
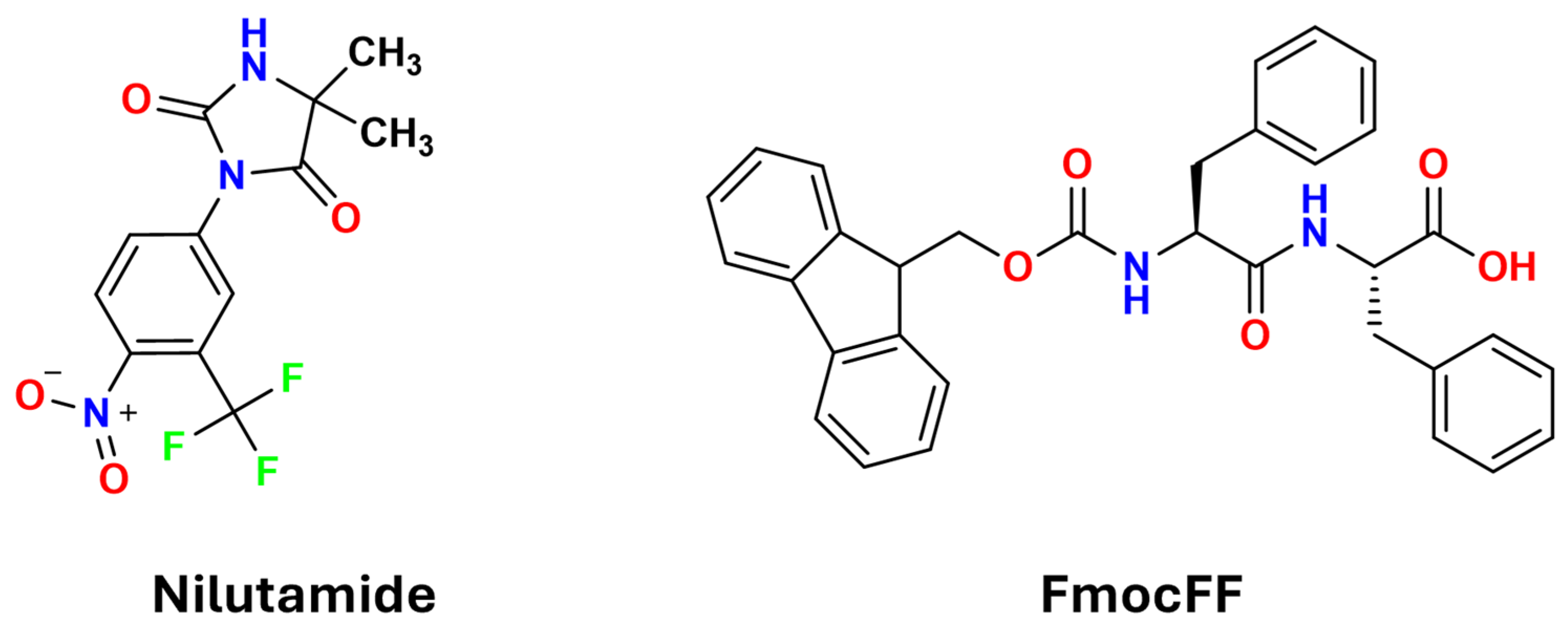
Polymorphic Control in Pharmaceutical Gel-Mediated Crystallization: Exploiting Solvent–Gelator Synergy in FmocFF Organogels



Abstract

1. Introduction
2. Results and Discussion
2.1. Gel-Phase Crystallization of Nilutamide
2.2. Crystal Morphology and Habit Modification
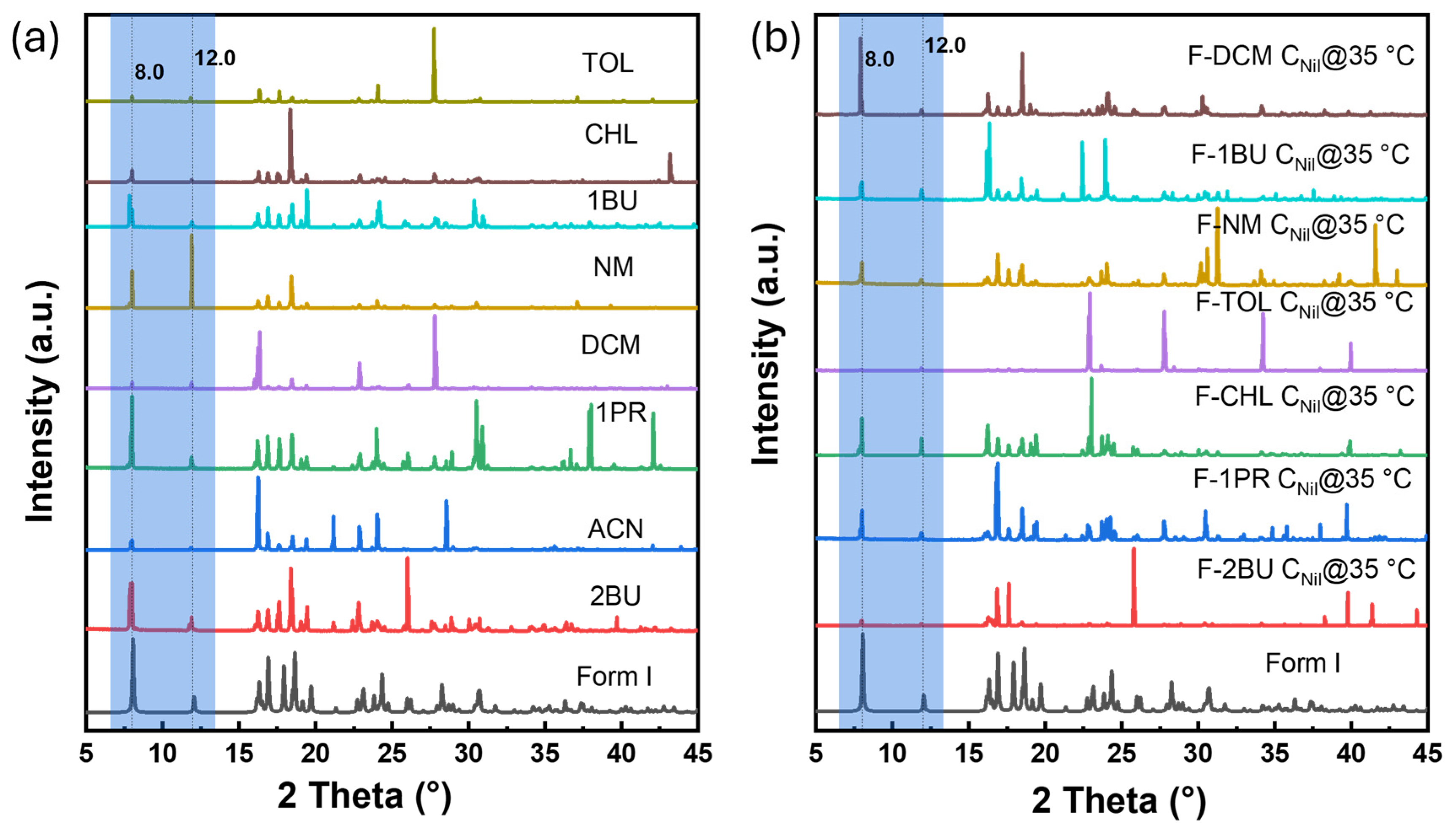
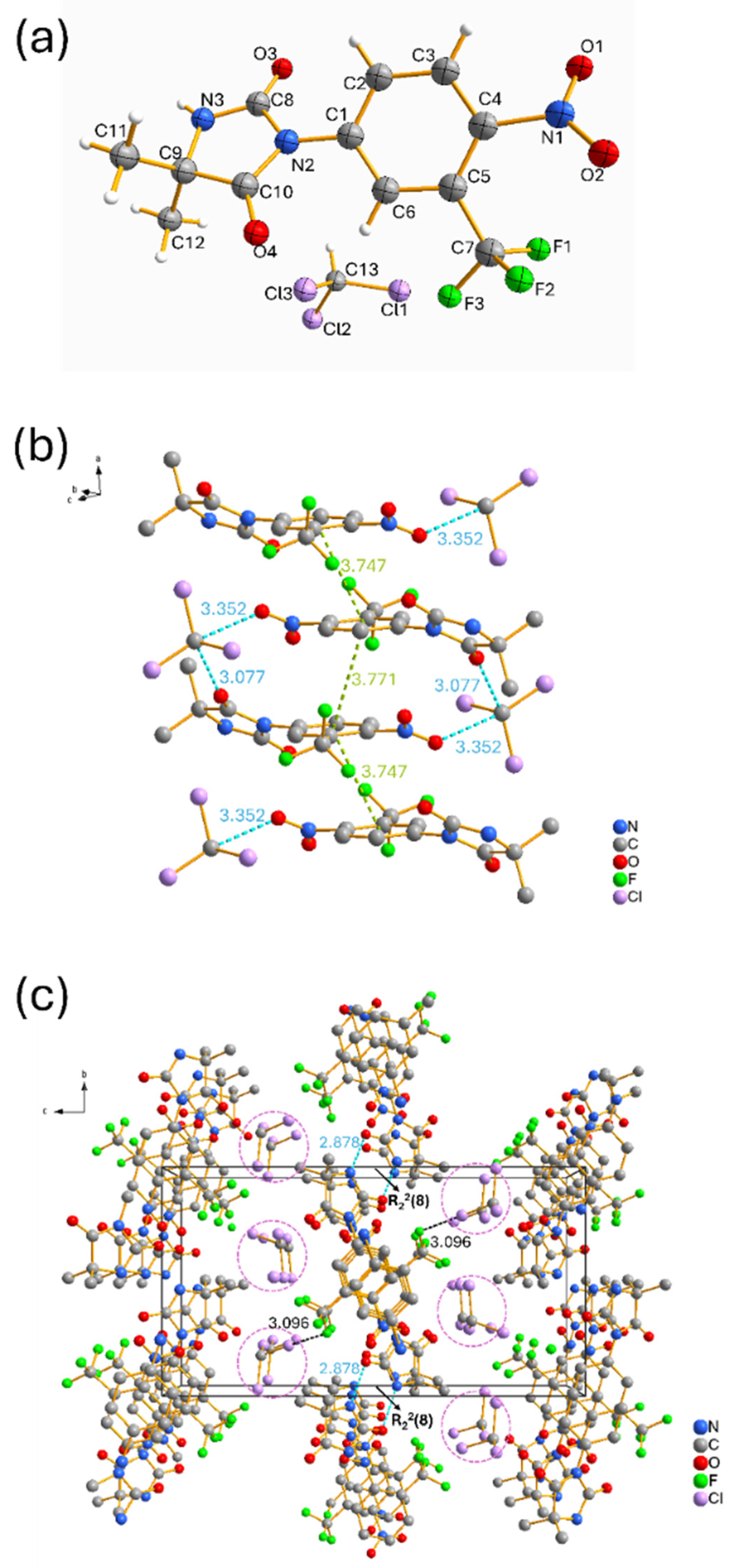
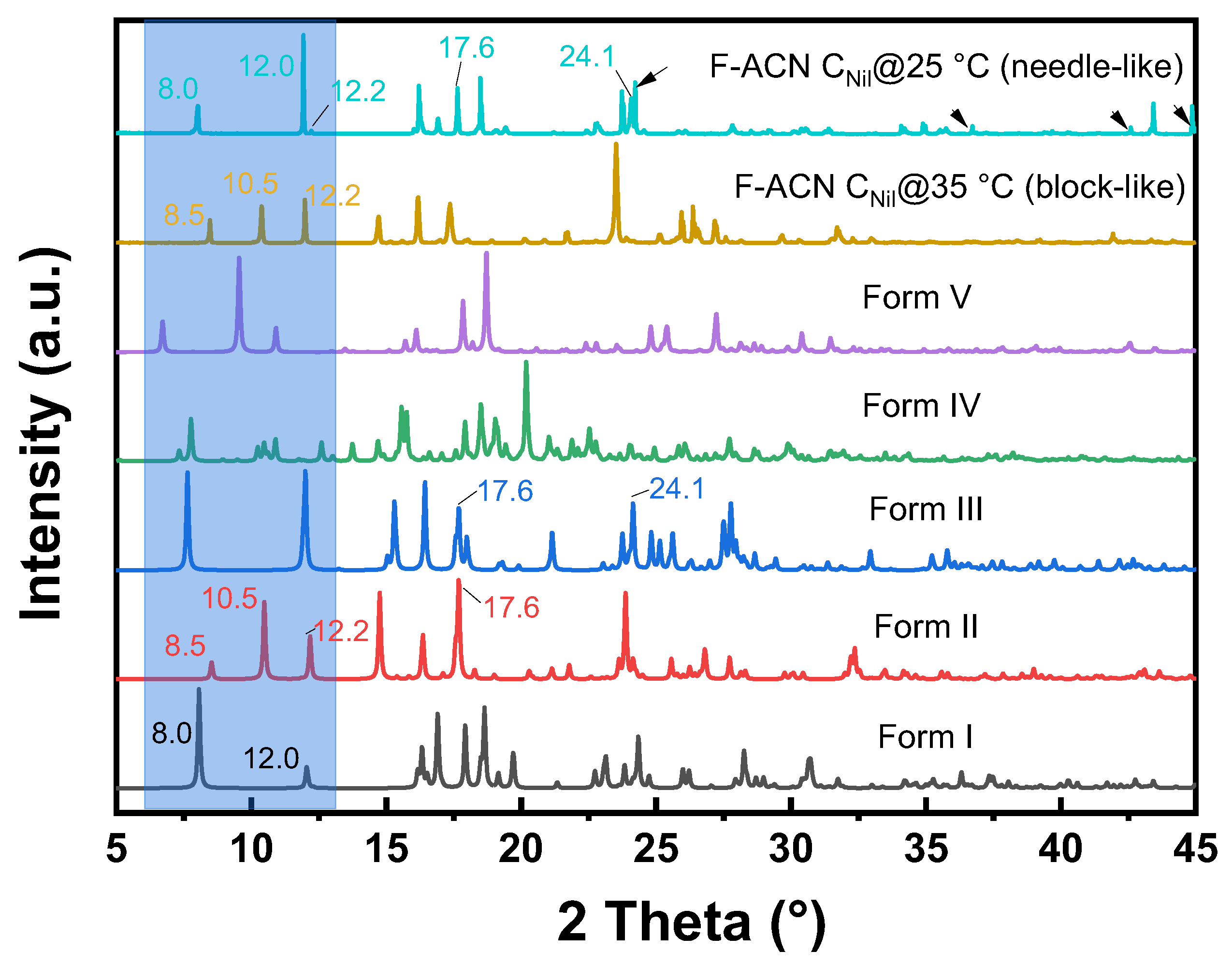
2.3. Polymorphic Identification and Novel Form Discovery
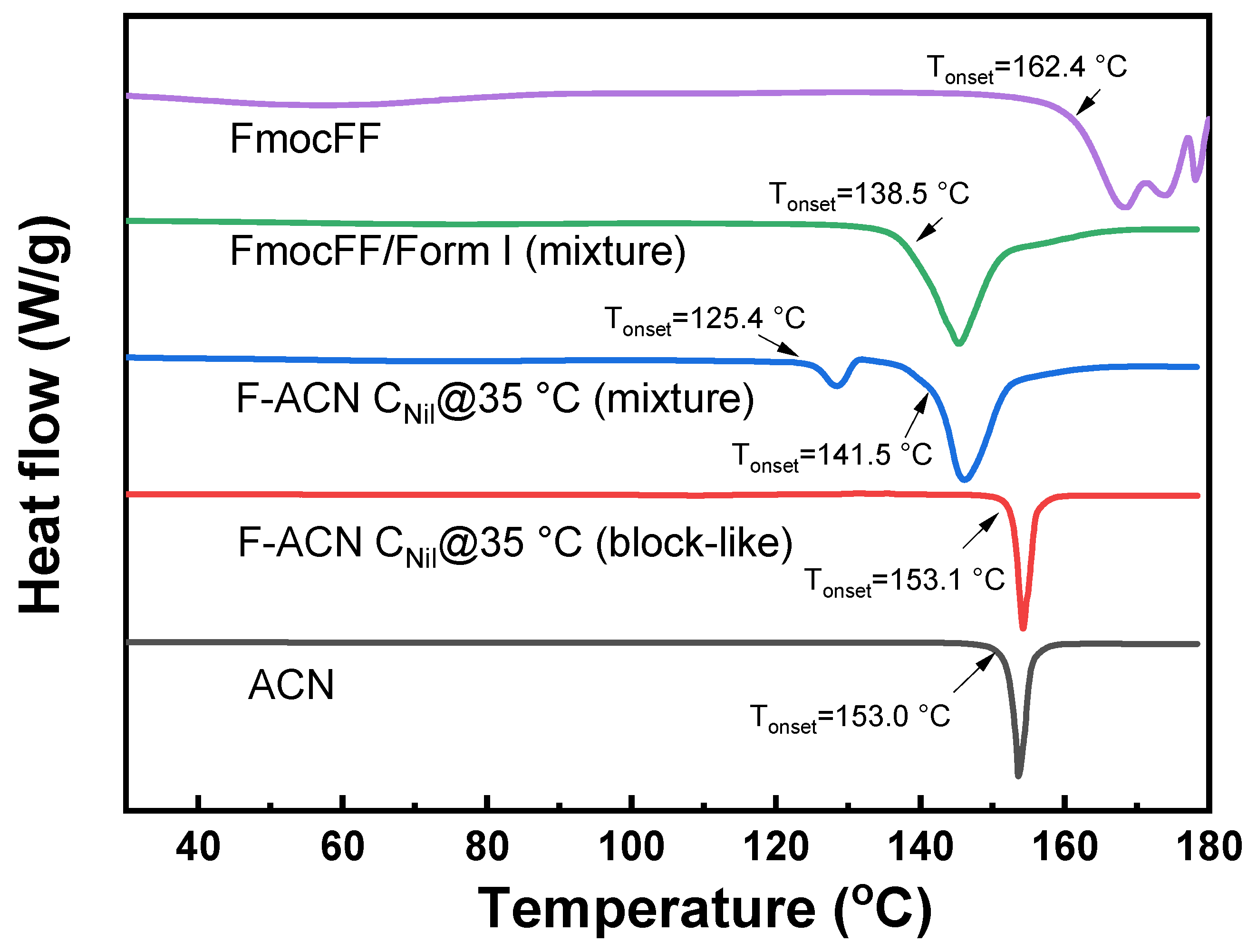
2.4. Thermal Analysis and Gelator–API Interactions
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Gel Preparation
4.2.1. Gel-Mediated Crystallization
4.2.2. Solution Crystallization
4.2.3. Empirical Solubility
4.3. Characterizations
4.3.1. Polarizing Optical Microscopy (POM)
4.3.2. X-Ray Powder Diffraction (XRPD)
4.3.3. Single-Crystal X-Ray Diffraction (SXRD)
4.3.4. Fourier-Transform Infrared Spectroscopy (FTIR)
4.3.5. Differential Scanning Calorimetry (DSC)
4.3.6. Second-Harmonic Generation Microscopy (SHG Microscopy)
4.3.7. Calculation Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, J.; Sarma, B.; Evans, J.M.B.; Myerson, A.S. Pharmaceutical Crystallization. Cryst. Growth Des. 2011, 11, 887–895. [Google Scholar] [CrossRef]
