Abstract
Corynebacterium glutamicum is an important industrial production strain that is widely used in amino acid fermentation, biopharmaceuticals, and other fields. It is particularly important to develop efficient genome editing methods for the targeted modification of C. glutamicum production strains. Currently, the gene editing system of C. glutamicum is inefficient and time-consuming. In this paper, we reported on a Francisella novicida (Fn) CRISPR-Cpf1-based system for genome editing. The system used linear DNA detached from the plasmid, and, with the assistance of the recombinase RecET, gene deletion was achieved by simultaneous electrotransformation of linear DNA with a plasmid carrying the FnCpf1 and crRNA expression cassette for double-strand breaks of the genome. Compared with previous all-in-one plasmids, this system reduced the time for one round of constructing recombinant plasmids and shortened the editing cycle by about 24 h. Finally, we successfully constructed an engineered strain (X−2) with high L-valine production by using the linear DNA-mediated gene deletion system. This method is of great importance for accelerating the process of metabolic engineering modification of C. glutamicum and its further application in high value-added products.
1. Introduction
Corynebacterium glutamicum, a novel chassis strain and heterologous protein-expressing host, is a green and efficient chassis microorganism for the production of numerous high value-added products such as bulk amino acids, fine chemicals, and nutraceuticals [1,2,3,4,5]. Since C. glutamicum does not produce an endotoxin itself, it is a non-pathogenic and safe strain [6] favored by researchers and producers. With the expansion of market demand, the market value of fermentation products of C. glutamicum has been increasing year by year. Given that C. glutamicum has a good protein expression system, it can be modified by metabolic engineering to screen high-yielding industrial strains and create higher economic value [7,8,9]. Since conventional strain improvement uses random mutagenesis, problems, such as growth defects, low tolerance, or the accumulation of by-products, can arise during the development of the strain [10]. With the development of molecular genetics and whole-genome sequencing, rational metabolic engineering approaches have been applied to modify specific genes in the strain genome with the help of gene editing techniques to improve the production of strain metabolites. Currently, there are still a number of challenges regarding the genetic manipulation of C. glutamicum [3,11], which lags far behind Escherichia coli. C. glutamicum mainly relies on the traditional gene editing method involving the Pk18mobsacB system [12], which is based on the kanamycin resistance gene and the levansucrase encoding gene sacB as screening markers, and it achieves traceless editing of target genes on the genome by two rounds of homologous recombination, commonly used for gene deletion, insertion, and targeted mutagenesis. However, the following problems arise in practical applications: first, because the sacB gene is prone to inactivation, it results in poor lethality and a high revertant mutation rate, thereby reducing the efficiency of the second round of screening [13]. Second, when deleting certain genes affecting growth, almost all of them revert to the original strain after the second round of exchange, making it extremely difficult to screen the target strain. Moreover, the target sites cannot be precisely selected when multi-copy integration of the genome is performed. This results in the disadvantages of time-consuming, inefficient, and complicated screening of transformants. Therefore, shortening the time of strain transformation and improving the efficiency of recombinant strain screening are urgent issues to be solved.
To address this issue, fast, developed, clustered regularly interspaced short palindromic repeat (CRISPR) genome editing technologies have been applied in C. glutamicum and other species [14]. The CRISPR interference (CRISPRi) technology was established in C. glutamicum before CRISPR-based genome editing [15]. Except for SpCas9 and FnCpf1, many CRISPR-Cas systems for microbial genome editing, such as Cas12f1 [16,17,18], have been reported on and studied. The applicable CRISPR systems in Corynebacterium glutamicum are SpCas9 and FnCpf1. FnCpf1 has greater target specificity than SpCas9. FnCpf1 is a single-stranded RNA-guided endonuclease that lacks tracrRNA and utilizes the T-rich PAM to make DNA double-stranded breaks by staggered cleavage at the ends [19,20,21]. The advantages of CRISPR/Cpf1 precision and specificity in gene editing make it more conducive to overcoming some of the limitations of CRISPR-Cas9; applications in rice, mouse, and human cells are all largely free of off-target effects [22,23,24]. The Red recombination system and the RecET recombination system are both typical recombination systems derived from E. coli. The RecET recombination system consists of two phage-encoded proteins, RecE and RecT [25]. It has been noted that integrating a single copy of recET in the genome can play a role in recombination [26]. It was also shown that plasmid overexpression of recET can be used in the study of the gene editing system of C. glutamicum ATCC14067, which led to a substantial increase in editing efficiency [24]. Based on previous studies showing that RecET plays a more effective role compared to other homologous recombinases, including Red, RecET and RecA, the editing efficiency is usually enhanced by exogenous recombinases in the C. glutamicum gene editing system [27,28]. The RecET-Cre/loxP gene editing system of C. glutamicum ATCC14067 employed the use of plasmid overexpression, Ptrc promoter, to induce the expression of recET, while adding inducers during the preparation of competent cells, resulting in a substantial increase in editing efficiency. [29] Although the integration of a single copy of recET in the genome could also provide a degree of recombination, it was cumbersome that recET needed to be deleted again before the strain could be industrially produced, thereby extending the time for strain transformation [28]. It was shown that the C. glutamicum genome lacked a typical crRNA sequence of Streptococcus pyogenes (SpCas9), causing a toxic effect of SpCas9 on cells when expressed in C. glutamicum [30]. Therefore, when using SpCas9, researchers found that nonspecific codon optimization [31], or control of SpCas9 expression was required to ensure smooth gene editing. Therefore, FnCpf1 protein became our first choice for developing C. glutamicum gene editing methods. A research team successfully constructed the CRISPR/Cpf1 system with an all-in-one single plasmid, and the efficiency of tiny DNA modification was 86–100% when single-stranded ssDNA recombination was utilized; the study also noted that when no recombinase was available, the gene editing efficiency of large fragments of double-stranded dsDNA was 5–15% [32]. Since then, some researchers have optimized a series of parameter optimizations (PAM sequence, spacer sequence, and repair template); the editing efficiency was only improved in a limited range in studies related to the absence of recombinase in this system. One study developed a multiple genome simultaneous editing strategy for the C. glutamicum ATCC14067 strain by optimizing the PAM sequence and the editable site range region CRISPR/Cpf1-RecT system, which resulted in the simultaneous editing of three genes and an editing efficiency of 93.6% when two genes were edited. The CRISPR/Cpf1-RecET system was used to delete 1 kb, 5 kb, and 20 kb DNA fragments from the genome with editing efficiencies of 79.6%, 91.3%, and 36.4%, respectively, thus achieving effective deletion of large segments of DNA in C. glutamicum [31]. Recently, our research team also developed a gene editing system for inducing double-strand breaks before recombination, which used an inducible promoter to express the FnCpf1 protein; the editing efficiency was improved by adding inducers and extending the electrotransformation recovery time (5 h) [33]. Currently, the all-in-one single-plasmid system (SpCas9/FnCpf1, sgRNA, and homologous sequences expressed in the same vector) suffers from plasmid size limitation. It was also difficult to re-express the recombinase system, and without recombinase involved in recombination, the efficiency of gene editing is extremely low. This system complicates the operation by placing homologous recombinant fragments on the plasmid, which not only makes the vector construction more difficult, but also consumes more time and labor [32].
In this study, we used a constitutive promoter to express FnCpf1, shortened the strain screening time, simplified the construction steps of the plasmid (P-Cpf1-crRNA) carrying the FnCpf1 and crRNA expression cassette for double-strand breaks (DSBs) of the genome, detached the DNA recombinant fragment (reusable) from the plasmid, and explored the feasibility of a linear DNA-mediated CRISPR-Cpf1 gene editing system. First, in order to express the exogenous recombinase protein RecET, we constructed a plasmid (P-Cpf1-crRNA) for DSBs and established a linear DNA fragment recombination-mediated CRISPR-Cpf1 system; however, studies showed that this system could only be used for gene deletion and that the editing efficiency was low. Then, in order to further improve the deletion efficiency, this study optimized the cleavage system by enhancing Cpf1 genomic cleavage using screening promoters and optimizing the crRNA spacer sequence. We optimized the gene recombination by introducing the exogenous recombinase RecET as well as by optimizing the length of the homologous arms, the amount of linear DNA added, and the recovery time, simplified the operation process, and shortened the gene editing time. Finally, the established linear DNA fragment-mediated CRISPR-Cpf1 system was successfully applied to the metabolic engineering transformation of L-valine-producing strains. Compared with the previous all-in-one single plasmid system (FnCpf1, crRNA and recombinant DNA fragment in the same vector), the system constructed in this study contains a plasmid (P-Cpf1-crRNA) for DSBs and a linear DNA fragment detached from the plasmid. As a result, this study’s advantages include a shorter editing cycle and a roughly 24 h reduction in the time needed to create a single round of recombinant plasmid. When the same deletion or integration site is selected for metabolic engineering modification, the plasmid (P-Cpf1-crRNA) for DSBs designed in this study can be saved and reused and editing operation can be completed again only by preparing different recombinant DNA fragments. Finally, we successfully constructed an engineered strain (X−2) with high L-valine production by using the linear DNA-mediated gene deletion system.
2. Materials and Methods
2.1. Materials
The primers used for gene cloning and genomic recombination are listed in Supplementary Table S1. The strains and plasmids used in this study are listed in Supplementary Table S3. PrimerSTAR® HS DNA polymerase was purchased from Takara Bio. Inc. (Dalian, China). The 2×Rapid Taq Mix and ClonExpress® II One Step Cloning Kit were obtained from Vazyme Bio. Inc. (Nanjing, China). Brain Heart Infusion Broth (BHIB) was obtained from BD Difco™ Inc. (New York, NY, USA).
2.2. Strains and Culture
The strains used in this study are listed in Supplementary Table S2. C. glutamicum ATCC13032 was cultured overnight at 32 °C and 220 rpm using brain-heart-infusion-supplemented (BHIS) medium (37 g/L BHIS medium and 91 g/L sorbitol); E. coli DH5α was used as a host for plasmid conservation using LB medium (10 g/L peptone, 5 g/L yeast extract, and 10 g/L NaCl) at 37 °C, and 220 rpm overnight; kanamycin at working concentrations of 50 μg/mL was used for E. coli and 10 μg/mL was used for C. glutamicum; chloramphenicol was used at working concentrations of 25 μg/mL for E. coli and 10 μg/mL for C. glutamicum; pectinomycin was used at working concentrations of 50 μg/mL for E. coli and 200 μg/mL for C. glutamicum.
2.3. Construction of Plasmids
Based on the principle of homologous recombination, the in vitro recombination system was prepared using linearized vectors and purified DNA fragments with the help of homologous recombinase (ClonExpress® II One Step Cloning Kit), which was transformed into E. coli DH5α competent cells by a chemical transformation method to screen the recombinant plasmids. The plasmids used in this study are listed in Table S3.
Construction of plasmid pEC-XK99E-recET carrying recET and the chloramphenicol-resistance gene: pEC-XK99E was linearized and the recET fragment was amplified with primers ET-1/ET-2 using the E. coli genome as the template.
