes used in the food industry, such as ripening, salting, fermentation, and marination, can increase the likelihood of biogenic amine (BA) formation [
19]. Histamine, tyramine, putrescine, cadaverine, spermine, spermidine, phenylethylamine, and tryptamine are common BAs detected in food. It has been reported that consuming foods with high BA concentrations (>1000 mg/kg of food) may cause allergic symptoms such as nausea, headaches, and changes in blood pressure. Nevertheless, histamine and tyramine are the most toxic and relevant biogenic amines for ensuring food safety, according to the EFSA Journal (2011). Their consumption may cause severe acute allergic-like responses [
18].
Several studies have been conducted to produce bacterial laccases through fermentation processes for different purposes due to their numerous advantages. Many of these MCO enzymes have been heterologously expressed in different bacteria [
3,
13,
14,
20].
In addition to LAB laccases’ ability to degrade BAs, laccase enzymes have numerous other applications. These include bioremediation of organic compounds found in industry, such as benzylic alcohols, aromatic polycyclic hydrocarbons, and pesticides [
21], bleaching and degradation of azo colorants and textile effluents [
9,
10], synthesis of polymers, creation of new bioactive compounds and antibiotics [
22,
23], biosensor development [
24], and various applications in the food industry, including waste depuration, juice clarification, and wine stabilization [
25,
26].
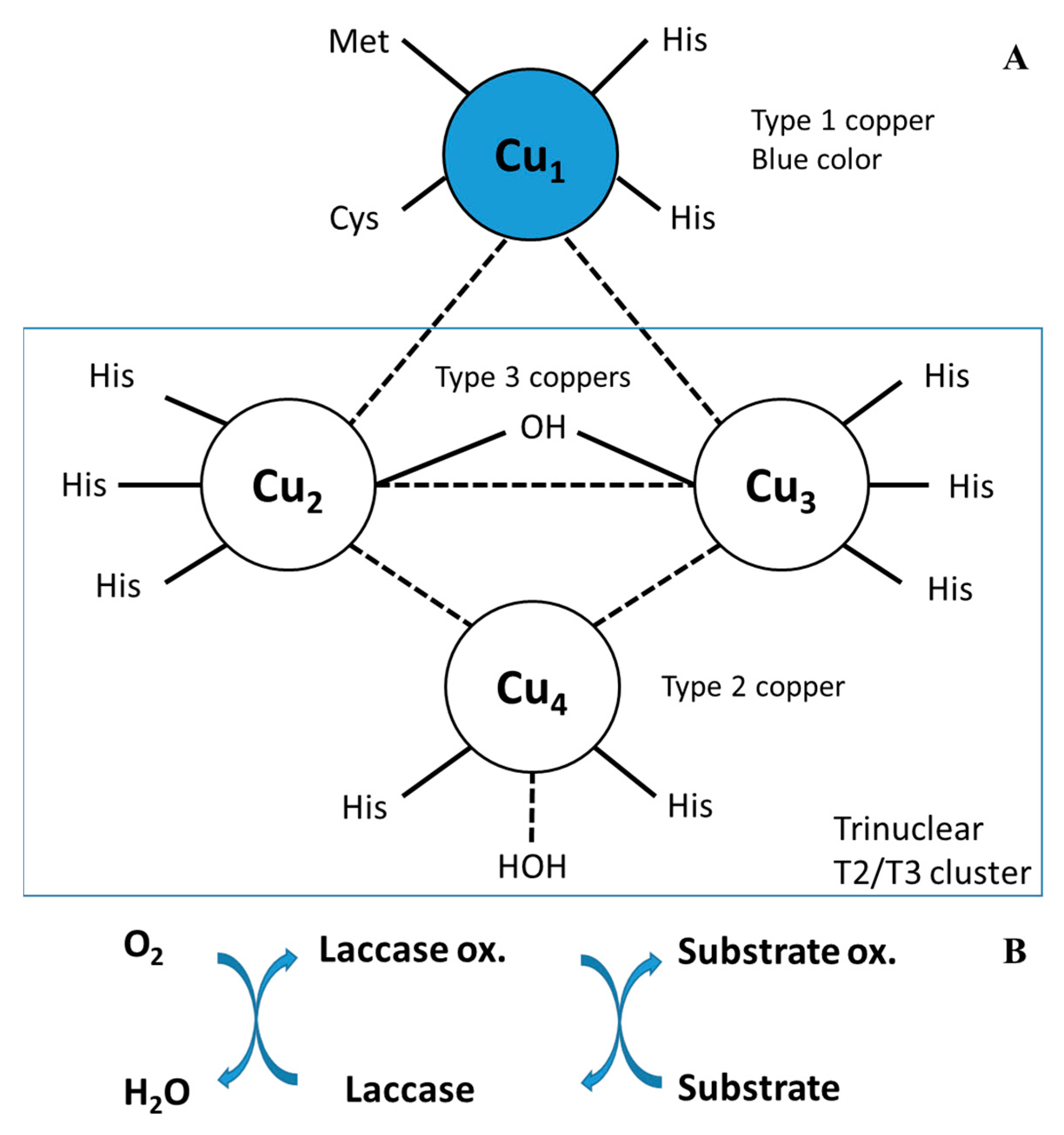
Regarding their chemical structure, laccases can bind up to four copper ions, which play a crucial role in their catalytic activity [
27]. Additionally, these enzymes contain three types of copper ions with different magnetic and spectroscopic properties. Copper ions are bound in copper centers or sites and classified as type 1 (T1), type 2 (T2), or double type 3 (T3). T1 copper, found in T1 sites, displays electronic absorbance at 600 nm and electro-paramagnetic resonance (EPR), resulting in the blue color of blue laccases. The T2 site is a mononuclear center containing T2 copper that is EPR detectable but colorless. The T3 site, comprised of two closely linked T3 copper molecules, exhibits weak absorbance near the UV spectrum (around 330 nm) and is not identifiable by EPR. Additionally, a trinuclear cluster (T2/T3) is formed by one T2 and two T3 copper ions. The T1 site, situated adjacent to the enzyme surface, facilitates the transfer of electrons from the substrate to the T2/T3 cluster. Four electrons originating from the T1 site reduce molecular oxygen, yielding two water molecules.
Figure 1 illustrates the copper sites within the well-characterized CotA laccase found in
Bacillus subtilis [
28,
29], as well as the laccase-mediated oxidation reaction of substrates.