- Lee, E.H. A Practical Guide to Pharmaceutical Polymorph Screening & Selection. Asian J. Pharm. Sci. 2014, 9, 163–175. [Google Scholar] [CrossRef]
- Gao, Z.; Rohani, S.; Gong, J.; Wang, J. Recent Developments in the Crystallization Process: Toward the Pharmaceutical Industry. Engineering 2017, 3, 343–353. [Google Scholar] [CrossRef]
- Singhal, D. Drug Polymorphism and Dosage Form Design: A Practical Perspective. Adv. Drug Deliv. Rev. 2004, 56, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Steed, J.W. Anion-Tuned Supramolecular Gels: A Natural Evolution from Urea Supramolecular Chemistry. Chem. Soc. Rev. 2010, 39, 3686. [Google Scholar] [CrossRef]
- Kumar, D.K.; Steed, J.W. Supramolecular Gel Phase Crystallization: Orthogonal Self-Assembly under Non-Equilibrium Conditions. Chem. Soc. Rev. 2014, 43, 2080–2088. [Google Scholar] [CrossRef]
- Sharma, H.; Kalita, B.K.; Pathak, D.; Sarma, B. Low Molecular Weight Supramolecular Gels as a Crystallization Matrix. Cryst. Growth Des. 2023, 24, 17–37. [Google Scholar] [CrossRef]
- Rahim, M.A.; Hata, Y.; Björnmalm, M.; Ju, Y.; Caruso, F. Supramolecular Metal–Phenolic Gels for the Crystallization of Active Pharmaceutical Ingredients. Small 2018, 14, 1801202. [Google Scholar] [CrossRef]