The plasmid Pk18mobsacB-Δcgl1890::kan* was constructed to screen for the model strain (QC-1), which was used for the rapid identification of gene deletion. The Pk18mobsacB plasmid was first linearized, and the upstream and downstream homologous arms were amplified using the C. glutamicum genome as the template and primers KA-1/KA-2 and KA-9/KA-10; the overlapping fragments of promoter and kanamycin-resistance genes were amplified using Pk18mobsacB as the template and primers KA-3/KA-4 and KA-7/KA-8. The insertion sequence derived from xylA was amplified with primers KA-5/KA-6.
The plasmid pXMJ19-PlacM-Cpf1-crRNAxylA (called plc-crRNAxylA) was constructed as follows: the plasmid pXMJ19 was linearized; PUC57-Cpf1 was used as a template; the PlacM and Cpf1 fragment were amplified with primers CPF-3/CPF-2, and the original Ptac in pXMJ19 was replaced with PlacM to obtain plasmid pXMJ19-PlacM-Cpf1. The insertion of miniGene (crRNAxylA) into it yielded plc-crRNAxylA.
For the construction of plasmid pXMJ19-Ptuf-Cpf1-crRNAxylA (called ptc-crRNAxylA), the plasmid pXMJ19-Ptuf-Cpf1 was obtained by linearizing the existing pXT01 plasmid and amplifying the FnCpf1 fragment with primers CPF-1/CPF-2 using PUC57-Cpf1 as the template. The plasmid ptc-crRNAxylA was obtained in the same way as above.
Plasmids pXMJ19-Ptuf-Cpf1-crRNAΔalaT and pXMJ19-Ptuf-Cpf1-crRNAΔavtA were constructed as follows: pXMJ19-Ptuf-Cpf1 was linearized, and crRNAalaT or crRNAavtA (miniGene) were homologously recombined with it.
2.4. Preparation of Linear DNA Fragments
The model strain (QC-1) genome was used as the template, amplified with primers CZ-1/CZ-2 and CZ-3/CZ-4, overlapping PCR, and then obtained for the preparation of the deletion linear DNA fragment (534 bp exogenous sequence from xylA). The C. glutamicum genome was used as the template, with primers AT-1/AT-2; AT-3/AT-4, TA-1/TA-2, and TA-3/TA-4 were overlapped by PCR amplification to obtain the linear DNA fragments for knockdown of alaT and avtA, respectively.
2.5. Preparation and Transformation of Chemotransformed Competent Cells
E. coli DH5α competent cells were prepared by CaCl2-mediated chemotransformation. The in vitro recombinant system was prepared and then mixed with the competent cells, placed on ice for 20 min, heat-treated at 42 °C for 1 min, recovered in a shaker at 37 °C for 1 h, spread on LB medium plates with corresponding antibiotic resistance selection, incubated at 37 °C for 12 h, and then identified by PCR.
2.6. Competent Cells Preparation and Electrotransformation
Competent cells medium was prepared by dissolving 3.75 g of BHIB and 9.1 g of sorbitol (BHIS) in water (final volume of 80 mL) and sterilization at 121 °C for 20 min. Glycine (2.5 g) was dissolved in 20 mL of deionized water with 0.1 mL of Tween 80 and then added to the sterilized medium by filtration through a membrane to remove any microbial contaminants. The C. glutamicum strain preserved at −80 °C was inoculated in BHIS solid-culture plates by delineating three zones of purification and incubated overnight at 32 °C. Single colonies grown in the three zones were then inoculated into tubes of BHIS liquid medium and incubated at 32 °C and 220 rpm on a shaker for 15 h. The strains were inoculated at 1.5% inoculum into a 500 mL triangular flask of BHIS liquid medium (100 mL loading) and incubated at 30 °C and 200 rpm on a shaker for 7–10 h. When the OD600 reached 0.4–0.8, the strains were left to stand in an ice bath for 20 min. C. glutamicum strains containing pEC-XK99E-recET were incubated at an OD600 of approximately 0.1 with the addition of the inducer IPTG (1 mM). The strains were washed three times by centrifugation through 10% glycerol (pre-cooled). Finally, the strain cells were resuspended with 2 mL of 10% glycerol (pre-cooled), aliquoted, and stored at −80 °C for backup.
The constructed recombinant plasmid (500 ng)/dsDNA fragment (1 μg) was mixed with C. glutamicum competent cells and added to a pre-cooled electrotransfer cup in an ice bath for 20 min before electrotransformation at 2500 V. A 900 μL aliquot of pre-warmed BHIS recovery solution was added and heat-shocked in a water bath at 46 °C for 6 min, followed by incubation at 32 °C for 2 h at 200 rpm, and then spread onto BHIS solid plates containing antibiotics and incubated overnight at 32 °C.
2.7. RecET-Assisted CRISPR-Cpf1 Genome Editing Procedure Using Linear DNA
The two-plasmid gene editing system was established with one plasmid(pEC-XK99E-recET) having spectinomycin resistance and the other plasmid(P-Cpf1-crRNA) having chloramphenicol resistance. The plasmid P-Cpf1-crRNA was constructed to verify the cleavage system, and then prepared recombinant DNA repair fragments. Next, the plasmid pEC-XK99E-recET expressing the recombinant enzyme was constructed, and the plasmid was transformed into the strain to be modified, which was further prepared into competent cells to finally obtain Corynebacterium glutamicum containing the recombinant enzyme. For gene editing, the plasmid(P-Cpf1-crRNA) for DSBs and recombinant fragment were electrotransformed together into the starting strain competent cells, resuscitated, and spread on solid BHIS containing chloramphenicol and spectinomycin, incubated overnight at 32 °C, and colonies were identified by PCR to screen the correct target strain. Ultimately, the plasmid was discarded. The cycle time of one gene editing was N + 3 days.
2.8. Plasmid Loss
The recombinant strains containing plasmids identified by PCR were transferred to tubes without added antibiotics, incubated for approximately 4 h, then diluted and spread onto plates that did not contain antibiotics, followed by counter-spotting onto no-antibiotics plates and added to the corresponding antibiotic-containing plates and incubated overnight at 32 °C. The strains that grew on the no-antibiotics plates but not on the plates containing antibiotics were the recombinant strains with successful plasmid loss.
2.9. Determination of Genome Editing Efficiency
To facilitate the determination of genome editing efficiency, a model strain QC-1 was constructed, and a deletion of the designed DNA sequence at the genome level resulted in kanamycin-resistant strains. Therefore, the number of transformants was counted on kanamycin agar plates, and the editing efficiency was determined by calculating the proportion (%) of kanamycin-resistant cells.
2.10. Fermentor (5 L) Culture
The seed medium was prepared in a 500 mL flask at 32 °C with 30 mL of medium containing (per liter) glucose 35 g, yeast extract (Oxoid Ltd., London, UK) 5 g, corn steep liquor (Julong Ltd., Pingdingshan, China) 30 mL, KH2PO4 1.5 g/L, MgSO4·7H2O 0.5 g/L, VB1 1.0 mg/L, FeSO4·7H2O 10 mg/L, MnSO4·H2O 10 mg/L, VH 0.2 mg/L, pH 7.0–7.2, 0.1 MPa, at 121 °C, 20 min, with shaking at 220 rpm.
The fermentation medium comprised: glucose 100.0 g/L, corn pulp 35.0 mL/L, (NH4)2SO4 10.0 g/L, soybean meal hydrolysate 25.0 mL/L, KH2PO4 2.0 g/L, MgSO4·7H2O 0.7 g/L, VB1(Vitamin B1, Thiamine) 1.0 mg/L, FeSO4·7H2O 10 mg/L, MnSO4·H2O 10 mg/L, VH(Vitamin H, Biotin) 0.1 mg/L, pH 7.0–7.2, 0.1 MPa, 121 °C, 20 min.
The slant-activated organisms were connected to seed triangle flasks (1 L) and incubated at 32 °C and 220 rpm for 16 h. The primary seeds were then connected to secondary seed jars at 10%, filled with 2 L of liquid, and incubated at 32 °C until OD600 ≈ 14. The secondary seeds were connected to 5 L fermenters at 15% inoculum, and a fresh medium was added. The fermentation conditions were: a temperature of 32 °C; dissolved oxygen at 20–30%; continuous flow with 25% ammonia and a pH of approximately 7.0; a supplement of 80% glucose solution at a specific flow acceleration rate, and residual sugar 1–1.5%.
2.11. Substrate and Product Analysis
Cell growth was monitored by measuring the absorbance at 600 nm (OD600). The glucose concentration was measured using an SBA-40E biological sensor (Shandong Academy of Sciences, Jinan, China).
The prepared samples were analyzed by high-performance liquid chromatography (HPLC) using an Agilent 1100 series. For the determination of L-valine and L-alanine, the supernatant of the fermentation broth was centrifuged and passed through a 0.22 μm membrane filter. 2,4-Dinitrofluorobenzene (DNFB) was used as a derivatizer. Chromatographic separation conditions: The column (4.6 mm × 150 mm, 5 μm) was an Agilent ZORBAX Eclipse AAA (Agilent Technologies Inc., Santa Clara, CA, USA), the mobile phase was acetonitrile–sodium acetate buffer, the flow rate was 1.0 mL/min, the column temperature was 33 °C, and the detection wavelength was 360 nm.
3. Results
3.1. Construction of the Preliminary CRISPR-Cpf1 Gene Editing System
To facilitate the validation and statistical analysis of the CRISPR-Cpf1 system for C. glutamicum ATCC13032 gene editing efficiency, we first constructed the model strain (QC-1) and selected a rigorous screening marker. Commonly used screening markers are the kanamycin resistance gene (kanR) and upp (uracil phosphoribosyltransferase is encoded by the gene of upp in C. glutamicum, and it can convert 5-fluorouracil to 5-fluoro-dUMP, which inhibits thymidylate synthase, leading to cell death) [26]. When using upp as a screening marker, a CGXII basic medium is used, which is not rich enough in nutrients to meet the needs for the rapid growth of C. glutamicum, resulting in a whole culture process greater than 48 h, thus affecting the screening time and efficiency. However, when kanR is used as a screening marker, the use of a nutrient-rich BHIS medium can shorten the incubation process to approximately 18 h.