Additionally, laccases possess a considerable capacity for oxidation of numerous substrates, utilizing molecular oxygen as an electron acceptor that is readily available. Laccase oxidation capacity, however, relies on the difference in redox potential between the substrate and the T1 copper atom [
30]. Oxidation mechanisms may also be dependent on the stability and redox potential of the intermediate radicals generated and the steric hindrance between substrates and the laccase T1 site.
Some studies have suggested that certain laccases need mediator compounds to facilitate the oxidation of non-phenolic substrates. Mediator compounds, which are typically small, low-molecular-weight molecules, act as electron shuttles between the substrate and the laccase. The mediator compounds are initially oxidized by the laccases and then in turn oxidize the substrate. The first mediator used in the laccase mediator system (LMS) for pulp delignification was 2,2′-azinobis(3-ethylbenzothiazoline-6-sulphonic acid (ABTS), which is now considered a canonical substrate for laccases [
31]. Apart from ABTS, the most commonly used mediators are 2,6-dimethylphenol (2,-DMP), syringaldazine (SGZ), acetosyringone (ACS), guaiacol, and L-DOPA [
32].
Mediator compounds have been shown to promote the laccase catalysis of non-phenolic substrates. Several N-heterocyclic compounds have been utilized as effective mediators for the oxidation of non-phenolic substrates. However, Camarero et al. (2005) suggested that the use of naturally occurring mediators could be of considerable interest due to their economic and environmental benefits [
33].
Some years later, Guan et al. (2018) reported that LMSs comprise three primary groups of mediators: synthetic mediators, natural mediators, and polyoxometalates (POM) [
28]. While synthetic mediators have been utilized in lignin degradation, dye colorization, and polycyclic aromatic hydrocarbon (PAH) oxidation, their application presents certain limitations, such as the high costs associated with employing them at industrial scales, the potential formation of toxic derivatives which may inactivate laccases, and their poor regeneration capacity. On the other hand, natural mediators derived from plants and industrial by-products are regarded as a cost-effective and environmentally friendly substitute for synthetic mediators. The majority of natural mediators are phenolic compounds with low chemical potential, and thus they can be swiftly oxidized by laccases. In this context, Giménez et al. (2022) reported that several phenolic compounds, including caftaric acid, catechin, epicatechin, and gallic acid, can serve as laccase substrates [
34]. These substrates may act as mediators in the oxidation reactions of non-phenolic substrates.
This study presents the biochemistry characterization of three new recombinant LAB laccases derived from Pediococcus parvulus ENOLAB 3909, Lacticaseibacillus paracasei ENOLAB 4314 (formerly known as Lactobacillus paracasei), and Lactococcus lactis ENOLAB 5298. Additionally, it explores their effectiveness in degrading BAs with and without mediators, including natural and synthetic compounds.
2. Materials and Methods
2.1. Strains and Plasmids
The Pediococcus parvulus ENOLAB 3909 strain was isolated from wine. It was kindly provided by A. Querol from the Institute of Agrochemistry and Food Technology (IATA) at the Spanish National Research Council (CSIC).
The Lactobacillus paracasei 4314 strain was isolated by the ENOLAB group from red wine of Origen Appellation (D.O.) Jumilla.
The Lactococcus lactis 5298 strain was isolated from cheese. It was kindly provided by A. Picon of the National Institute of Agricultural Research and Technology, which is part of the Spanish National Research Council (CSIC).
Escherichia coli DH5α was used for cloning and propagation of the putative laccase genes of the three previous strains using the plasmid pET-28a(+) from Novagen. E. coli BL21(DE3) with the pGro7 plasmid was provided by R. Muñoz from the Institute of Food Science, Technology, and Nutrition (ICTAN), and it was used for laccase expression. The pGro7 plasmid encodes for two chaperones (GroES–GroEL) that assist in protein folding.
2.2. E. coli Routine Growth Conditions
E. coli cells were cultured at 37 °C in Luria–Bertani (LB) medium (either broth or agar) [
35]. The pET-28a(+) DH5α transformed cells were cultured in LB supplemented with 50 µg mL
−1 kanamycin, while pET-28a(+) pGro7 BL21 transformant cells were grown in the same LB medium with 50 µg mL
−1 kanamycin (Kn) and 20 µg mL
−1 cloramphenicol (Cm).
Pediococcus parvulus 3909 and Lacticaseibacillus paracasei 4314 were statically cultivated in MRS medium with cysteine HCl (0.5 g/L) (Scharlau S.L., Barcelona, Spain) at pH = 6.5 and 28 °C, while Lactococcus lactis 5298 was grown in M17 medium (Oxoid Ltd., Basingstoke, UK) at 37 °C.
2.3. Gene Cloning, Plasmid Construction, and Expression Conditions
Table 1 shows the primers used for amplifying the three laccase genes as well as the specific restriction enzymes for cloning these genes into
E. coli DH5α.
PCR was carried out using native FIREPol DNA polymerase (Solis Biodyne, Tartu, Estonia) in an Eppendorf thermocycler; the reaction mixture contained 1 µL DNA, 0.5 µL Taq polymerase FIREPol, 1 µL dNTPs mixture (0.2 mM each one), 1 µL of forward and reverse primers (1 mM), 4 µL MgCl
2 (2 mM), 5 µL buffer Taq 10X (FIREPol), and 36.5 µL MilliU water. The PCR thermal profile setup was detailed in Callejón et al. (2017) [
13]. It involved initial denaturation (95 °C for 5 min), 35 cycles of denaturation (94 °C for 1 min), primer annealing (58 °C for
P. parvulus and
L. lactis and 54 °C for
L. paracasei for 1 min), and extension (72 °C for 1 min). Finally, reactions were completed with 5 min elongation time at 72 °C followed by cooling to 10 °C.
Laccase genes were cloned in pET-28a(+) using 50 ng of vector and a ratio of 20:1 insert/vector with the aid of a T4 DNA ligase enzyme from Nzytech (Lisbon, Portugal) for P. parvulus and L. paracasei and a 15:1 ratio with a T4 DNA ligase from ThermoFisher (Waltham, MA, USA) for L. lactis. One microliter of the ligation reaction was inserted in E. coli DH5α competent cells (50 µL) by electroporation. Electroporation conditions were 1.7 mV, 25 µF, and 200 Ω, and the equipment employed was from BioRad (Madrid, Spain). After electroporation, the cells were suspended in 1mL of SOC medium and incubated at 37 °C for one hour. To assess the viability of the cells post-electroporation, dilutions of the transformation mix were plated on LB agar. Additionally, LB + Kn (50 µg mL−1) plates were used to isolate presumed transformants.
