


Profiling the Genomes and Secreted Effector Proteins in Phytopythium vexans Global Strains


Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Extraction and Genome Sequencing
2.2. Genome Assembly
2.3. Phylogenetic Analyses
2.4. Genome Annotation
2.5. Identification of Putative Effectors
3. Results
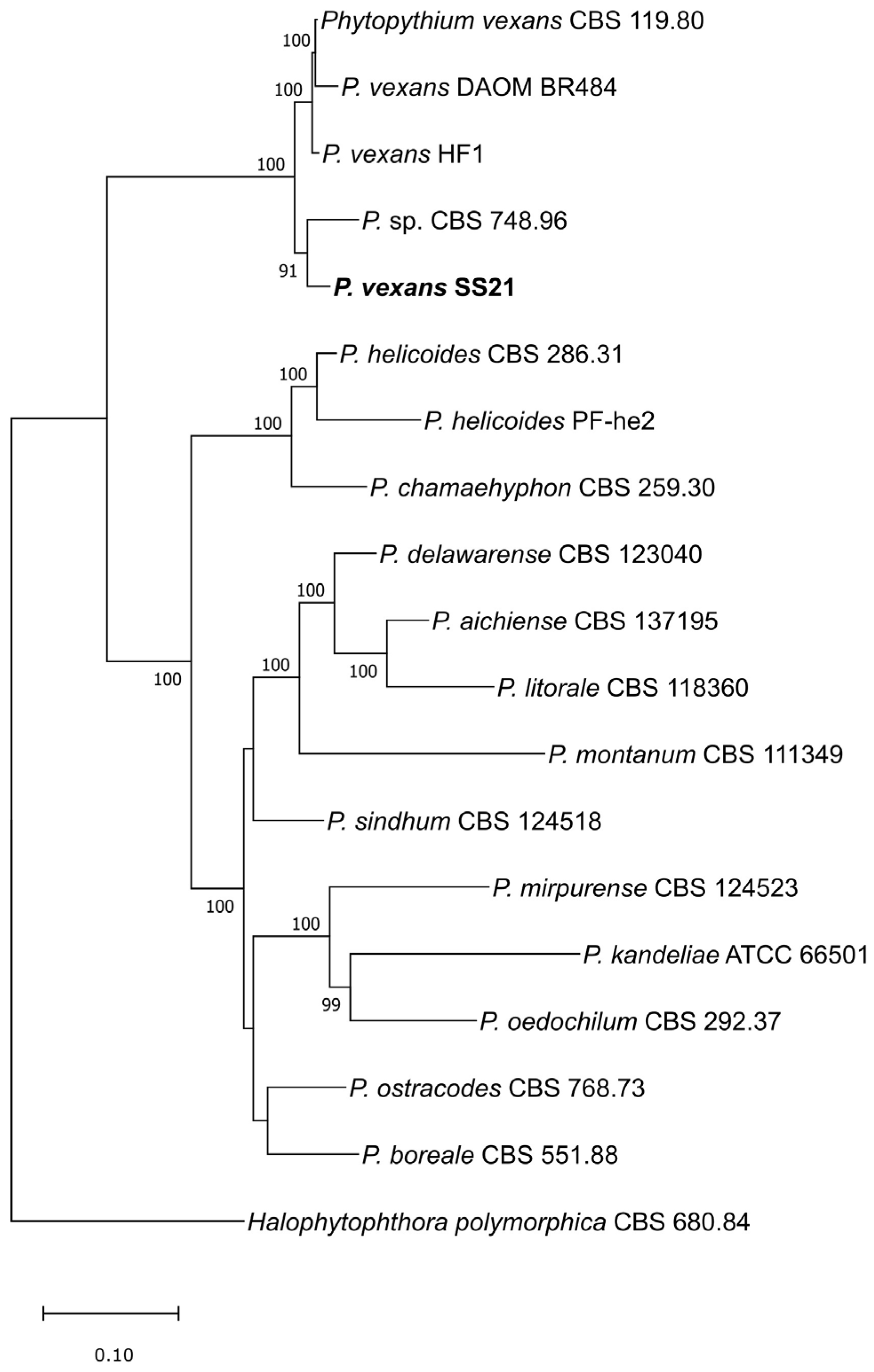
3.1. Genome Assembly, Gene Modeling and Phylogeny
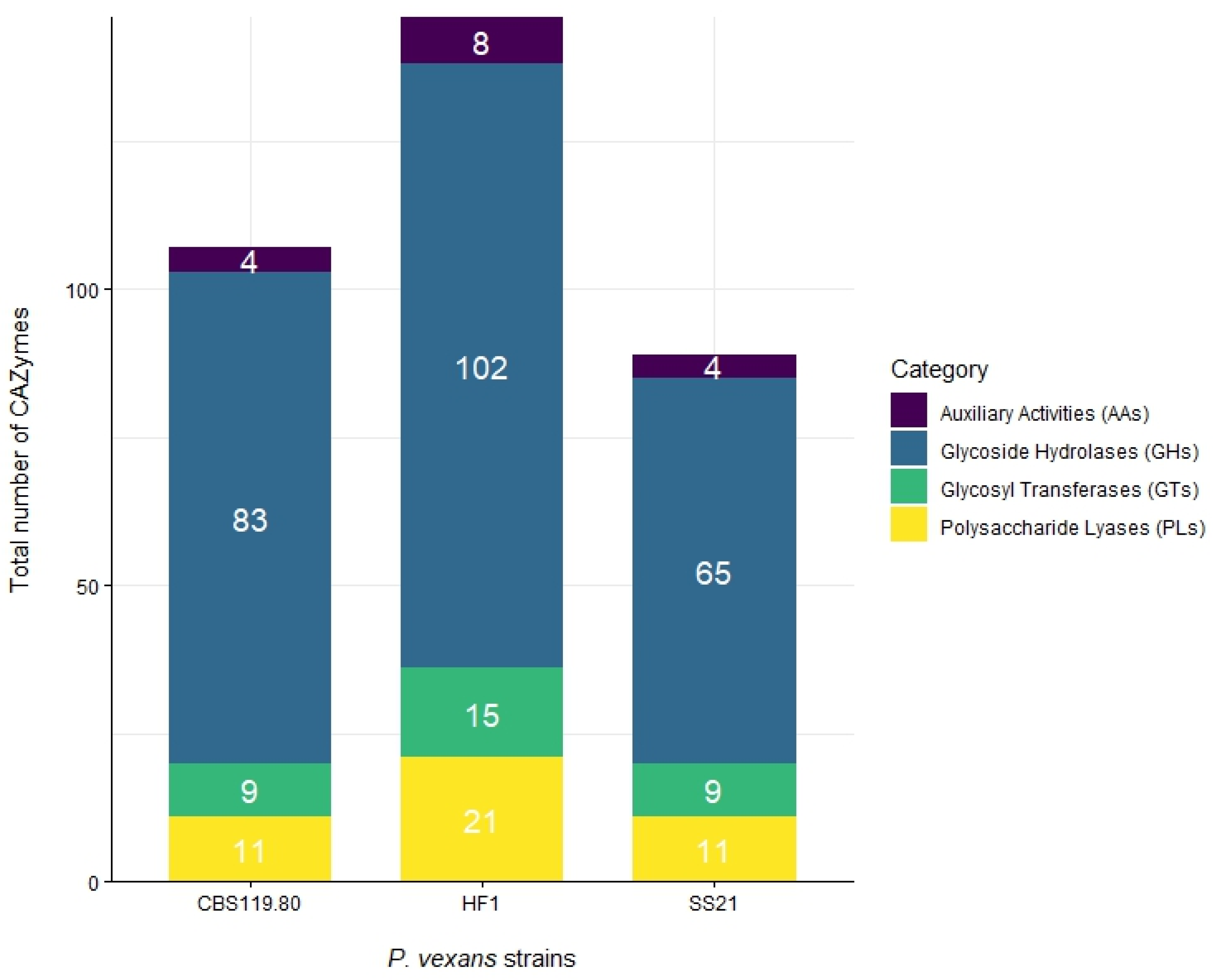
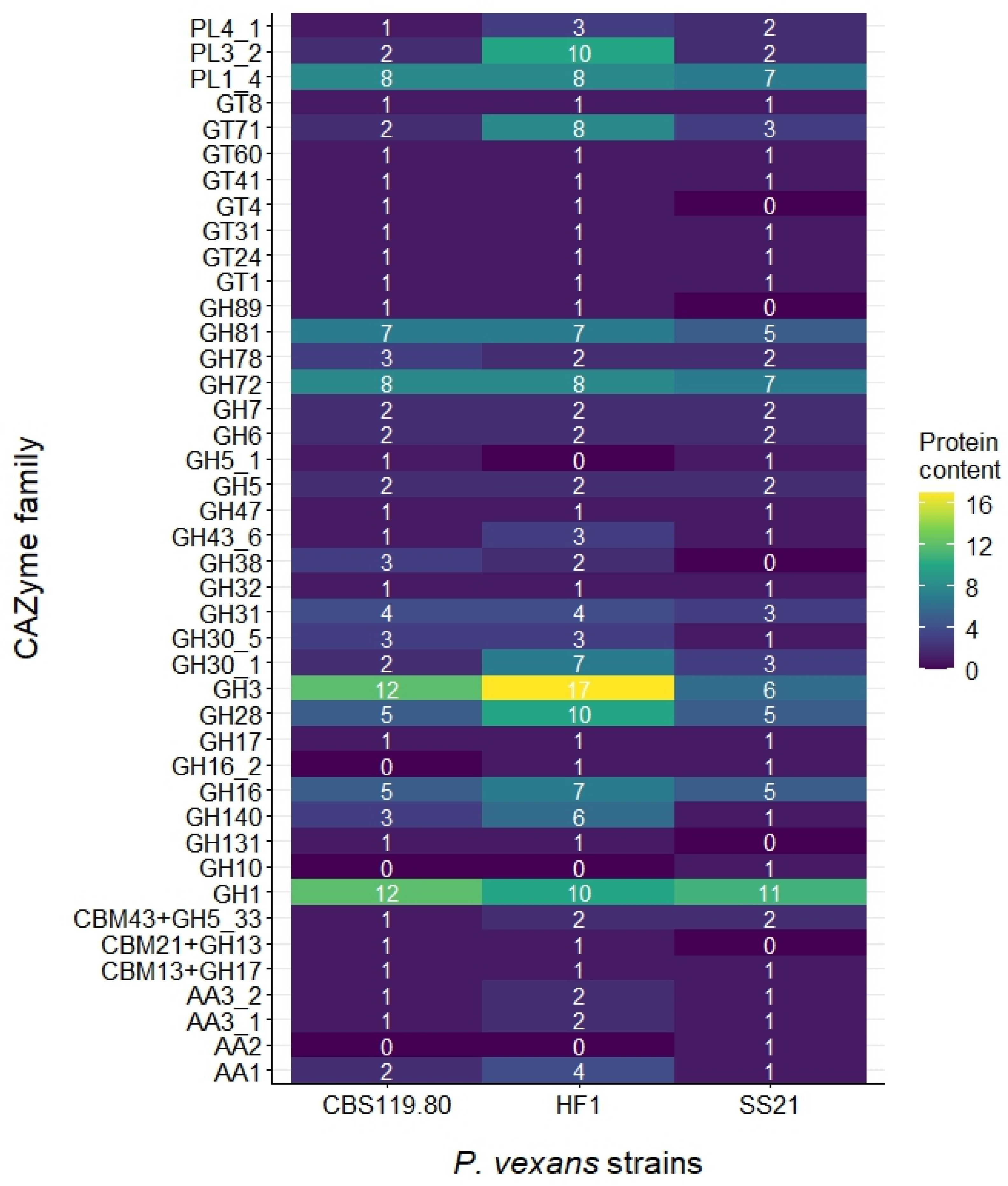
3.2. Secretome Analysis
3.3. Microbe-Associated Molecular Pattern (MAMP) Genes
3.4. Apoplastic Effectors Profiling
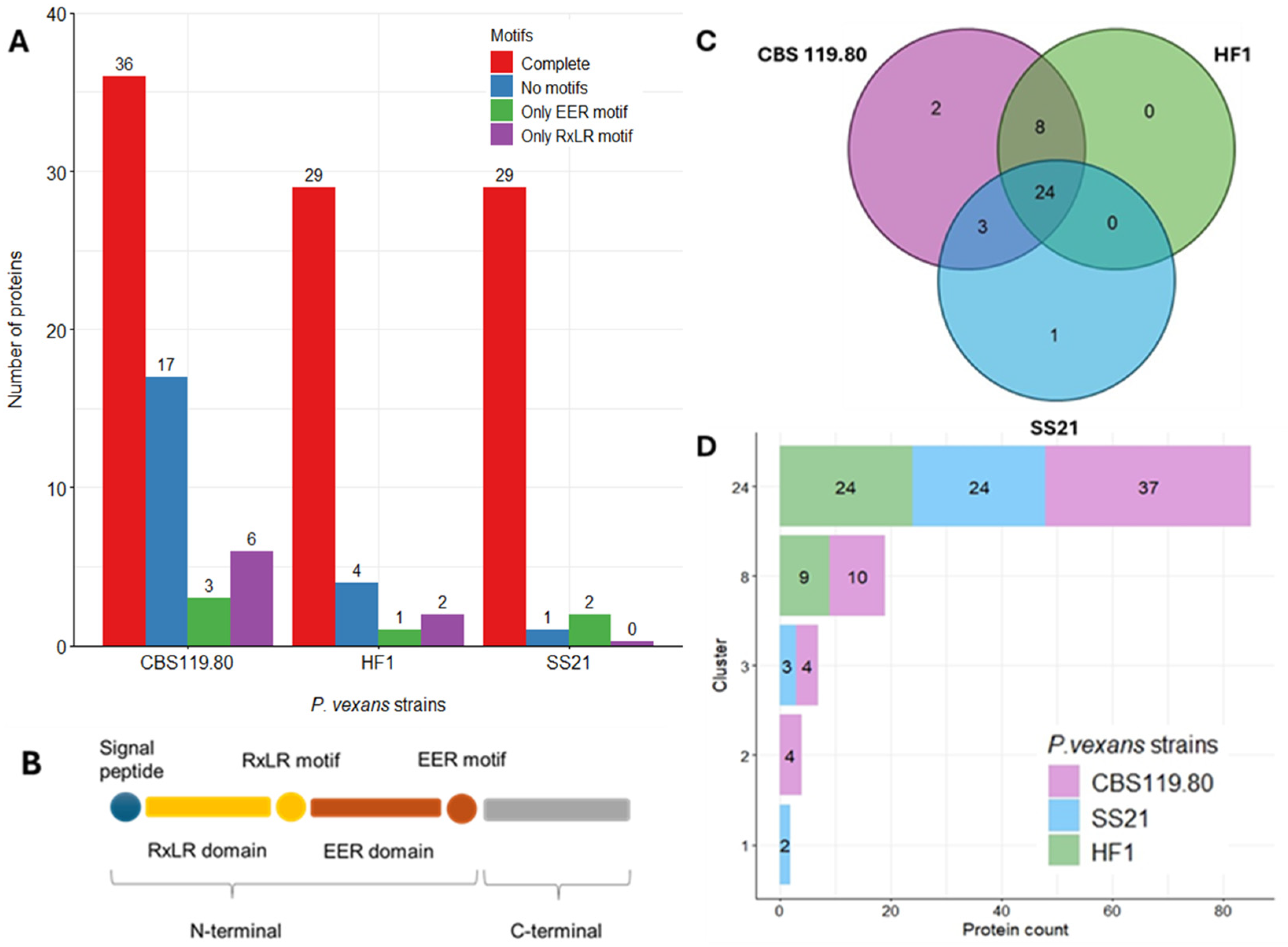
3.5. Cytoplasmic Effectors Profiling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghimire, B.; Baysal-Gurel, F. A diagnostic guide to Phytopythium helicoides and Phytopythium vexans causing root and crown rot diseases. Plant Health Prog. 2023, 24, 527–538. [Google Scholar] [CrossRef]
- Jabiri, S.; Lahlali, R.; Bahra, C.; Bendriss Amraoui, M.; Tahiri, A.; Amiri, S. First report of Phytopythium vexans associated with dieback disease of apple trees in Morocco. J. Plant Pathol. 2020, 102, 1319. [Google Scholar] [CrossRef]
- Zeng, H.C.; Ho, H.H.; Zheng, F.C. Pythium vexans causing patch canker of rubber trees on Hainan Island, China. Mycopathologia 2005, 159, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Pan, X.; Kong, B.; Cun, H.; Li, N.; He, Y.; Ma, J.; Zhang, Y.; Ma, Y.; Cao, K. First report of apple root rot caused by Phytopythium vexans in China. Plant Dis. 2022, 106, 3002. [Google Scholar] [CrossRef]