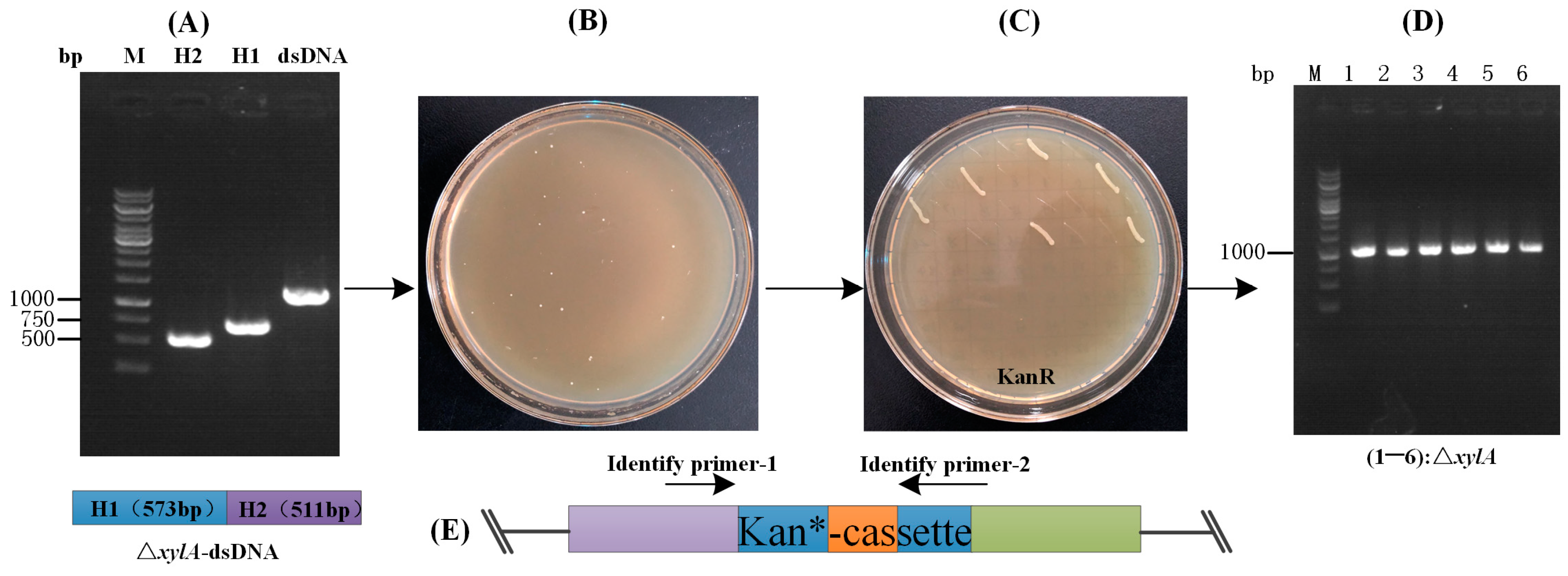
First, we designed a deletion repair strategy and constructed a model strain (QC-1) for rapid validation and statistics of gene deletion efficiency. Previously, a study designed the kanamycin resistance in the Cre/loxP system of the C. glutamicum ATCC14067 expression cassette, and when the target gene was correctly deleted, the strain exhibited kanR [27]. Therefore, drawing on the concept of this study, an insertionally inactivated kanamycin resistance gene was integrated at the cgl1890 locus of C. glutamicum, and kanamycin resistance was restored by deleting the exogenous base sequence (534 bp, derived from xylA) in the kanamycin resistance gene (Figure 1). The model strain (QC-1) can be used to screen colonies with the correct gene deletion. This is because the removal of the 543 bp DNA fragment from the kan*-cassette caused the strain to develop kanamycin resistance. In addition, we used colony PCR to verify that the results of the 543 bp DNA fragment deletion were consistent with the results of colony growth on agar plates supplemented with kanamycin, demonstrating the feasibility of using kanamycin as a screening marker for genome editing efficiency statistics. Accordingly, the purpose of constructing the model strain QC-1 and counting the number of transformants (kanR CFU) using this strain in the study was to eliminate the step of identifying positive transformants by PCR each time, which can provide a more convenient and efficient method of validating the statistics for a future series of cleavage and recombination optimization studies designed to examine whether the optimization has a positive impact on the excision effect and recombination effect of the editing system. The method was used to examine whether the optimization positively affects the cleavage and recombination of the editing system, while providing a reference basis.
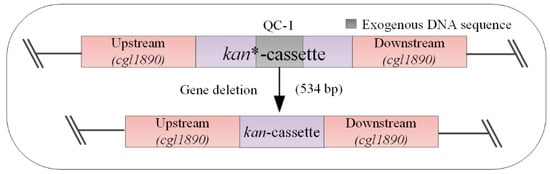
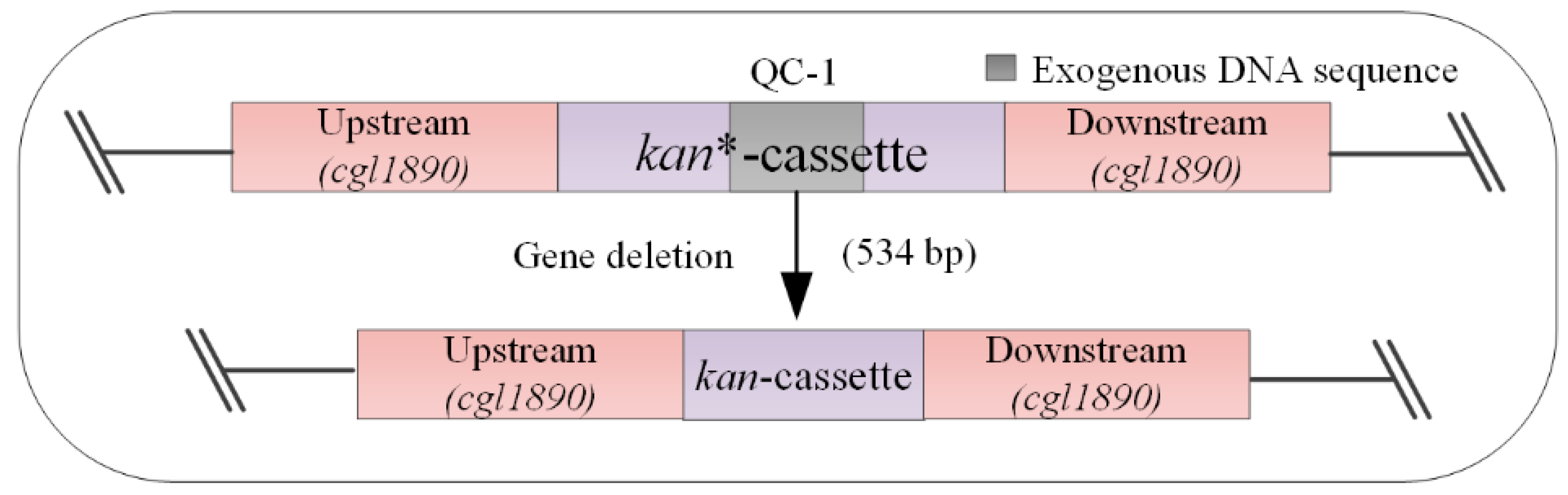
Figure 1.
Construction and working principle of the model strain (QC-1). When gene deletion is performed, the dsDNA fragment repairs the inactivated kanR gene (the gene is inactivated by insertion of an exogenous DNA sequence of 534 bp (from xylA) into the kanR gene). If correctly edited, the strain will be kanamycin-resistant and can be grown in a solid medium containing kanamycin. kanR: kanamycin resistance; kan*: inactivated kanamycin resistance gene.
The CRISPR-Cas system is guided by a segment of guide RNA that recognizes the PAM (protospacer adjacent motif) of the target gene locus, then binds to a nucleic acid endonuclease protein to achieve specific DNA-targeted cleavage and homologous recombination of double-strand break DBS (double-strand-breaks) using linear DNA template repair [30,32]. First, the efficiency of gene editing was verified in the absence of recombinase (Figure 2A). The one-plasmid pXMJ19-PlacM-Cpf1-crRNAxylA (called plc-crRNAxylA) gene editing system was first established in this study. The plasmid (plc-crRNAxylA) having chloramphenicol resistance was first constructed to verify the cleavage system following the plasmid design method shown in Figure 3A. Then, we prepared recombinant DNA repair fragments (containing an upstream homologous arm and a downstream homologous arm of approximately 500 bp) for deletion of the exogenous xylA sequence (534 bp) in the model strain (Figure 1). Competent cells of the model strain (QC-1) were also prepared. For gene editing, the plasmid (P-Cpf1-crRNA) for the DSBs and recombinant fragment were electrotransformed together into the QC-1 strain competent cells, resuscitated, and spread on solid BHIS containing chloramphenicol, incubated overnight at 32 °C, and then paired to in a solid kanamycin resistance medium, and colonies were identified by PCR to screen the correct target strain. Ultimately, the plasmid was discarded. The cycle time of one gene editing was N + 3 days. Compared with previous all-in-one plasmids, this system reduced the time for one round of constructing recombinant plasmids and shortened the editing cycle (Figure 3B). However, as verified by electrotransformation, no transformants were found to grow on the kanamycin medium plates. Similarly, in the absence of recombinase, the dsDNA editing efficiency of our experimental results was lower than that of previous researchers [32]. Our analysis hypothesized that it was the lack of recombinase facilitation in C. glutamicum that prevented the initially established CRISPR-Cpf1 system from being used for gene deletion. Therefore, in the next optimization process, we tried to establish the two-plasmid gene editing system (Figure 2B) with one plasmid(pEC-XK99E-recET) having spectinomycin resistance and the other plasmid (P-Cpf1-crRNA) having chloramphenicol resistance to promote recombination and improve the editing efficiency [13].