Putative transformants were verified through sequencing of the corresponding laccase gene sequence. The confirmed transformant(s) were then cultured in 10 mL of LB + Kn broth at 37 °C overnight. Next, the plasmid pET-28a(+) was extracted from the cultivated cells using a Miniprep kit from Metabion (Planegg, Germany). Subsequently, one microliter of the extracted plasmid was added to 50 µL of E. coli BL21 pGro7 competent cells to transform them via electroporation under identical conditions to those followed with DH5α. The transformation mixture was plated on LB + Cm (20 µg/mL) and LB + Kn + Cm in parallel to obtain transformants carrying both vectors.
The amplified fragment with T7 promoter and terminator primers confirmed several colonies as true transformants via sequencing. After verification, a subset of the clones was cultured in 20 mL of Terrific Broth (TB) medium with Kn (50 µg mL−1), Cm (20 µg mL−1), CuCl2 (0.2 mM), and arabinose (2 mg mL−1) and maintained at 37 °C and 120 rpm. The purpose was to assess the expression of laccases in these clones. Once the cell growth reached 0.6 OD, 1 mM of isopropyl-β-D-1-thiogalactopyranoside (IPTG) was added to 10 mL of the culture medium as a laccase inducer. The other 10 mL of the culture remained untreated (induction control). Both cultures were maintained at 20 °C and 120 rpm for the first four hours, followed by static incubation at 20 °C overnight. The following day, the cells underwent centrifugation and were subsequently washed with a phosphate buffer with a pH of 7.4. They were then frozen at a temperature of −80 °C for an hour and subsequently disrupted with glass beads measuring 0.1 mm in diameter. Finally, they were centrifuged again to obtain supernatants which were then used to evaluate protein concentration and laccase activity toward ABTS.
Protein concentration was determined utilizing the bicinchoninic acid (BCA) method [
36] and Pierce (Rockford, IL, USA) TM BCA kit. Activity measurement was conducted according to Callejón et al. (2014), using a standard reaction buffer (SRB) containing 50 mM sodium acetate pH 4.0, 0.1 mM CuSO
4, and 2 mM ABTS as substrate [
15]. ABTS oxidation was evaluated on the FLUOstar OPTIMA spectrophotometer (BMG LABTECH, Ortenberg, Germany) by measuring the increase in OD
420, as described by Olmeda et al. (2021) [
3]. The clone with the highest activity-to-concentration ratio was selected for subsequent procedures and assays.
To obtain the necessary quantity and concentration of protein for purification and further analysis, the selected clone was cultivated in a larger-volume culture. Consequently, transformed
E. coli BL21 cells were grown in 2L TB medium, and the recombinant laccases were expressed by the addition of IPTG at the same concentration as used in the small-scale cultures. Cloning and laccase expression protocols utilized in this study were thoroughly outlined by Callejón et al. (2017) and were executed with a reduction of CuCl
2 concentration from 1 mM to 0.2 mM [
13]. All other conditions were upheld.
2.4. Purification of Laccases
The purification process for all laccase proteins is summarized below. Firstly, overnight-induced cells were washed with phosphate buffer at pH 7.4 and kept at −80 °C for 1 h. Afterward, the cells were suspended in 10 mL of lysis buffer consisting of 1 mM PMSF, 10 µg/mL RNAse, 5 µg/mL lysozyme, 1 mM MgCl
2, 0.1% Tween 20, and 1 pill of cOmplete 1X from Roche, all dissolved in a 5 mM imidazole buffer. The crude laccase extract was obtained via mechanical rupture, using 1 g of 0.1 mm diameter glass beads from Biospec products per 1 mL of the cell lysis buffer. The extract underwent centrifugation at 13,000×
g revolutions per minute for 5 min at 4 °C in a PrismR centrifuge (Labnet International Inc., Corning, NY, USA). It was then dialyzed with 5 mM imidazole buffer (50 mM NaH
2PO
4, 300 mM NaCl, 0.05% Tween 20, and 5 mM imidazole, pH 8.0). This proceeded overnight and was subsequently centrifuged again, following the procedure outlined in Callejón et al. (2017) [
13].
Metal-chelating chromatography was employed using a Ni
+2-NTA agarose column to purify the proteins. Laccase proteins were successfully eluted from the column using 250 mM imidazole elution buffer, which shared the same composition as 5 mM imidazole but with a higher concentration of 250 mM. Different fractions were collected and analyzed through SDS 7.5% PAGE protein gel electrophoresis to identify those with high purity and concentration. The fractions were tested for laccase activity using the same procedure outlined for the crude extract obtained in smaller volumes (refer to
Section 2.3). The fractions with the highest concentration of protein and activity were combined and dialyzed with a 14 kDa cellulose membrane (Merck group, Darmstadt, Germany) against a phosphate buffer at pH 7.4 along with 0.05% Tween 20 at 4° and agitated overnight. Following dialysis, the solution was retrieved and centrifuged at 10,000 rpm for 10 min. The supernatant was separated and then aliquoted for further laccase biochemical characterization. Protein concentration and activity were measured using the method described in
Section 2.3.