- Smith, D.K. Supramolecular Gels—A Panorama of Low-Molecular-Weight Gelators from Ancient Origins to next-Generation Technologies. Soft Matter 2024, 20, 10–70. [Google Scholar] [CrossRef]
- Contreras-Montoya, R.; Álvarez de Cienfuegos, L.; Gavira, J.A.; Steed, J.W.; Gavira, A.; Contreras-Montoya, R. Supramolecular Gels: A Versatile Crystallization Toolbox. Chem. Soc. Rev. 2024, 53, 10604–10619. [Google Scholar] [CrossRef]
- Zhang, Q.; Yan, Y.; Xu, Y.; Zhang, X.; Steed, J.W. Selective Crystallization of Pyrazinamide Polymorphs in Supramolecular Gels: Synergistic Selectivity by Mimetic Gelator and Solvent. J. Colloid. Interface Sci. 2025, 687, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Artusio, F.; Castellví, A.; Sacristán, A.; Pisano, R.; Gavira, J.A. Agarose Gel as a Medium for Growing and Tailoring Protein Crystals. Cryst. Growth Des. 2020, 20, 5564–5571. [Google Scholar] [CrossRef]
- Mullin, J.W. Crystal Growth. In Crystallization; Mullin, J.W.B.T.-C., Fourth, E., Eds.; Elsevier: Oxford, UK, 2001; pp. 216–288. ISBN 978-0-7506-4833-2. [Google Scholar]
- Wang, C.; Zhou, L.; Zhang, X.; Yang, Y.; Yin, Q.; Roberts, K.J. The Role of Solvent Composition and Polymorph Surface Chemistry in the Solution-Mediated Phase Transformation Process of Cefaclor. Ind. Eng. Chem. Res. 2018, 57, 16925–16933. [Google Scholar] [CrossRef]