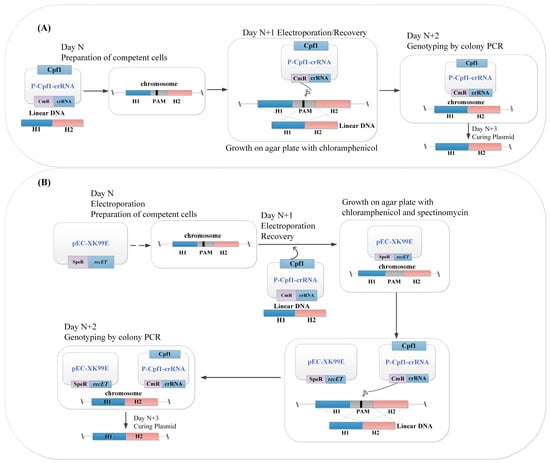
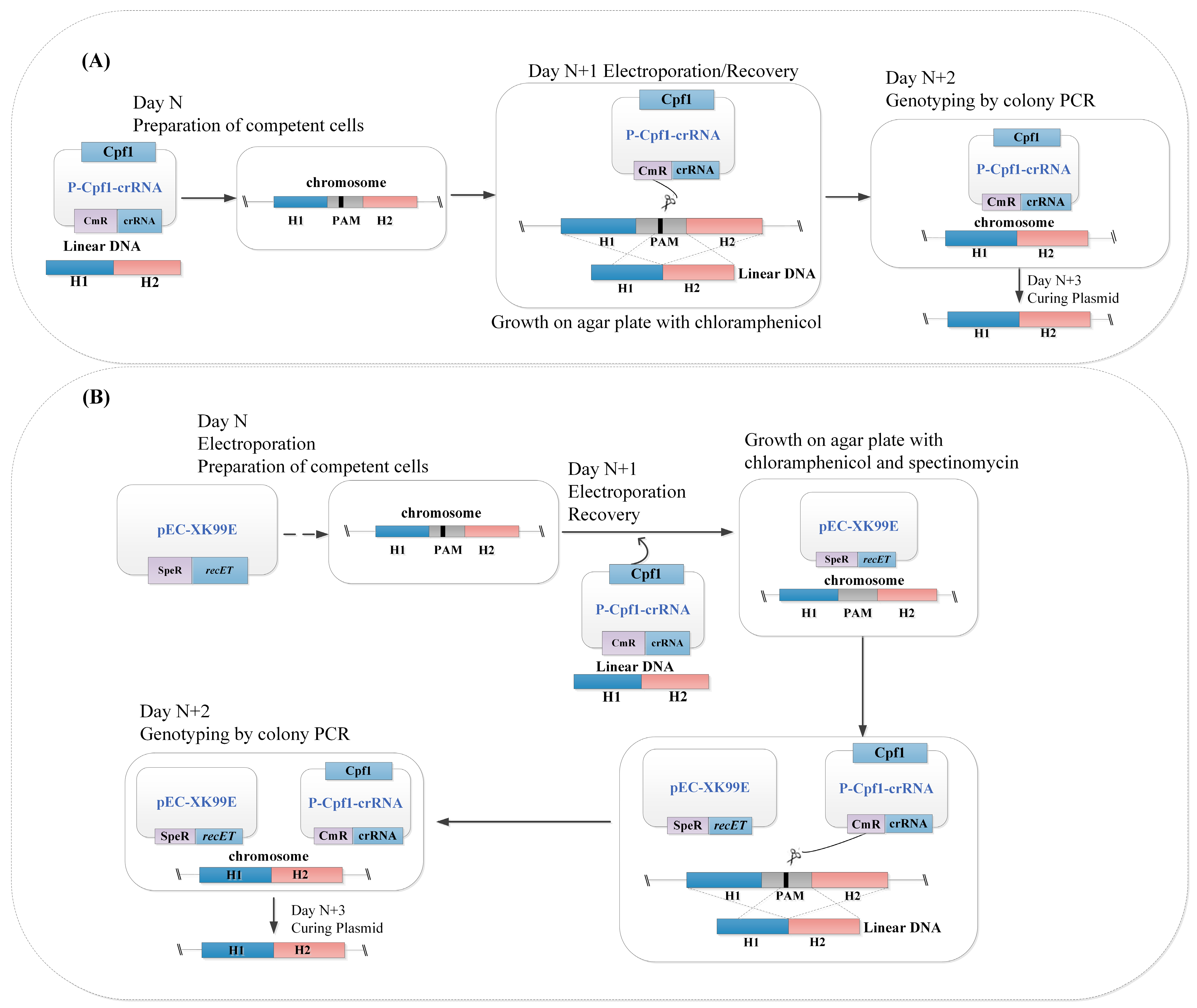
Figure 2.
(A) Flowchart of the one-plasmid (P-Cpf1-crRNA) gene editing system; and (B) flowchart of the RecET-assisted CRISPR-Cpf1 gene editing system using linear DNA in C. glutamicum. Plasmid P-Cpf1-crRNA was electrotransformed simultaneously with linear DNA into competent cells induced to express recET, and other operations were performed as above. RecET: recombinant and repair proteins derived from E. coli. P-Cpf1: expression of Cpf1 using promoter Ptuf or PlacM; speR: spectinomycin resistance; cmR: chloramphenicol resistance. H1/H2: upstream/downstream homologous regions, respectively; crRNA: a matured CRISPR RNA that contains 24 bp targeting sequences; Cpf1: Cpf1 derived from F. novicida (NC_008601); PAM: a T-rich protospacer adjacent motif; N: preparation time of competent cells.
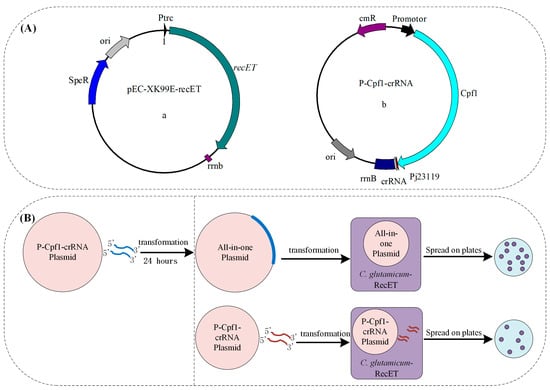
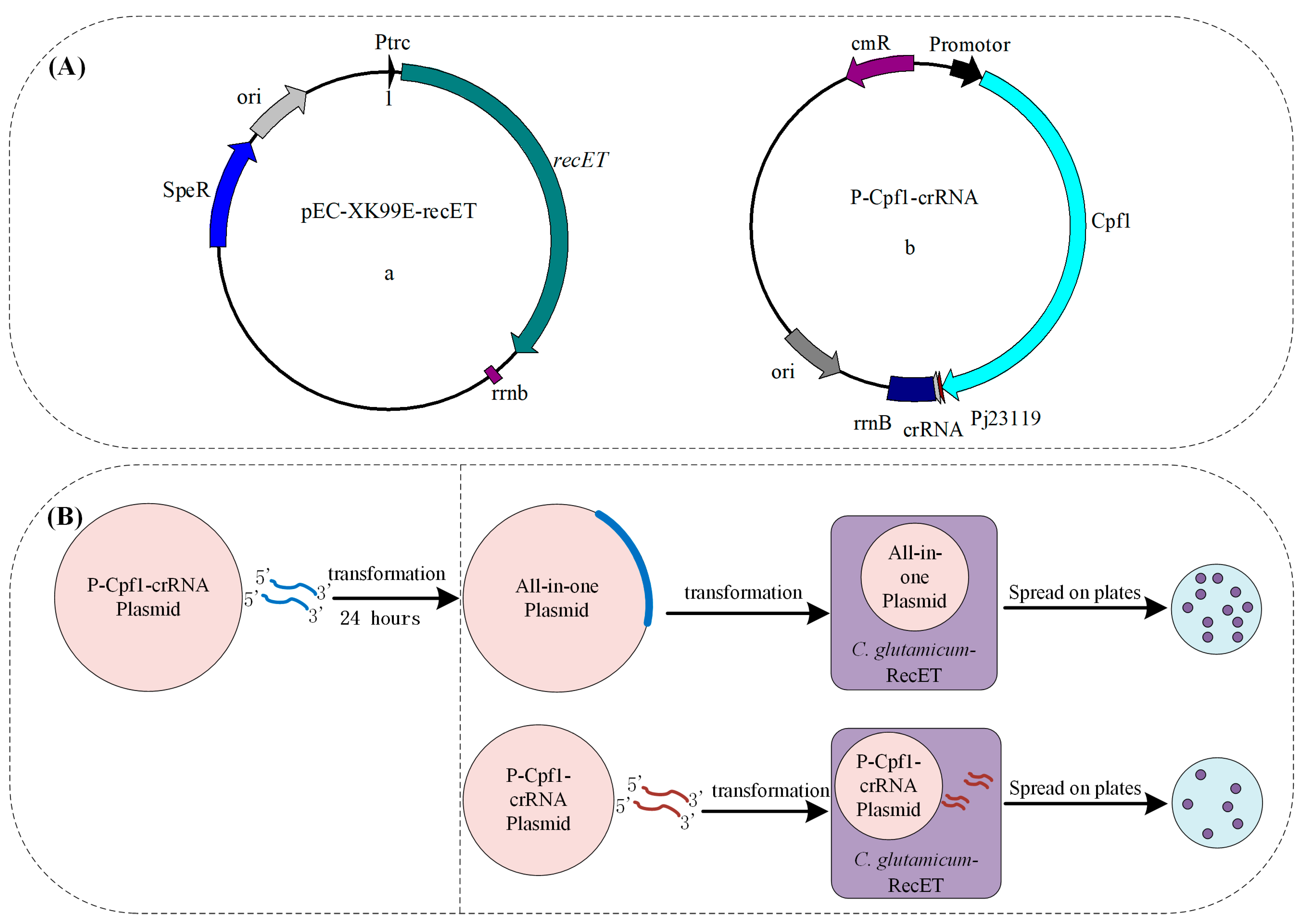
Figure 3.
(A) Schematic diagram of the plasmid for induced expression of recombinase RecET (a); the plasmid (P-Cpf1-crRNA) carrying the FnCpf1 and crRNA expression cassette for DSBs that can be used with linear DNA for genome editing (b). Pj23119: a synthetic constitutive expression promoter (B). Compared with previous all-in-one plasmids, this system reduced the time for one round of constructing recombinant plasmids and shortened the editing cycle.
3.2. Optimization of the CRISPR-Cpf1 Gene Editing System
The factors affecting the gene editing efficiency of the CRISPR-Cas system mainly include the cleavage system and the recombination system [34].
3.2.1. Optimization of the Cleavage System
In the CRISPR-Cas system, the Cas-crRNA complex binds target DNA to induce DNA DSBs, which are lethal to most strains that lack a non-homologous end-joining mechanism and hence lead to a dramatic decrease in cell survival [28]. Therefore, the targeting efficiency of crRNA cleavage can be reflected by the number of surviving transformants in strain cells [24]. The CRISPR system can use DSBs as a selection pressure. If a low-targeting efficiency crRNA is selected it will lead to a high false-positive rate, resulting in the survival of many wild-type cells in the population. Therefore, it is important to use a recombinant plasmid with a good cleavage effect to improve the cleavage system of the target site. In this study, the promoter Pj23119 and crRNA were inserted in the vector pXMJ19 to construct a plasmid (P-Cpf1-crRNA) for DSBs. The cleavage system was improved by promoter optimization and crRNA optimization, and the plasmid was transformed into a model strain (QC-1). The number of surviving transformants (colony-forming units, c.f.u) was used to measure whether the plasmid had a good cleavage system, thus further improving the screening efficiency of the target strain.