2.5. Laccase Biochemical Characterization
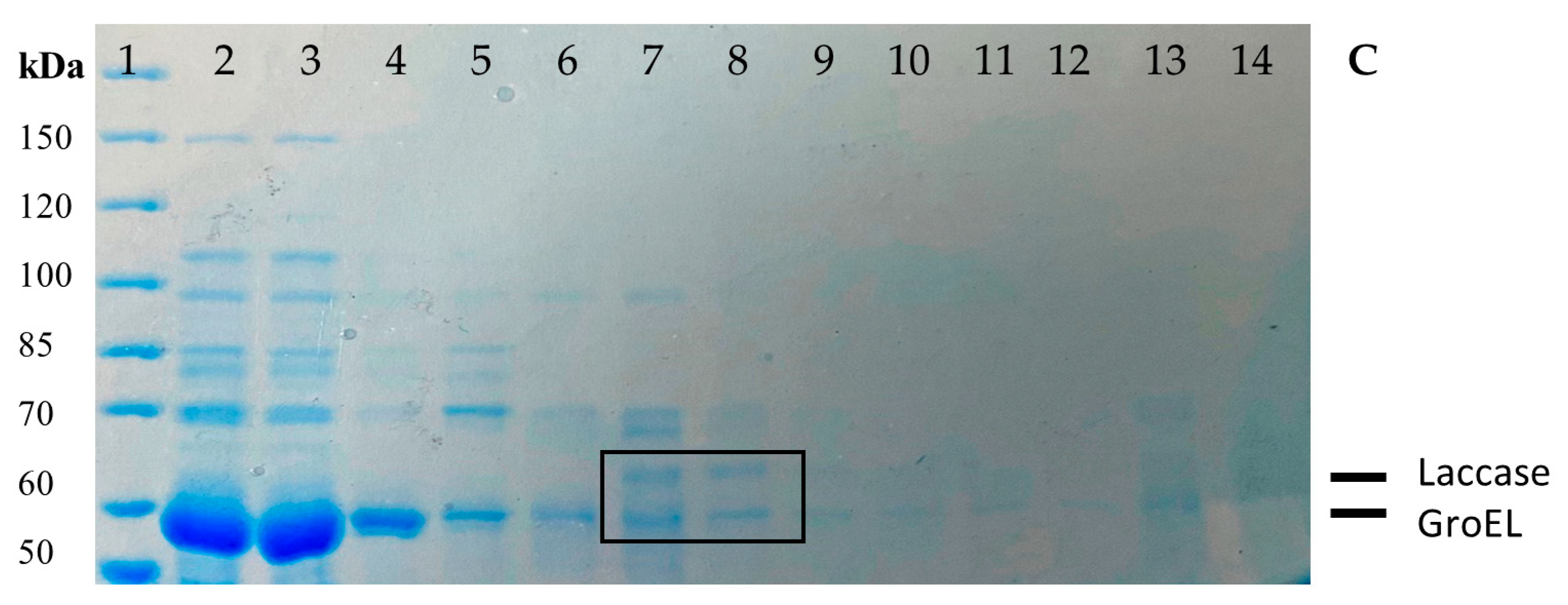
The relative molecular weight of the denatured proteins was assessed by comparing their migration positions with the molecular weight marker (PAGE ruler unstained) in an SDS 7.5% PAGE after purification.
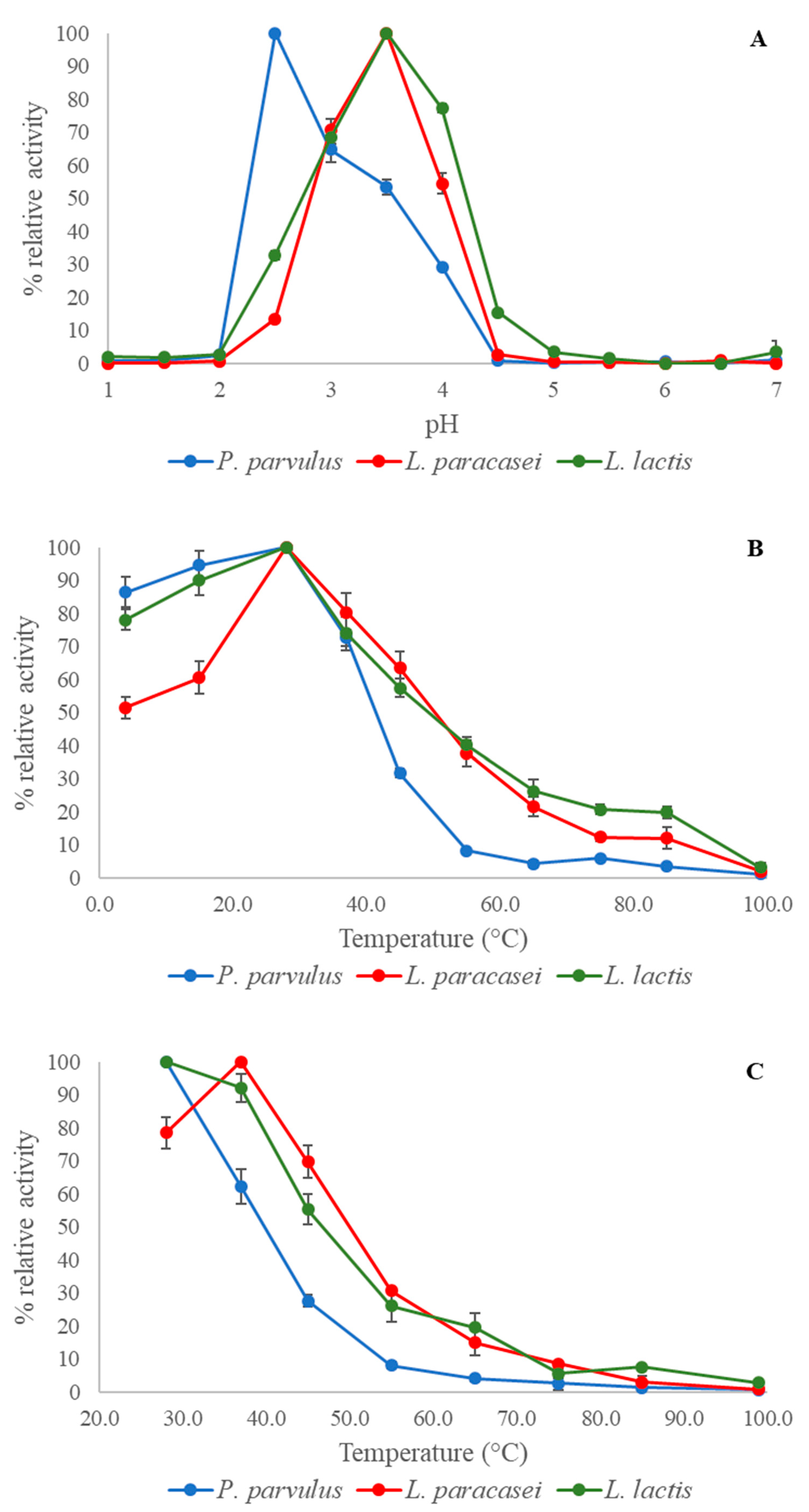
To assess the impact of pH on laccase activity, 200 μL of 50 mM HCl-KCl buffer + 0.1 mM CuSO
4 were utilized at pH values ranging from 1.0 to 2.0. Similarly, citrate–phosphate buffer + 0.1 mM CuSO
4 was used at pH values ranging from 2.5 to 9.0 with 2 mM ABTS, following the method outlined in Olmeda et al. (2021) [
3]. The relevant laccase concentration was carefully prepared and the mixture was subsequently placed in a 96-well microplate. The ABTS oxidation kinetics at the OD
420 were recorded for 3 min, as previously described. Reactions were conducted in triplicate, and enzymatic activity was calculated as a relative percentage of the maximum value achieved at optimal pH (% relative activity).