- RodrÍGuez-PadrÓN, C.; Siverio, F.; PÉRez-Sierra, A.; RodrÍGuez, A. Isolation and pathogenicity of Phytophthora species and Phytopythium vexans recovered from avocado orchards in the Canary Islands, including Phytophthora niederhauserii as a new pathogen of avocado. Phytopathol. Mediterr. 2018, 57, 89–106. [Google Scholar] [CrossRef]
- Benfradj, N.; Migliorini, D.; Luchi, N.; Santini, A.; Boughalleb-M’Hamdi, N. Occurrence of Pythium and Phytopythium species isolated from citrus trees infected with gummosis disease in tunisia. Arch. Phytopathol. Plant Prot. 2017, 50, 286–302. [Google Scholar] [CrossRef]
- Mannai, S.; Benfradj, N.; Boughalleb-M’Hamdi, N. Occurrence of Globisporangium and Phytopythium species associated with apple and peach seedlings decline in Tunisian nurseries. J. Plant Pathol. 2024, 106, 643–655. [Google Scholar] [CrossRef]
- Vawdrey, L.L.; Langdon, P.; Martin, T. Incidence and pathogenicity of Phytophthora palmivora and Pythium vexans associated with durian decline in far northern Queensland. Australas. Plant Pathol. 2005, 34, 127–128. [Google Scholar] [CrossRef]
- Thao, L.D.; Hien, L.T.; Liem, N.V.; Thanh, H.M.; Khanh, T.N.; Binh, V.T.P.; Trang, T.T.T.; Anh, P.T.; Tu, T.T. First report of Phytopythium vexans causing root rot disease on durian in Vietnam. New Dis. Rep. 2020, 41, 2. [Google Scholar] [CrossRef]