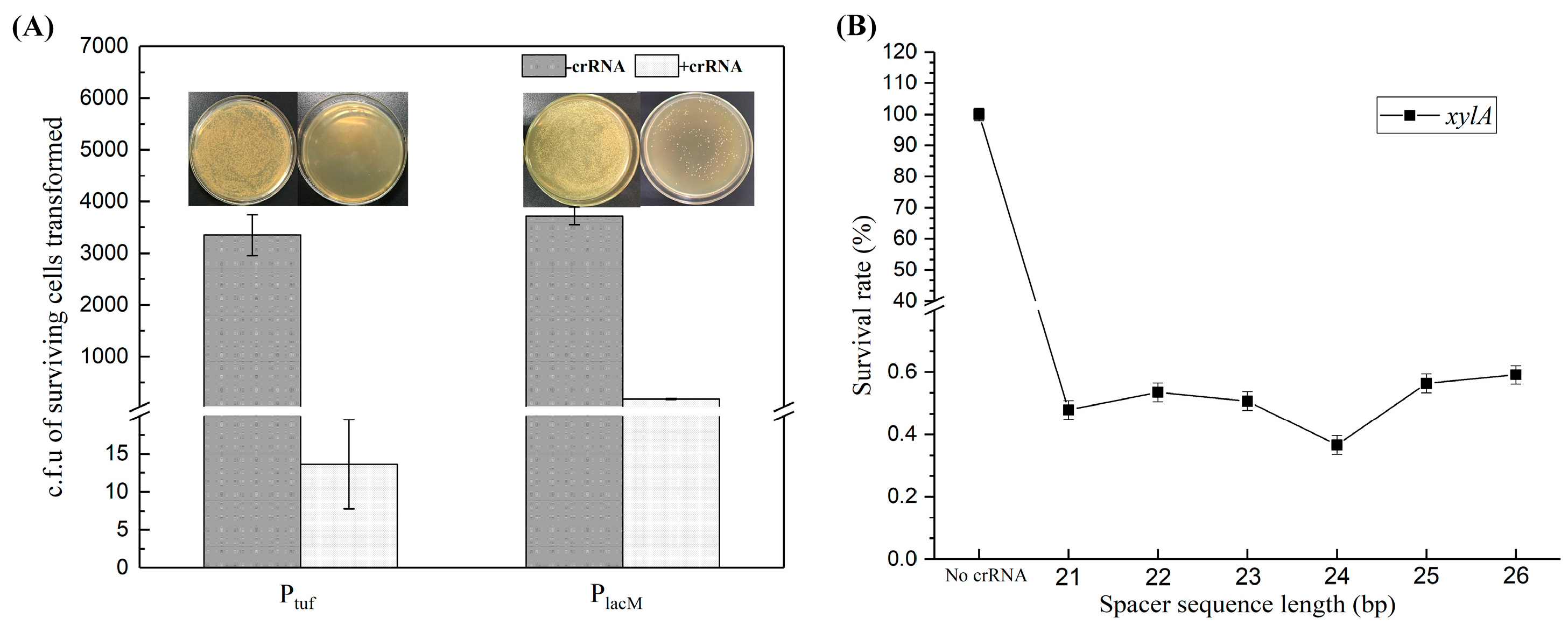
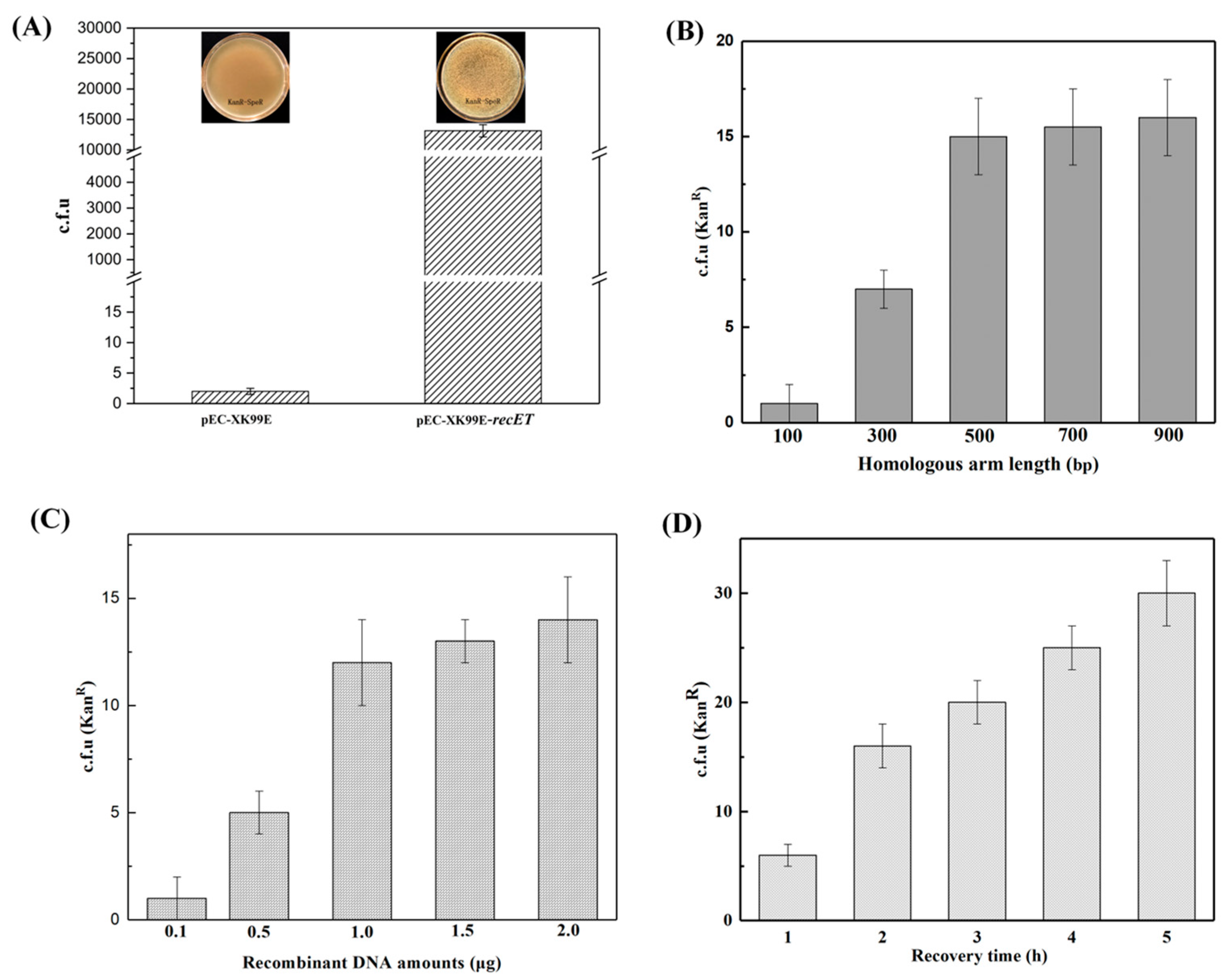
First, the promoter for expressing FnCpf1 protein was optimized. We selected the strong promoter Ptuf to replace the PlacM promoter to construct the plasmid pXMJ19-Ptuf-Cpf1-crRNAxylA (called ptc-crRNAxylA) [28], and we used pXMJ19-Ptuf-Cpf1 without crRNA and pXMJ19-PlacM-Cpf1 as the control group. The two plasmids were transformed into the model strain (QC-1) competent cells to compare the above two plasmids. The experimental results showed that the number of surviving cells (c.f.u) in the Ptuf and PlacM control groups were 3.35 × 103 ± 397 and 3.72 × 103 ± 171, respectively (Figure 4A). This indicates that the plasmid pXMJ19-PCpf1 does not have a cleavage role in the absence of crRNA; while in the presence of crRNA, the number of surviving cells in the Ptuf and PlacM groups decreased dramatically to 14 ± 6 and 185 ± 10, respectively (Figure 4A). It can be seen that both ptc-crRNAxylA and plc-crRNAxylA could have a cleavage role, but the number of transformants that missed cleavage in the Ptuf group was significantly less than that in the PlacM group, the cleavage effect of the former was more significant, and the Ptuf promoter exhibited a good cleavage effect, indicating that the Ptuf promoter was better for the expression of FnCpf1.
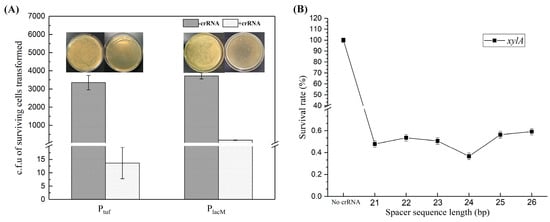
Figure 4.
Improved cleavage system by promoter optimization (A); and crRNA optimization (B). The number of surviving transformants was used to measure whether the plasmid had a good cleavage system. c.f.u: colony-forming units. (A) Comparison of the cutting effect of plasmids pXMJ19-Ptuf-Cpf1-crRNAxylA and pXMJ19-PlaM-Cpf1-crRNAxylA. To optimize the cleavage system and to verify the effect of promoters tuf and lacM on the expression of Cpf1, the number of transformants with and without crRNA was determined in plates without antibiotics, respectively. (B) The spacer sequence-length optimization of crRNAs for FnCpf1-mediated gene editing in C. glutamicum on target gene xylA with different spacer sequences. The spacer sequences are also shown in Supplementary File: Table S4.
To determine the optimal length of the crRNA spacer sequence, crRNAs targeting C. glutamicum xlyA genes were constructed with 21–26 bp spacers (Figure 4B). A series of crRNAs in the presence of the 5′-TTTA-3′ PAM sequence, were designed to cleave xlyA for measuring the cleavage efficiency of the CRISPR-Cpf1 system. The crRNAs containing 21–26 bp spacer sequences were inserted into the ptc-crRNAΔxylA vector. Then, the C. glutamicum cells containing the ptc-crRNAΔxylA were spread on plates in the absence of antibiotics. The targeting effect of the crRNA can be calculated based on the number of colonies. As shown in Figure 4B, the survival rate of cells with the crRNA targeting xlyA was 0.37 ± 0.05% when the spacer length was 24 bp, while survival rates of cells with other crRNAs containing 21–23 bp or 25–26 bp spacer sequences were 0.48–0.59%. The results showed that a 24 bp spacer sequence had the highest efficiency for gene editing using FnCpf1.
3.2.2. Optimization of the Recombination System
When DSBs are unable be repaired, we can use a donor DNA as the repair template by homologous recombination (HR) [28]. Therefore, when target gene deletion is performed, recombination repair with dsDNA as a donor is required. In response to the low gene editing efficiency in study 2.1, this study comprehensively optimized the gene editing system constructed above to improve the editing efficiency of linear DNA by introducing the exogenous recombinase RecET as well as by optimizing the length of the homologous arms, the recombinant DNA amounts, and the recovery time to promote gene recombination.
First, we optimized the recombination system using recombinase RecET. The phage recombinant protein recombination system is a technique that promotes homologous recombination of linear DNA or oligonucleotides. The gene editing system of C. glutamicum can also improve the efficiency of gene editing with the help of recombinase. In the gene editing system of C. glutamicum, RecET protein has been mostly studied for its efficient recombination role [13]. Additionally, the recombination system is influenced by various other factors, such as the recombinant DNA amounts for electrotransformation, the length of the designed homologous arm, and the electrotransformation recovery time [2]. Since the endogenous recombination pathways in C. glutamicum are inefficient, it is hard to accomplish gene deletion without the introduction of recombinases in the C. glutamicum genome. Some studies have shown that the presence of RecET in competent cells dramatically increased transformant survival and improved gene editing efficiency [31]. Hence, we constructed plasmid pEC-XK99E-recET expressing recombinase and induced expression of recET with the Ptrc promoter and transformed this plasmid into the strains to be modified. The strains were further prepared into competent cells, and C. glutamicum capable of expressing the recombinant enzyme was obtained in order to improve the efficiency of its gene editing (Figure 3A). Then, we prepared the competent cells of the model strain (QC-1) containing pEC-XK99E-recET (the inducer was isopropylthio-β-D-galactoside (IPTG), 1 mM). In verifying the recombinogenic effect of RecET, donor DNA was electrotransformed into competent cells of the model strain (QC-1) containing RecET, and the number of transformants in the presence or absence of recombinase was counted and compared (Figure 5A).