Laccase kinetic parameters (k0.5, kcat, kcat/cat 0.5, and specific activity toward ABTS) were determined at room temperature using various concentrations of ABTS (0.01–2 mM). Assays were carried out in a 200 μL reaction mixture that contained 50 mM citrate–phosphate buffer optimized for each laccase pH. The ABTS oxidation reaction commenced upon the inclusion of the appropriate enzyme. Kinetic reactions were monitored through OD420 measurements and performed in triplicate. Negative controls replicated the reactions without the enzyme. Michaelis–Menten or sigmoidal kinetics were used to evaluate kinetics data, and Vmax (ΔOD/min) kinetic constants were computed utilizing GraphPad 9.2.0 software. The specific enzymatic activity was determined by calculating the Vmax of the reactions and the protein concentration and was expressed as units per milligram (U/mg). One unit refers to the fraction of the active enzyme amount that catalyzes the oxidation of 1 µmol of substrate per minute, relative to the mass of protein in milligrams. The extinction coefficient ε420 = 36,000 M−1 cm−1 was used for ABTS oxidation.
The impact of temperature on laccase activity was assessed by adding 2 mM ABTS to 200 μL of 50 mM citrate–phosphate buffer at each laccase optimum pH. The buffer was then heated to a specific temperature between 4 and 99 °C for 5 min before introducing the enzyme to initiate ABTS oxidation at each temperature. The reaction lasted for 3 min before being halted with the addition of 10 mM sodium azide, and the OD420 was recorded.
Laccase thermal stability analysis was conducted in the temperature range of 28 to 99 °C using a method similar to the temperature effect procedure. However, there was one difference: the buffer was pre-incubated with the enzyme at the specific temperature for 5 min. Following this, the ABTS reaction mixture was kept at room temperature for 5 min, and subsequently 2 mM ABTS was added to initiate the oxidation reaction, which continued for 3 min at room temperature. Finally, the reaction was halted by adding 10 mM sodium azide and the OD420 was measured. The level of activity toward ABTS at each temperature were calculated for both the optimum temperature and thermal stability experiments. The maximum value obtained in each experiment was considered as 100% activity for calculation.
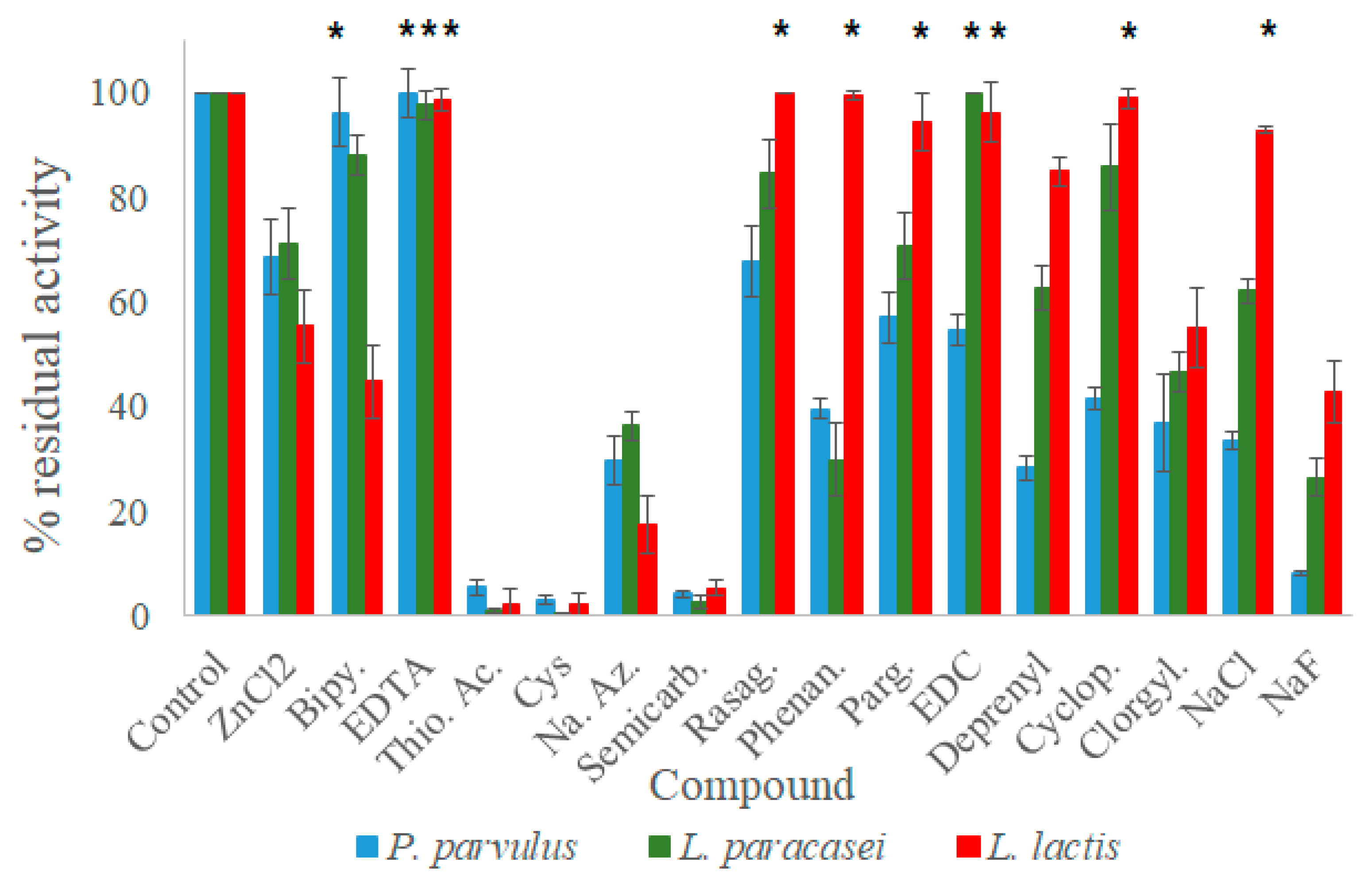
Given that these laccases could have the ability to oxidize amines, the effect of some known inhibitors for amine oxidases and laccases was studied. Following the procedures outlined by Callejón et al. (2017) and Olmeda et al. (2121) [
3,
13], experimental evaluations were conducted. Some of the compounds function as metal-chelating agents for the T1 copper atom. Examples include 2,2′-bipyridil, phenanthroline, and EDTA. Other compounds impact the enzyme’s oxidizing potential. These include cyclopropylamine, deprenyl, rasagiline, and sodium azide, which work to block oxygen’s electron transfer. In addition, clorgyline and pargyline are specific inhibitors of amine oxidase, while N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC) is a modifying agent used for carboxyl group involvement in either the catalytic process or substrate binding [
37]. Semicarbazide, on the other hand, inhibits a specific subgroup of copper-dependent amine oxidases [
14]. Cysteine and thioglycolic acid are reducing agents that produce stable copper complexes with laccases [
38]. Finally, sodium fluoride and chloride halides, in addition to zinc chloride, are reported as laccase inhibitors because they bind to the T2 copper and prevent oxygen reduction [
39]. The assays for laccase activity were devised by dispensing 196 μL of citrate–phosphate buffer at the optimal pH of each laccase into a 96-well microplate, then adding 1 mM of each inhibitor and enzyme. After an incubation time of 10 min, enzymatic reactions were initiated by adding 2 mM ABTS. After three minutes, the reactions were halted with the addition of 10 mM sodium azide. The FLUOstar OPTIMA spectrophotometer recorded the OD
420 value. A control reaction was carried out without any inhibitor and served as the 100% benchmark of relative enzymatic activity. The impact of inhibitors on maximum activity was calculated accordingly.