- Ivors, K.L.; Abad, Z.G.; Benson, D.M. Evaluating the pathogenicity of Pythium vexans isolates from fraser fir in North Carolina. Plant Health Prog. 2008, 9, 8. [Google Scholar] [CrossRef]
- Baysal-Gurel, F.; Liyanapathiranage, P.; Panth, M.; Avin, F.A.; Simmons, T. First report of Phytopythium vexans causing root and crown rot on flowering cherry in Tennessee. Plant Dis. 2021, 105, 232. [Google Scholar] [CrossRef]
- Panth, M.; Baysal-Gurel, F.; Avin, F.A.; Simmons, T. Identification and chemical and biological management of Phytopythium vexans, the causal agent of Phytopythium root and crown rot of woody ornamentals. Plant Dis. 2021, 105, 1091–1100. [Google Scholar] [CrossRef]
- Linde, C.; Kemp, G.H.J.; Wingfleld, M.J. Pythium and Phytophthora species associated with eucalypts and pines in South Africa. Eur. J. For. Pathol. 1994, 24, 345–356. [Google Scholar] [CrossRef]
- Spies, C.F.J.; Mazzola, M.; McLeod, A. Characterisation and detection of Pythium and Phytophthora species associated with grapevines in South Africa. Eur. J. Plant Pathol. 2011, 131, 103–119. [Google Scholar] [CrossRef]
- Mosca, S.; Aci, M.M.; Procopio, G.; Vadalà, V.; Vizzari, G.; Francomano, E.; Mohamed, N.Z.; Li Destri Nicosia, M.G.; Agosteo, G.E.; Spadaro, D.; et al. Integrated analyses of the plant and soil microbiome identify Phytopythium vexans as agent of the Kiwifruit Vine Decline Syndrome. Plant Soil 2024, 509, 729–742. [Google Scholar] [CrossRef]