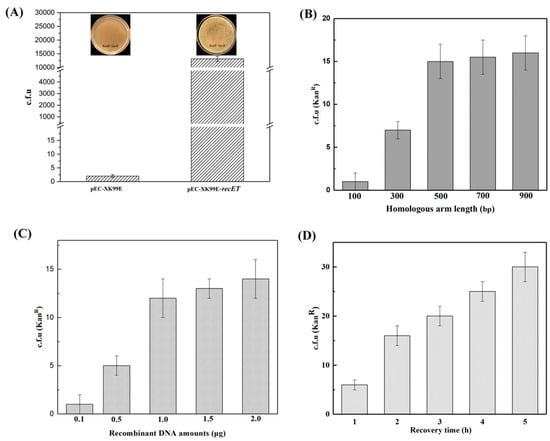
Figure 5.
To improve the editing efficiency of linear DNA, gene recombination was promoted by introducing the exogenous recombinase RecET (A); and optimizing the length of the homologous arm (B); the recombinant DNA amounts (C); and the strain recovery time (D).
Second, we optimized the homologous arm length [1]. The length of the homology arm of the repair template affects the efficiency of homologous recombination to a certain degree [1,35]. In this study, homologous arms of different sizes (100, 300, 500, 700, and 900 bp) were selected for overlapping PCR in the construction of linear DNA repair templates. First, competent cells of the model strain (QC-1) containing the recombinant enzyme RecET were prepared, and 1 μg of linear DNA fragment and 500 ng of plasmid (pCpf1-crRNAxylA) for DSBs were simultaneously electrotransformed into competent cells of the model strain (QC-1) containing RecET (pEC-XK99E-recET) according to the gene editing procedure (Figure 2B). The number of transformants on medium plates containing kanamycin showed an increasing trend upon increasing the length of the homologous arm of the repair template. A sufficient number of transformants could be sufficient for further screening of the target strains. Therefore, the optimal homology arm length of 500 bp was determined based on the experimental results (Figure 5B).
Then, the amount of linear DNA addition was optimized. We selected the optimal homology arm length of 500 bp to construct the linear DNA. Transformation was performed as above, and the plasmid (pCpf1-crRNAxylA) for DSBs was transformed into RecET-containing competent cells of the model strain (QC-1) together with different recombinant DNA amounts (0.1, 0.5, 1.0, 1.5, and 2.0 μg). The number of transformants on kanamycin medium plates was counted in five groups to verify the effect of the amount of repair template addition on recombination system. The experimental results are shown in Figure 5. The number of transformants on kanamycin plates increased with the recombinant DNA amounts. The optimal amount of linear DNA addition was ultimately determined to be 1.0 μg (Figure 5C).
Finally, we optimized the recovery time of electrotransformation. After electrotransformation, the recovery time of the strain in a liquid medium affects the recombination system between the linear DNA and the target site. According to the results of the above study, linear DNA (1 μg recombinant DNA amounts, 500 bp homologous arm) and a plasmid (pCpf1-crRNAxylA) for DSBs were transformed simultaneously into RecET-containing competent cells of the model strain (QC-1). The number of transformants on kanamycin plates at different recovery times was counted. The experimental results are shown in Figure 5. The number of transformants with kanamycin resistance on the plates increased with the increase in the resuscitation time. The choice of 4 h was sufficient for the actual bacteriophage recovery (Figure 5D).
3.3. Statistics of the Linear DNA-Mediated CRISPR-Cpf1 Gene Editing System
Using the facilitation effect of RecET and Ptuf on the CRISPR-Cpf1 gene editing system, this study aimed to perform gene deletion with linear DNA detached from plasmids (Figure 2B). Based on the results of the above optimization study, a linear DNA with a homology arm of 500 bp was prepared and electrotransformed into competent cells of strain QC-1 without recET, and no colonies were found to grow in plates of medium containing kanamycin after nightly incubation. while electrotransformation into competent cells containing recET resulted in approximately 4 × 104 transformants. This indicates that the presence of recET greatly facilitates the genetic recombination performed by the linear DNA fragment. Next, the plasmid (Ptc-crRNAxylA) for DSBs was constructed (Figure 3A). The linear DNA (1 μg) and Ptc-crRNAxylA plasmid (500 ng) were electrotransformed together into competent cells of the model strain (QC-1) containing pEC-XK99E-recET, resuscitated, and then spread on chloramphenicol-resistant plates, incubated overnight, and single colonies (Figure 6B) were paired onto kanamycin resistance plates to calculate the gene deletion efficiency (Figure 6C). Finally, the colonies were identified by PCR and screened for the correct positive strain (Figure 6D). The editing efficiency of Ptc-crRNAxylA was 30.4 ± 1.6% (Table 1). However, the experimental results showed that the linear DNA could not be successfully genomically integrated. Therefore, the method can only be used for gene deletion in C. glutamicum. Then, to verify the application of this system, L-valine-producing strains were constructed.
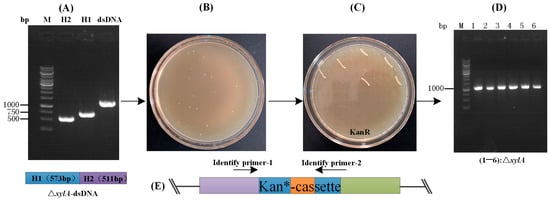
Figure 6.
Validation of the gene deletion efficiency of the optimized CRISPR-Cpf1 system: (A) preparation of linear DNA: upstream homology arm H1 (573 bp) and downstream homology arm H2 (511 bp) of the ΔxylA locus; (B) linear DNA-mediated gene deletion of ΔxylA; (C) screening of recombinant strains after ΔxylA using kanR as a screening marker; and (D) primers JD-1/JD-2 were used to verify kanR strains by PCR, and the recombinant strains were then sequenced. M: DNA marker; 1–6: Target strains. (E) purple indicates “Upstream (cgl1890)”, green indicates “Downstream (cgl1890)”, blue indicates kan-cassette, and orange indicates exogenous DNA sequences.

Table 1.
Editing efficiency of the model strain (QC-1).
3.4. Construction of an L-Valine-Producing Strain
L-alanine is a by-product of the fermentation process of C. glutamicum strain XQ. Two L-alanine synthesis pathways exist in C. glutamicum for the conversion of pyruvate to L-alanine catalyzed by either alanine aminotransferase alaT or avtA [36]. Therefore, we started from a mutagenic strain XQ with high L-valine production and double deletion of both alaT and avtA to block the L-alanine synthesis pathway and reduce the consumption of the key substrate pyruvate by L-alanine (by-product).
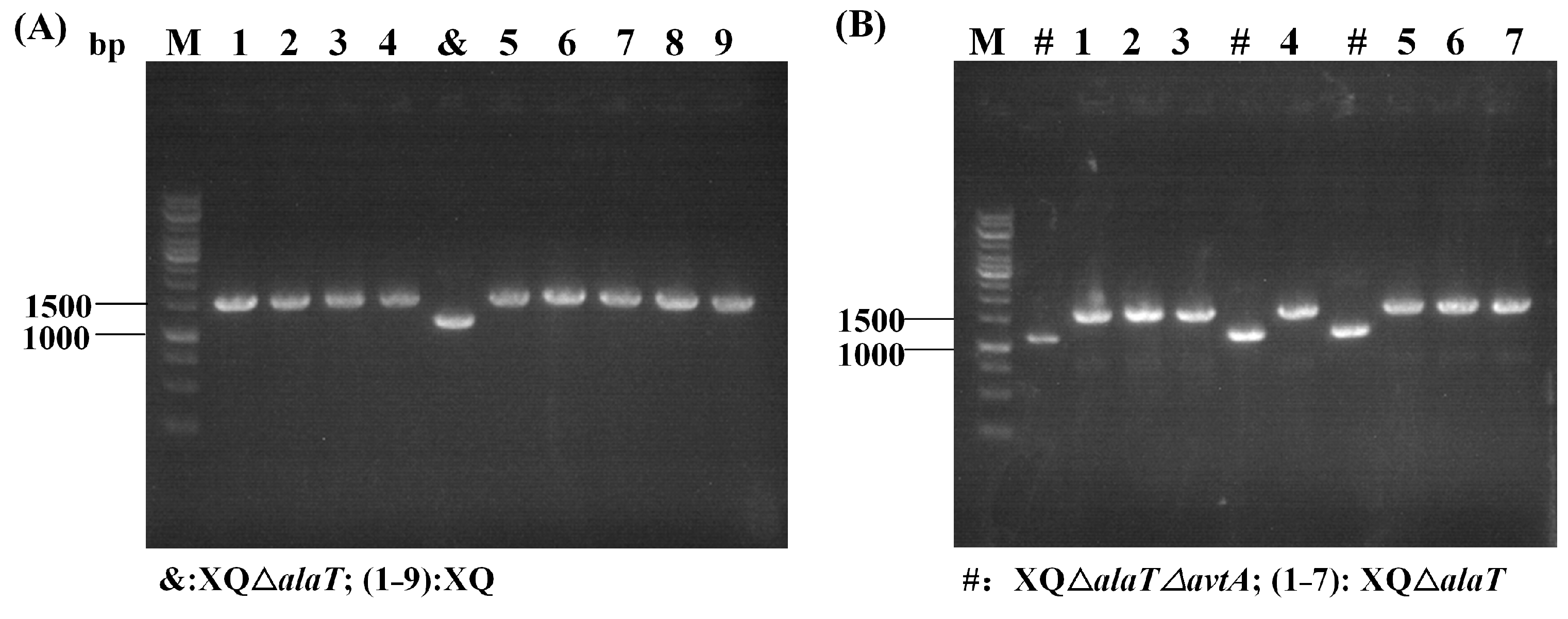
During the transformation of the strain, the RecET-assisted CRISPR-Cpf1 gene editing system established in this study was utilized. Two L-valine recombinant strains, XQΔalaT (X−1) and XQΔalaTΔavtA (X−2), were constructed using the strategy of deletion with linear DNA fragments and plasmid (P-Cpf1-crRNA) for DSBs, and the genetic screening efficiency was determined to be 10% and 30%, respectively (Figure 7 and Table 2). The correct target strains were then sequenced, and the two plasmids pEC-XK99E-recET and pXMJ19-Ptuf-Cpf1-crRNA were lost. Finally, the recombinant strain X−2 was verified by fermentation in a 5 L fermenter, and the L-valine yield was measured.
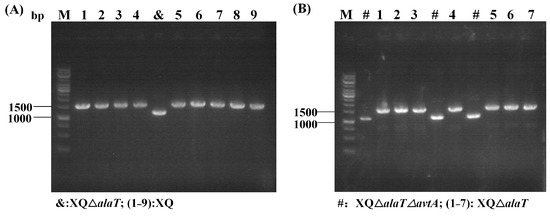
Figure 7.
Gene editing efficiency for the construction of L-valine recombinant strains: (A) screening of target strain XVΔalaT (X−1); and (B) screening of target strain XVΔalaTΔavtA (X−2); M: DNA marker.