- Miller, J.M.; Collman, B.M.; Greene, L.R.; Grant, D.J.W.; Blackburn, A.C. Identifying the Stable Polymorph Early in the Drug Discovery–Development Process. Pharm. Dev. Technol. 2005, 10, 291–297. [Google Scholar] [PubMed]
- Wang, C.; Ma, C.Y.; Hong, R.S.; Turner, T.D.; Rosbottom, I.; Sheikh, A.Y.; Yin, Q.; Roberts, K.J. Influence of Solvent Selection on the Crystallizability and Polymorphic Selectivity Associated with the Formation of the “Disappeared” Form I Polymorph of Ritonavir. Mol. Pharm. 2024, 21, 3525–3539. [Google Scholar] [CrossRef]
- Turner, T.D.; Corzo, D.M.C.; Toroz, D.; Curtis, A.; Dos Santos, M.M.; Hammond, R.B.; Lai, X.; Roberts, K.J. The Influence of Solution Environment on the Nucleation Kinetics and Crystallisability of Para-Aminobenzoic Acid. Phys. Chem. Chem. Phys. 2016, 18, 27507–27520. [Google Scholar] [CrossRef]
- Moris, M.; Van Den Eede, M.-P.; Koeckelberghs, G.; Deschaume, O.; Bartic, C.; Clays, K.; Van Cleuvenbergen, S.; Verbiest, T. Solvent Role in the Self-Assembly of Poly(3-Alkylthiophene): A Harmonic Light Scattering Study. Macromolecules 2021, 54, 2477–2484. [Google Scholar] [CrossRef]
- Saikia, B.; Chen, D.; de Coene, Y.; Van Cleuvenbergen, S. Organogels of FmocFF: Exploring the Solvent-Dependent Gelmorphic Behavior. Gels 2024, 10, 749. [Google Scholar] [CrossRef]