2.6. Epicatechin Oxidation Measurements
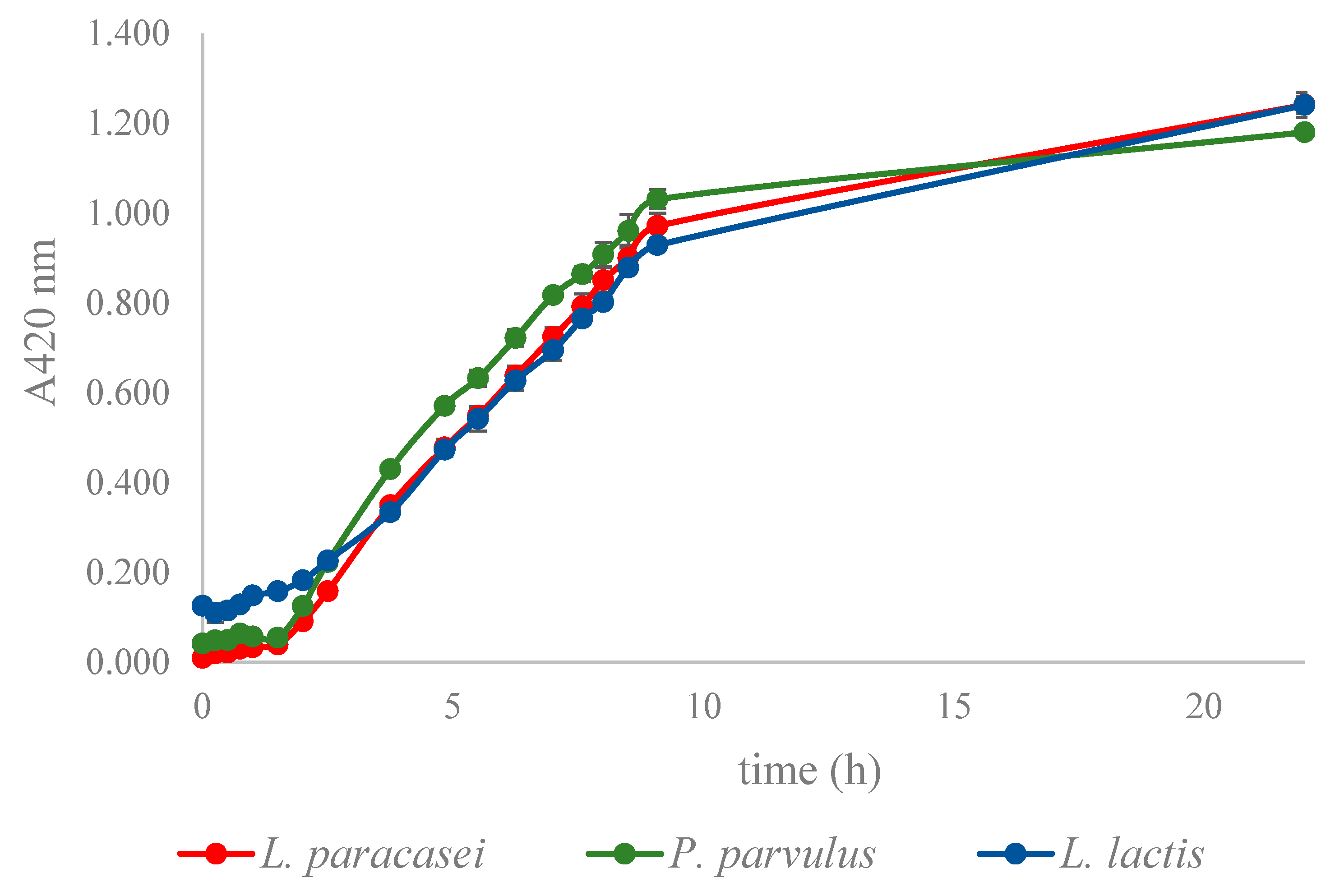
The quantitative degradation of 2 mM epicatechin by LAB laccases in 50 mM acetate buffer pH 4 was followed through the optical density measurement at 420 nm for almost 24 h.
2.7. Degradation of Biogenic Amines
Degradation reactions were carried out using 25 μg of laccase in a reaction solution comprising 50 mM acetate buffer, 0.1 mM CuSO
4, and 150 mg/L biogenic amines (BAs), similar to previous studies conducted with other LAB laccases [
3,
13,
14]. Additionally, either 2 mM of mediator compounds (ABTS or epicathechin) were added to the reaction mixture or the absence of mediators was maintained. Histamine, tyramine, putrescine, and phenylethylamine (PEA) were chosen for the assays because of their toxicity or high levels in foods. Additionally, the influence of laccase on dopamine was examined due to their similar chemical structures. ABTS was utilized as the mediator for the bacterial laccases because it is a canonical substrate for these enzymes. Furthermore, epicathechin was tested as a potential natural mediator since it is a flavan-3-ol commonly present in wine and certain vegetable foods [
40]. The reactions were incubated for 24 h and were terminated with 10 mM sodium azide. Next, the samples were filtered using 0.22 µm nylon membranes (Fisher, Alcobendas, Spain) and then directly exposed to a derivatizing reagent, o-phthaldialdehyde (OPA), in the chromatograph injector, in accordance with Callejón et al. (2016) [
14]. Residual amine concentration was determined using reverse-phase liquid chromatography with fluorescence detection (LC-FLD) on an HPLC Agilent 1200SL system equipped with a 150 × 4.6 mm Poroshell 120 EC-C18 2.7 μm column (Agilent, Santa Clara, CA, USA) and a fluorescent detector. Subsequently, the biodegradation potential of three bacterial laccases for biogenic amines was assessed by comparing the percentage of amine degradation in reactions with laccase to those without laccase (reaction controls).