- Polat, Z.; Awan, Q.N.; Hussain, M.; Akgül, D.S. First report of Phytopythium vexans causing root and collar rot of kiwifruit in Turkey. Plant Dis. 2017, 101, 1058. [Google Scholar] [CrossRef]
- Pánek, M.; Střížková, I. A comparison of the virulence of selected Pythium, Globisporangium, Phytopythium and Phytophthora species against strawberry plants. J. Plant Dis. Prot. 2021, 128, 1447–1458. [Google Scholar] [CrossRef]
- Browne, E.Y.; Edwards, S.G.; Nellist, C.F. First report of Phytopythium vexans and Phytopythium litorale associated with root rot symptoms on red raspberry (Rubus idaeus). New Dis. Rep. 2023, 48, e12197. [Google Scholar] [CrossRef]
- Nzungize, J.; Geps, P.; Buruchara, R.; Buah, S.; Ragama, P.; Busogoro, J.P.; Baudin, J.P. Pathogenic and molecular characterization of Pythium species inducing root rot symptoms of common bean in Rwanda. Afr. J. Microbiol. Res. 2011, 5, 1169–1181. [Google Scholar] [CrossRef]
- Villanueva, O.; Ellouze, W. First report of a Canadian isolate of Phytopythium vexans causing root rot disease on apple and peach under laboratory conditions. New Dis. Rep. 2023, 48, e12195. [Google Scholar] [CrossRef]
- Baysal-Gurel, F.; Ghimire, B. Impact of Phytopythium vexans on plant health: Hosts, symptoms, detection, and management. Plant Health Cases 2023, 2023, phcs20230019. [Google Scholar] [CrossRef]