- Luo, J.; Beer, T.M.; Graff, J.N. Treatment of Nonmetastatic Castration-Resistant Prostate Cancer. Oncology 2016, 30, 336–344. [Google Scholar]
- Kassouf, W.; Tangury, S.; Aprikian, A.G. Nilutamide as Second Line Hormone Therapy for Prostate Cancer After Androgen Ablation Fails. J. Urol. 2003, 169, 1742–1744. [Google Scholar] [CrossRef]
- Surov, A.O.; Voronin, A.P.; Drozd, K.V.; Gruzdev, M.S.; Perlovich, G.L.; Prashanth, J.; Balasubramanian, S. Polymorphic Forms of Antiandrogenic Drug Nilutamide: Structural and Thermodynamic Aspects. Phys. Chem. Chem. Phys. 2021, 23, 9695–9708. [Google Scholar] [CrossRef] [PubMed]
- Prashanth, J.; Surov, A.O.; Drozd, K.V.; Perlovich, G.L.; Balasubramanian, S. Polymorphs, Cocrystal and Hydrate of Nilutamide. CrystEngComm 2023, 25, 3501–3513. [Google Scholar] [CrossRef]
- Dok, A.R.; Legat, T.; de Coene, Y.; van der Veen, M.A.; Verbiest, T.; Van Cleuvenbergen, S. Nonlinear Optical Probes of Nucleation and Crystal Growth: Recent Progress and Future Prospects. J. Mater. Chem. C 2021, 9, 11553–11568. [Google Scholar] [CrossRef]
- Van Cleuvenbergen, S.; Hennrich, G.; Willot, P.; Koeckelberghs, G.; Clays, K.; Verbiest, T.; Van Der Veen, M.A. All Optical Determination of Microscopic and Macroscopic Structure of Chiral, Polar Microcrystals from Achiral, Nonpolar Molecules. J. Phys. Chem. C 2012, 116, 12219–12225. [Google Scholar] [CrossRef]