2.8. Statistical Analyses
All data are reported as the mean value ± SD. The ROUT method was used to analyze any outliers among the replicates. One-way ANOVA was used to compare group means, followed by a Dunnett’s multiple comparison test between the control and each inhibitor for the analysis of inhibitor compounds on laccases. These analyses were carried out using the GraphPad Prism statistical program 9.2.0 (Dotmatics, Boston, MA, USA).
4. Discussion
The nucleotide sequences of the three recombinant laccases from
P. parvulus ENOLAB3909,
L. paracasei ENOLAB4314, and
L. lactis ENOLAB5298 reveal that they belong to subfamily J—Bacterial CueO (similar to the CueO laccase from
E. coli) of the multicopper oxidase superfamily, according to the Laccase Engineering Database (
https://lcced.biocatnet.de/, accessed 2 November 2023). Additionally, the multiple sequence alignment and conservation among these and other bacterial laccases show high scores, as well as significant conservation of the motifs that form the four copper ligands, which is typical of blue laccases. Both
L. paracasei and
P. parvulus laccases displayed a blue color that is characteristic of blue multicopper oxidases, caused by their absorbance peak at 600 nm. This is due to the Cu type 1 situated in the T1 site. However,
L. lactis laccase did not demonstrate any apparent blue color, possibly due to its low protein concentration after purification. According to Morozova et al. [
42],
P. ostreatus produces two laccase isoforms. The POXA1b isoform is induced by copper ions and contains four copper ions in its active site, with an absorption maximum at 605 nm. In contrast, the POXA1w isoform lacks an absorption maximum in the blue region of the spectrum. Enzymes without a maximum at 600 nm are often referred to as laccase-like enzymes due to their inherent catalytic activity, like typical “blue” laccases. However, its homology with the rest of the LAB laccases confirms it as a blue laccase. In addition, the three laccases exhibited oxidation activity toward the canonical substrate ABTS, thus confirming their proper classification as laccases.
The molecular weights of the bacterial laccases under consideration are comparable to those of other recently described laccases, such as those from
L. plantarum J16 (59.3 kDa),
P. acidilactici CECT 5930 (60 kDa), and
P. pentosaceus 4816 (64 kDa). Moreover, these weights are similar to those of Cot A laccases from
B. subtilis 168 (66 kDa) [
43] and
B. subtilis WD23 (67.5 kDa) [
44].
The oxidation of the non-phenolic substrate ABTS exhibits a pH profile that monotonically decreases, with higher activity at lower pH values [
45]. As a result, the optimal pH range for ABTS oxidation by the three recombinant laccases is acidic, between 2.5 and 3.5, which is consistent with reported optimal pHs for other bacterial laccases, including
M. albomyces [
46],
L. plantarum [
14], and
P. pentosaceus [
3], which have optimum pHs of 3.5, and
P. acidilactici, which has an optimum pH of 4.0 [
13]. It is worth noting that
P. parvulus laccase demonstrated the highest activity toward ABTS at the most acidophilic pH. The optimal temperature for all three laccases indicated that they are mesophilic, resembling
P. acidilactici recombinant laccase (28 °C) [
13], but lower than other bacterial laccases like CotA from
Bacillus species (55–75 °C) [
47] and CueO from
E. coli (55 °C) [
48].
The limited thermal resistance of the three laccases at high temperatures had not been observed previously in other recombinant laccases. This finding may account for their lower specific activities toward ABTS at high temperatures compared to previously reported laccases. Nonetheless, the laccases evaluated in this study exhibited higher relative activities at 28 °C (their optimal temperature) than other LAB laccases, such as those from
L. plantarum J16 8944 [
14] and
P. pentosaceus 4816 [
3].
The three recombinant laccases exhibited sigmoidal kinetics on the artificial mediator ABTS, as previously observed for the
P. pentosaceus 4816 and
P. acidilactici 5930 laccases. This behavior has been documented in monomeric enzymes, whereby the substrate can bind to a second site alongside the active center. This is evident in the case of CotA [
49]. The K
0.5 value for
P. parvulus laccase equaled the reported K
m value for
L. plantarum J16 laccase [
14], while the k
0.5 value of
L. paracasei laccase was slightly higher than that reported for
P. acidilactici 5930 [
3].
Although
L. lactis laccase demonstrated a greater affinity for ABTS, it exhibited the lowest k
cat among the three laccases studied. Roskoski (2007) demonstrated that k
cat represents the maximum number of substrate molecules converted to product by an active site unit per unit time when the enzyme is saturated by the substrate [
50]. Furthermore, the k
cat/k
m value or specificity constant is useful to compare different substrates, as in the case of several laccases. Based on the principle that the substrate with the highest k
cat/k
m value indicates the best substrate for an enzyme, it can be deduced that ABTS is a better substrate for
P. parvulus than for
L. lactis or
L. paracasei laccases. This deduction is supported by the fact that
P. parvulus laccase exhibited the highest specificity constant toward ABTS, with an intermediate value between those of the
P. pentosaceus and
P. acidilactici laccases [
3].