- Bozkurt, T.O.; Kamoun, S. The plant-pathogen haustorial interface at a glance. J. Cell Sci. 2020, 133, jcs237958. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, S. A catalogue of the effector secretome of plant pathogenic oomycetes. Annu. Rev. Phytopathol. 2006, 44, 41–60. [Google Scholar] [CrossRef]
- Kamoun, S. The Secretome of Plant-Associated Fungi and Oomycetes. Plant Relatsh. 2009, 5, 173–180. [Google Scholar] [CrossRef]
- Sonah, H.; Deshmukh, R.K.; Bélanger, R.R. Computational prediction of effector proteins in fungi: Opportunities and challenges. Front. Plant Sci. 2016, 7, 126. [Google Scholar] [CrossRef] [PubMed]
- Armitage, A.D.; Lysøe, E.; Nellist, C.F.; Lewis, L.A.; Cano, L.M.; Harrison, R.J.; Brurberg, M.B. Bioinformatic characterisation of the effector repertoire of the strawberry pathogen Phytophthora cactorum. PLoS ONE 2018, 13, e0202305. [Google Scholar] [CrossRef]
- Villanueva, O.; Nguyen, H.D.T.; Ellouze, W. Comparative genomic and secretome analysis of Phytophthora capsici strains: Exploring pathogenicity and evolutionary dynamics. Agronomy 2024, 14, 2623. [Google Scholar] [CrossRef]
- Adhikari, B.N.; Hamilton, J.P.; Zerillo, M.M.; Tisserat, N.; Lévesque, C.A.; Buell, C.R. Comparative genomics reveals insight into virulence strategies of plant pathogenic oomycetes. PLoS ONE 2013, 8, e75072. [Google Scholar] [CrossRef]