- van Cleuvenbergen, S.; Depotter, G.; Clays, K.; Kędziora, P. Second-Order NLO Response in Chiral Ferroelectric Liquid Crystals: Molecular and Bulk Consideration. J. Mol. Liq. 2021, 326, 115328. [Google Scholar] [CrossRef]
- Galindo, J.M.; Tardío, C.; Saikia, B.; Van Cleuvenbergen, S.; Torres-Moya, I. Recent Insights about the Role of Gels in Organic Photonics and Electronics. Gels 2023, 9, 875. [Google Scholar] [CrossRef]
- Diffraction RO Rigaku Oxford Diffraction. CrysAlisPro Software System, Version 1.171.37.35; Rigaku Corporation: Akishima, Japan, 2014.
- Sheldrick, G.M. Crystal Structure Solution with ShelXT. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- de Coene, Y.; Jooken, S.; Deschaume, O.; Van Steenbergen, V.; Vanden Berghe, P.; Van den Haute, C.; Baekelandt, V.; Callewaert, G.; Van Cleuvenbergen, S.; Verbiest, T.; et al. Label-Free Imaging of Membrane Potentials by Intramembrane Field Modulation, Assessed by Second Harmonic Generation Microscopy. Small 2022, 18, e2200205. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organogel Label | Solvent | FmocFF Concentration (% w/v) |
|---|---|---|
| F-NM | Nitromethane | 10 |
| F-1PR | 1-Propanol | 15 |