The three laccases’ most effective inhibitory compounds were semicarbazide, a carbonyl-modifying compound that inhibits copper-dependent monoamine oxidases [
13], as well as cysteine and thioglycolic acid, reducing agents of laccases that can form stable copper complexes [
38]. These three inhibitory compounds almost entirely nullified the enzymatic activity of the three LAB laccases toward ABTS, thus proving their potential to halt oxidation reactions of other substrates. Prior studies have already documented the impact of these inhibitors on laccases from various bacterial sources, including
P. pentosaceus 4816 and
P. acidilactici 5930 laccases,
B. vallismortis [
51],
B. licheniformis [
52], and
T. versicolor [
38]. An additional finding from our study is that sodium azide, an electron-transfer blocking agent for oxygen [
3], and NaF, which binds to the T2 site and prevents oxygen reduction [
3], were highly effective inhibitors of our laccases, reducing activity by 70–80%. Other halides that have the potential to substitute Cu
+2, such as NaCl and ZnCl
2, exhibited lower inhibition than NaF. Even
L. lactis laccase showed resistance to NaCl. Maalej-Kammoun et al. (2009) reported inhibition of
Trametes sp. laccase for NaCl, starting from 100 mM up to higher concentrations [
53]. This suggests that the
L. lactis laccase is insensitive to this inhibitor at 1 mM.
EDC is a carboxyl-group modifying agent that has been reported to strongly inhibit the catalytic activity of
L. plantarum J16 and
P. acidilactici laccases. However, our findings showed inhibition only for
P. parvulus. The non-inhibition by EDC of
L. paracasei and
L. lactis laccases suggests the absence of carboxyl groups in the substrate binding region, as proposed for fungal laccases [
37].
Additionally, variations were observed among metal-chelating agents that facilitate the removal of bound copper ions from enzymes, including EDTA, bipyridyl, and phenanthroline. EDTA did not show any inhibitory effects on any of the three laccases due to its low affinity to copper, as with other bacterial laccases such as
L. plantarum J16 [
14], and
P. acidilactici [
13]. However, it did cause around 50% inhibition of
P. pentosaceus laccase activity toward ABTS [
3], indicating slight differences in characteristics among laccases of species belonging to the same genus. Maalej-Kammoun et al. (2009) reported the inhibition of laccase from a
Trametes sp. strain by 10 mM EDTA [
53], a concentration ten times higher than that used in our study (1 mM).
Both
P. pentosaceus and
P. acidilactici laccases displayed a similar sensitivity toward bipyridyl, a laccase inhibitor, as
L. lactis laccase [
3]. However, the
P. parvulus and
L. paracasei laccases exhibited high resistance to the same inhibitor. Phenanthroline caused a significant reduction (over 60%) in the activity of laccases from
P. parvulus and
L. paracasei, lower than that observed in
P. pentosaceus, P. acidilactici, and
L. plantarum, which showed almost complete activity loss [
14].
Given that we are examining the potential of the presented LAB laccases to degrade biogenic amines, it was insightful to assess the impacts of certain amine oxidase inhibitors. In this regard, flavine amine oxidase inhibitors, including pargyline and clorgyline, were tested. Pargyline led to a reduction in activity of approximately 40% in
L. paracasei and
P. parvulus laccases, similar to its effect on
P. pentosaceus laccase [
3]. However, it did not significantly affect
L. lactis laccase, consistent with the behavior of
L. plantarum laccase [
14]. Meanwhile, clorgyline caused a reduction in activity of around 50% for all three laccases, similar to the effect seen in
P. acidilactici [
3].
Callejón et al. (2016) found that
L. plantarum laccase has a greater ability to degrade dopamine and tyramine when compared to other biogenic amines. This is attributed to the phenolic structure present in both amines [
14]. Additionally, Xu and Fang (2019) reported that
L. fermentum recombinant laccase can degrade histamine and tyramine by 51.6% and 40.9%, respectively, as well as other BAs such as tryptamine, PEA, putrescine, cadaverine, and spermidine [
16]. Ni et al. (2022) demonstrated that a salt-tolerant, mutant recombinant laccase from
B. amyloliquefaciens effectively degraded histamine, tyramine, and putrescine by 25%, 20%, and 7%, respectively, in a 50 mM sodium phosphate buffer (pH 4.5) [
54]. Recently, Wang and colleagues (2022) reported the ability of
L. sakei recombinant laccase to degrade histamine and tyramine in the absence of mediators at various ethanol concentrations, with degradation rates of approximately 20% and 25%, respectively, when exposed to 20% ethanol, and in food matrices such as surimi and grape juice [
20].
Compared to previous investigations, the three LAB laccases in this study degraded dopamine without mediators. However, only
L. lactis laccase demonstrated significant potential to degrade tyramine, in contrast to the poor performance of
P. parvulus laccase. Additionally, these two laccases displayed potential to degrade PEA, while only
P. parvulus laccase showed potential to degrade putrescine. The two laccases’ capacity to degrade BAs was found to be comparable to that of
L. fermentum laccase [
16]. Our findings additionally demonstrate that phenolic amines are better substrates for LAB laccases compared to non-phenolic ones, as documented by Olmeda et al. (2021) [
3]. Similar to earlier research, the non-phenolic ABTS substrate improved the dopamine and tyramine degradation of both
P. parvulus and
L. paracasei LAB laccases, serving as an artificial mediator [
3,
13]. Surprisingly, the activity of
L. lactis laccase in degrading tyramine was reduced by ABTS. This phenomenon has not been observed in other bacterial laccases and requires further explanation in future studies.
It is widely acknowledged that laccases can potentially oxidize non-phenolic substrates via mediators that generate highly reactive cation radicals, enabling final substrate oxidation [
55]. Our findings indicate that epicatechin, a natural phenolic compound found in wine, was not an effective mediator for degrading biogenic amines. However, the three LAB laccases demonstrated the ability to degrade epicatechin, based on the observed increase in OD at 420 nm. To the best of our knowledge, epicatechin had not been previously investigated as a mediator for the degradation of biogenic amines. As a result, only
P. parvulus laccase retained its capacity to break down putrescine, and
L. paracasei laccase saw a minor improvement in its ability to degrade PEA when exposed to epicatechin.
In conclusion, among the three LAB tested, L. paracasei laccase exhibited the highest protein concentration and specific activity toward ABTS, while L. lactis laccase demonstrated the highest affinity toward ABTS but the lowest kcat constant on this substrate. Additionally, L. lactis laccase showed the highest resistance to a larger number of inhibitors. The artificial mediator ABTS proved to be more effective for biogenic amine degradation than the natural mediator epicatechin. L. paracasei laccase exhibited the greatest tyramine degradation in the presence of ABTS, whereas L. lactis laccase exhibited the highest degradation of this amine in the absence of mediators. Additionally, P. parvulus laccase demonstrated the capability to degrade other harmful biogenic amines like putrescine and PEA, while L. lactis degraded PEA in the absence of mediators. This study provides novel insights and perspectives regarding the utilization and implementation of recombinant bacterial laccases in diverse reaction matrices, including food and beverage production such as wine, cider, and beer.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}