Table 2.
Editing efficiency of strains X−1 and X−2.
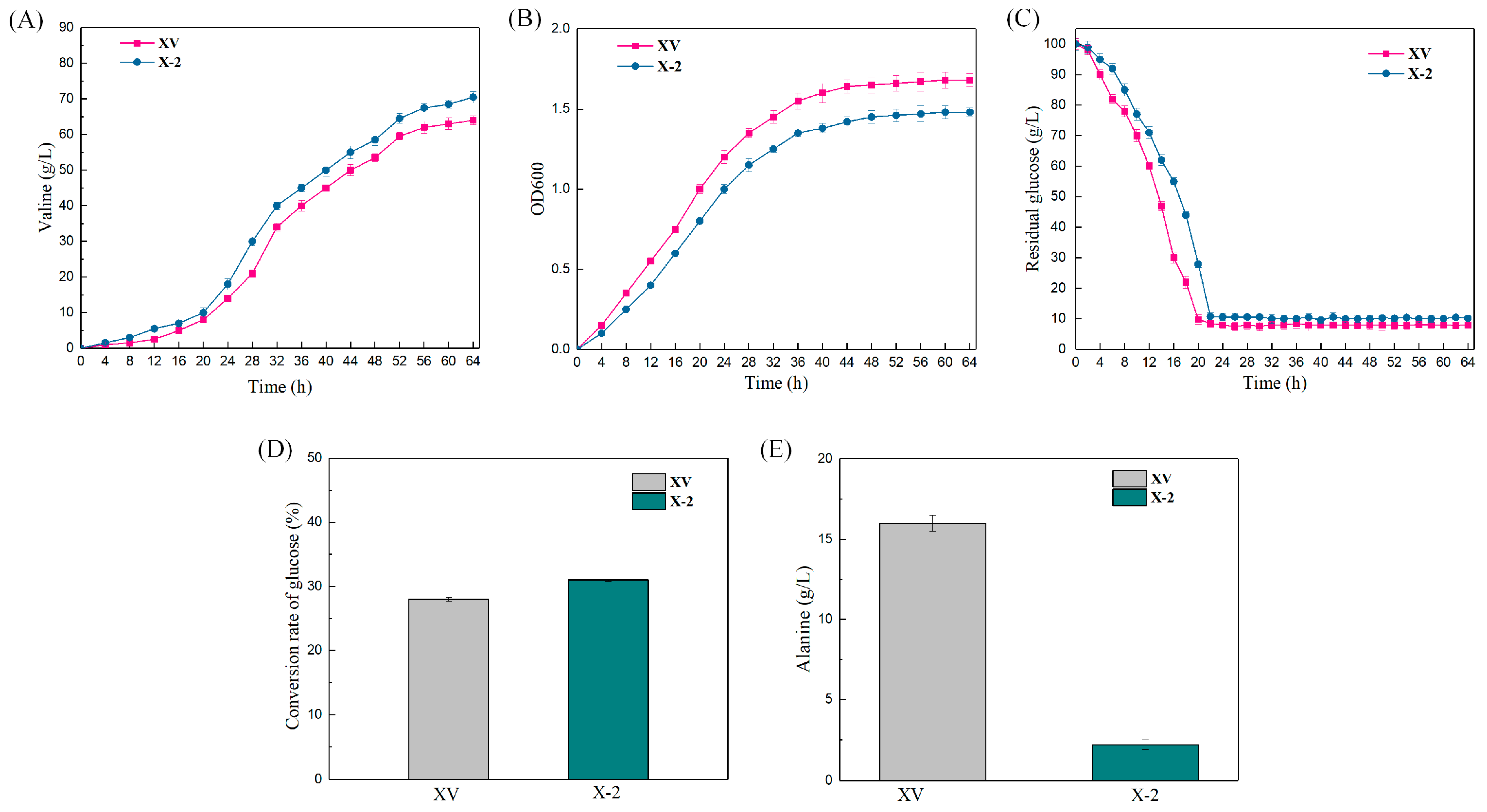
The production of L-valine, accumulation of L-alanine (by-product), OD600 of strain concentration, residual and sugar and sugar–acid conversion in the fermentation broth of XQ and XQΔalaTΔavtA (X−2) were measured by batch replenishment fermentation for 64 h (Figure 8). The experimental results showed that the trends of the fermentation process curves of strains XQ and X−2 were essentially the same, and the logarithmic growth period of the strain was 0–40 h. The growth of the strain stabilized after 40 h, but the growth rate of the genetically modified strains was reduced (Figure 8B). The content of glucose in the fermentation broth of strains XQ and X−2 was measured, and the amount of residual sugar decreased rapidly; after 20 h and 24 h, respectively, the residual sugar was less than 1%. Then, the residual sugar was controlled at 1–1.5% by the flow addition of an 80% glucose solution (Figure 8C). When alaT and avtA were double-deleted simultaneously, the accumulation of L-alanine (by-product) of strain X−2 was reduced from 16 ± 0.5 g/L to 2.2 ± 0.3 g/L (Figure 8E), which greatly promoted the synthesis of L-valine. After the growth of the strain entered the stable phase, L-valine production began to increase rapidly. The production of L-valine reached a maximum at 64 h. The L-valine production of X−2 increased from 64.0 ± 1.2 g/L to 70.5 ± 1.6 g/L, an increase of 10.2% (Figure 8A), and the sugar–acid conversion rate of strain X−2 also increased to 31.0% (Figure 8D). Thus, the linear DNA was used for gene deletion. The linear DNA can successfully construct industrial production strains, while the constructed L-valine recombinant strain can be used for industrial production.
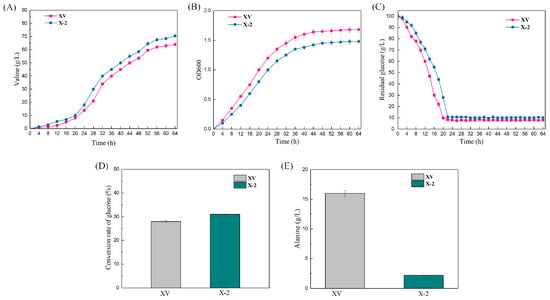
Figure 8.
Fermentation of recombinant L-valine-producing strains. The CRISPR-Cpf1 system established in this study was used to edit the genome of the starting strain XV to improve the efficiency of L-valine synthesis. The fermentation was carried out in a 5 L fermenter with supplemental fractionation, and the L-valine production (A); the biomass (optical density (OD) at 600 nm) (B); residual sugar amount (C); glucose conversion (D); and by-product L-alanine content (E) of the fermentation broth of the departure strains XV and X−2 (XVΔalaTΔavtA) were measured. The data were obtained from three parallel experiments, and the error bars represent standard deviations.
4. Discussion
CRISPR technology has been widely used as an efficient gene editing tool for the transformation of industrial microbial strains such as E. coli and Bacillus subtilis [37,38]. Currently, studies on the C. glutamicum CRIPSR-Cpf1 system have shown that the all-in-one single-plasmid system, which places FnCpf1, crRNA, and upstream and downstream homologous arms on the same plasmid, can be used for deletion and integration of large segments [32]. In this study, we introduced RecET recombinase to optimize the recombination system and constructed a RecET-mediated CRIPSR-Cpf1 gene editing system that can be used for efficient gene deletion. RecET plays a crucial role in homologous recombination of linear DNA. Rational expression of FnCpf1 can largely reduce the number of transformants with missed cuts.
For the expression of FnCpf1, we also tried dual plasmids expressing FnCpf1 and crRNA separately in the previous experiments. However, when the plasmids carrying FnCpf1 were transformed into the target strain, prepared into competent cells, and then subjected to gene editing, it was found that the number of missed cut false-positive transformants increased substantially, which seriously affected the cleavage lethality, probably due to the mutation of FnCpf1 gene resulting in gene-reduced editing efficiency [39]; Furthermore, when two plasmids carrying FnCpf1 and carrying crRNA were transformed simultaneously, the efficiency of electrotransformation decreased substantially, which could also negatively affect gene editing [40]. Therefore, to ensure a high transformation efficiency, this gene editing system was ultimately constructed by co-expression (co-transformation) of FnCpf1 and crRNA on the same plasmid. Compared with the existing gene editing methods of C. glutamicum [12,13,32], the constructed CRIPSR-Cpf1 system has the advantages of simplifying the operation process and improving the editing efficiency. The previously reported gene deletion or integration efficiencies of Corynebacterium glutamicum using the all-in-one single plasmid system were only 5–15% [32]. In contrast, the linear DNA recombineering system of this study increased the gene deletion efficiency to 30.4 ± 1.6%. The limitation of this system is that it can only be used for gene deletion and not for gene insertion, which needs to be further improved. The gene editing efficiency of the present study was relatively low compared to the two-plasmid system in which the repair template was attached to the plasmid [31], likely because a restriction repair system may have degraded the linear repair template prior to the genome repair process. Therefore, the method has broad application prospects for the transformation of C. glutamicum genomic strains.
In the absence of RecET, it is clear that relying only on the increase in plasmid copy numbers to promote homologous recombination cannot play a significant role in the improvement of gene editing efficiency. However, when RecET was added, gene recombination was promoted with the help of the recombinase RecET. When a single copy of recET is integrated into the genome, although it can play a limited role in recombination, it is necessary to delete the recET gene again before proceeding to industrial production of the strain, which will prolong the time of strain transformation [28].There are also studies that have greatly improved the efficiency of C. glutamicum genome editing with the induced expression of recET [27,28]. However, if recET is designed on the all-in-one plasmid, the number of bases of the plasmid exceeds 1.5 × 104 bp, which can affect the electrotransformation efficiency. Therefore, we used the Ptrc promoter to induce the expression of recET and constructed the vector pEC-XK99E-recET.
The gene editing efficiency also varied depending on the use of different ways of expressing FnCpf1. A previous study integrated the SpCas9 protein coding sequence in the genome and then deleted the protein after the strain was transformed, leading to a complicated operation [28]. In addition, when the SpCas9 protein was placed in the competent cells for expression, it was prone to mutation, which greatly reduced the cleavage effect [39]. Therefore, in this study, we expressed FnCpf1 with promoters Ptuf and PlacM, respectively, and the effect of Ptuf on cleavage lethality was much greater than that of PlacM. Given the good performance of RecET and Ptuf in gene editing, both were introduced into the linear DNA-mediated CRIPSR-Cpf1 system for gene deletion. It is undeniable that the current all-in-one system can be successfully used for gene deletion and integration, while the linear DNA system established in this study can only be used for gene deletion; the editing efficiency of the former is higher than that of the latter. However, there is ample scope for further improvement of the linear DNA-mediated CRIPSR-Cpf1 system. The use of linear DNA is still the focus of future development and optimization of the CRIPSR-Cpf1 system, because the use of recombinant fragments of the vector can reduce the number of plasmid constructs and the already constructed plasmid (P-Cpf1-crRNA) for DSBs can be reused in subsequent strain constructs, thus shortening of the strain screening time. Since the gene editing system in this study is limited to gene deletion, we should study the cleavage system and the recombination system more deeply, including recombinase from different sources and species, or for PAM, crRNA, etc., to lay a foundation for the future realization of genome insertion, and to further improve the editing efficiency of the CRISPR-Cas system in Corynebacterium glutamicum. In this study, the CRISPR-Cpf1 system was successfully applied to the transformation of L-valine-producing strains of C. glutamicum with the help of RecET, using linear DNA fragments for gene deletion, which reduced L-alanine (by-product) and increased the yield of L-valine. The development of efficient and convenient genome editing technologies for C. glutamicum has become a hot research topic. In addition to accelerating the process of metabolic engineering transformation of C. glutamicum, this should also have far-reaching implications in the application of high value-added products such as amino acids.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/fermentation10010031/s1. Sequence for the model strain (QC-1), Sequence for alaT and avtA, Table S1: Primers used in the study, Table S2: Strains used in the study, Table S3: Plasmids used in the study, Table S4: Spacer sequences used in the study of the crRNA optimization on target gene xylA.