| F-ACN | Acetonitrile | 15 |
| F-DCM | Dichloromethane | 10 |
| F-CHL | Chloroform | 15 |
| F-TOL | Toluene | 10 |
| F-1BU | 1-Butanol | 20 |
| F-2BU | 2-Butanol | 20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, D.; Robeyns, K.; Leyssens, T.; Saikia, B.; Van Cleuvenbergen, S. Polymorphic Control in Pharmaceutical Gel-Mediated Crystallization: Exploiting Solvent–Gelator Synergy in FmocFF Organogels. Gels 2025, 11, 509. https://doi.org/10.3390/gels11070509
Chen D, Robeyns K, Leyssens T, Saikia B, Van Cleuvenbergen S. Polymorphic Control in Pharmaceutical Gel-Mediated Crystallization: Exploiting Solvent–Gelator Synergy in FmocFF Organogels. Gels. 2025; 11(7):509. https://doi.org/10.3390/gels11070509
Chicago/Turabian StyleChen, Dong, Koen Robeyns, Tom Leyssens, Basanta Saikia, and Stijn Van Cleuvenbergen. 2025. "Polymorphic Control in Pharmaceutical Gel-Mediated Crystallization: Exploiting Solvent–Gelator Synergy in FmocFF Organogels" Gels 11, no. 7: 509. https://doi.org/10.3390/gels11070509
APA StyleChen, D., Robeyns, K., Leyssens, T., Saikia, B., & Van Cleuvenbergen, S. (2025). Polymorphic Control in Pharmaceutical Gel-Mediated Crystallization: Exploiting Solvent–Gelator Synergy in FmocFF Organogels. Gels, 11(7), 509. https://doi.org/10.3390/gels11070509