Author Contributions
Conceptualization, T.W., D.H., X.J. and S.L.; methodology, T.W., D.H. and Q.X.; software, S.G., L.H. and T.W.; validation, T.W. and D.H.; formal analysis, T.W. and X.J.; investigation, S.G. and J.Z.; writing—original draft preparation, T.W.; writing—review and editing, T.W. and D.H.; supervision, T.W.; project administration, D.H.; funding acquisition, T.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Science Foundation of Henan Province (222300420149).
InstitutionaL Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the datasets presented in this study are included in the article and supplementary materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bao, Z.; Cartinhour, S.; Swingle, B. Substrate and target sequence length influence RecTE(Psy) recombineering efficiency in Pseudomonas syringae. PLoS ONE 2012, 7, e50617. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Giesselmann, G.; Hoffmann, S.L.; Wittmann, C. Corynebacterium glutamicum for Sustainable Bioproduction: From Metabolic Physiology to Systems Metabolic Engineering. Adv. Biochem. Eng. Biotechnol. 2018, 162, 217–263. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, J.; Al Makishah, N.H.; Sun, X.; Wen, Z.; Jiang, Y.; Yang, S. Advances and Perspectives for Genome Editing Tools of Corynebacterium glutamicum. Front. Microbiol. 2021, 12, 654058. [Google Scholar] [CrossRef] [PubMed]
- Wendisch, V.F.; Bott, M.; Eikmanns, B.J. Metabolic engineering of Escherichia coli and Corynebacterium glutamicum for biotechnological production of organic acids and amino acids. Curr. Opin. Microbiol. 2006, 9, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Sun, G.; Zhang, X.; Nie, Q.; Zhao, Y.; Yang, J. Recent advances, challenges and metabolic engineering strategies in the biosynthesis of 3-hydroxypropionic acid. Biotechnol. Bioeng. 2022, 119, 2639–2668. [Google Scholar] [CrossRef] [PubMed]
- Kallscheuer, N.; Vogt, M.; Stenzel, A.; Gatgens, J.; Bott, M.; Marienhagen, J. Construction of a Corynebacterium glutamicum platform strain for the production of stilbenes and (2S)-flavanones. Metab. Eng. 2016, 38, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Gordillo Sierra, A.R.; Alper, H.S. Progress in the metabolic engineering of bio-based lactams and their omega-amino acids precursors. Biotechnol. Adv. 2020, 43, 107587. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, H.U. Systems strategies for developing industrial microbial strains. Nat. Biotechnol. 2015, 33, 1061–1072. [Google Scholar] [CrossRef]
- Srivastava, P.; Deb, J.K. Gene expression systems in corynebacteria. Protein Expr. Purif. 2005, 40, 221–229. [Google Scholar] [CrossRef]
- Vertes, A.A.; Inui, M.; Yukawa, H. Manipulating corynebacteria, from individual genes to chromosomes. Appl. Environ. Microbiol. 2005, 71, 7633–7642. [Google Scholar] [CrossRef]
- Yang, J.; Ma, X.; Wang, X.; Zhang, Z.; Wang, S.; Qin, H.; Mao, S.; Lu, F. Advances in gene editing of Corynebacterium glutamate. Sheng Wu Gong Cheng Xue Bao 2020, 36, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.; Tauch, A.; Jager, W.; Kalinowski, J.; Thierbach, G.; Puhler, A. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: Selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 1994, 145, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Wang, X.; Mao, Y.; Wang, Z.; Chen, T.; Zhao, X. Development of a markerless gene replacement system in Corynebacterium glutamicum using upp as a counter-selection marker. Biotechnol. Lett. 2015, 37, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cleto, S.; Jensen, J.V.; Wendisch, V.F.; Lu, T.K. Corynebacterium glutamicum Metabolic Engineering with CRISPR Interference (CRISPRi). ACS Synth. Biol. 2016, 5, 375–385. [Google Scholar] [CrossRef]
- Jeong, S.H.; Lee, H.J.; Lee, S.J. Recent Advances in CRISPR-Cas Technologies for Synthetic Biology. J. Microbiol. 2023, 61, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Okano, K.; Sato, Y.; Hizume, T.; Honda, K. Genome editing by miniature CRISPR/Cas12f1 enzyme in Escherichia coli. J. Biosci. Bioeng. 2021, 132, 120–124. [Google Scholar] [CrossRef]
- Yan, M.Y.; Yan, H.Q.; Ren, G.X.; Zhao, J.P.; Guo, X.P.; Sun, Y.C. CRISPR-Cas12a-Assisted Recombineering in Bacteria. Appl. Environ. Microbiol. 2017, 83, e00947-17. [Google Scholar] [CrossRef]
- Feng, X.; Zhao, D.; Zhang, X.; Ding, X.; Bi, C. CRISPR/Cas9 Assisted Multiplex Genome Editing Technique in Escherichia coli. Biotechnol. J. 2018, 13, e1700604. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef]
- Zetsche, B.; Heidenreich, M.; Mohanraju, P.; Fedorova, I.; Kneppers, J.; DeGennaro, E.M.; Winblad, N.; Choudhury, S.R.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol. 2017, 35, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ren, K.; Qiu, X.; Zheng, J.; Guo, M.; Guan, X.; Liu, H.; Li, N.; Zhang, B.; Yang, D.; et al. The crystal structure of Cpf1 in complex with CRISPR RNA. Nature 2016, 532, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liu, Q.; Shi, H.; Xie, J.; Zhang, Q.; Ouyang, Z.; Li, N.; Yang, Y.; Liu, Z.; Zhao, Y.; et al. Engineering CRISPR/Cpf1 with tRNA promotes genome editing capability in mammalian systems. Cell Mol. Life Sci. 2018, 75, 3593–3607. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Qin, R.; Li, H.; Li, D.; Li, L.; Wei, P.; Yang, J. Generation of targeted mutant rice using a CRISPR-Cpf1 system. Plant Biotechnol. J. 2017, 15, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.M.; Yu, D.; DiTizio, T.; Court, D.L. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. USA 2001, 98, 6742–6746. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Liu, J.; Guo, Y.; Fan, L.; Ni, X.; Zheng, X.; Wang, M.; Zheng, P.; Sun, J.; et al. MACBETH: Multiplex automated Corynebacterium glutamicum base editing method. Metab. Eng. 2018, 47, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, L.; Xie, S.; Zhao, N.; Han, S.; Lin, Y.; Zheng, S. Recombineering using RecET in Corynebacterium glutamicum ATCC14067 via a self-excisable cassette. Sci. Rep. 2017, 7, 7916. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hu, Q.; Zhang, Y.; Shi, R.; Chai, X.; Liu, Z.; Shang, X.; Zhang, Y.; Wen, T. A RecET-assisted CRISPR-Cas9 genome editing in Corynebacterium glutamicum. Microb. Cell Fact. 2018, 17, 63. [Google Scholar] [CrossRef]
- Luo, G.; Zhao, N.; Jiang, S.; Zheng, S. Application of RecET-Cre/loxP system in Corynebacterium glutamicum ATCC14067 for L-leucine production. Biotechnol. Lett. 2021, 43, 297–306. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Zhao, N.; Li, L.; Luo, G.; Xie, S.; Lin, Y.; Han, S.; Huang, Y.; Zheng, S. Multiplex gene editing and large DNA fragment deletion by the CRISPR/Cpf1-RecE/T system in Corynebacterium glutamicum. J. Ind. Microbiol. Biotechnol. 2020, 47, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Qian, F.; Yang, J.; Liu, Y.; Dong, F.; Xu, C.; Sun, B.; Chen, B.; Xu, X.; Li, Y.; et al. CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat. Commun. 2017, 8, 15179. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Wang, T.; Bo, T.; Cai, N.; Yuan, M.; Wu, C.; Jiang, H.; Peng, H.; Chen, N.; Li, Y. Enhanced production of D-pantothenic acid in Corynebacterium glutamicum using an efficient CRISPR-Cpf1 genome editing method. Microb. Cell Fact. 2023, 22, 3. [Google Scholar] [CrossRef] [PubMed]
- Garst, A.D.; Bassalo, M.C.; Pines, G.; Lynch, S.A.; Halweg-Edwards, A.L.; Liu, R.; Liang, L.; Wang, Z.; Zeitoun, R.; Alexander, W.G.; et al. Genome-wide mapping of mutations at single-nucleotide resolution for protein, metabolic and genome engineering. Nat. Biotechnol. 2017, 35, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Wang, X.; Sun, Y.; Dong, G.; Yang, Y.; Liu, X.; Bai, Z. Efficient gene editing in Corynebacterium glutamicum using the CRISPR/Cas9 system. Microb. Cell Fact. 2017, 16, 201. [Google Scholar] [CrossRef] [PubMed]
- Marienhagen, J.; Eggeling, L. Metabolic function of Corynebacterium glutamicum aminotransferases AlaT and AvtA and impact on L-valine production. Appl. Environ. Microbiol. 2008, 74, 7457–7462. [Google Scholar] [CrossRef] [PubMed]
- Altenbuchner, J. Editing of the Bacillus subtilis Genome by the CRISPR-Cas9 System. Appl. Environ. Microbiol. 2016, 82, 5421–5427. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lin, Z.; Huang, C.; Zhang, Y.; Wang, Z.; Tang, Y.J.; Chen, T.; Zhao, X. Metabolic engineering of Escherichia coli using CRISPR-Cas9 meditated genome editing. Metab. Eng. 2015, 31, 13–21. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Lu, Y.; Zheng, P.; Sun, J.; Ma, Y. Development of a CRISPR/Cas9 genome editing toolbox for Corynebacterium glutamicum. Microb. Cell Fact. 2017, 16, 205. [Google Scholar] [CrossRef]
- Cho, J.S.; Choi, K.R.; Prabowo, C.P.S.; Shin, J.H.; Yang, D.; Jang, J.; Lee, S.Y. CRISPR/Cas9-coupled recombineering for metabolic engineering of Corynebacterium glutamicum. Metab. Eng. 2017, 42, 157–167. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).