- Bozkurt, T.O.; Schornack, S.; Banfield, M.J.; Kamoun, S. Oomycetes, effectors, and all that jazz. Curr. Opin. Plant Biol. 2012, 15, 483–492. [Google Scholar] [CrossRef]
- Wilson, R.A.; McDowell, J.M. Recent advances in understanding of fungal and oomycete effectors. Curr. Opin. Plant Biol. 2022, 68, 102228. [Google Scholar] [CrossRef]
- McGowan, J.; Fitzpatrick, D.A. Genomic, network, and phylogenetic analysis of the oomycete effector arsenal. Msphere 2017, 2, 10-1128. [Google Scholar] [CrossRef]
- McGowan, J.; Fitzpatrick, D.A. Recent advances in oomycete genomics. In Advances in Genetics; Kumar, D., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 105, pp. 175–228. [Google Scholar]
- Mukhi, N.; Gorenkin, D.; Banfield, M.J. Exploring folds, evolution and host interactions: Understanding effector structure/function in disease and immunity. New Phytol. 2020, 227, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Gao, C.; Wang, T.; Chen, Y.; Cheng, Y.; Li, Z.; Chen, J.; Guo, L.; Sun, X.; Xu, J. Genome sequence resource for the ramie romycete pathogen Phytopythium vexans HF1. Mol. Plant-Microbe Interact. 2020, 33, 1270–1273. [Google Scholar] [CrossRef]
- Nguyen, H.D.T.; Dodge, A.; Dadej, K.; Rintoul, T.L.; Ponomareva, E.; Martin, F.N.; de Cock, A.W.A.M.; Lévesque, C.A.; Redhead, S.A.; Spies, C.F.J. Whole genome sequencing and phylogenomic analysis show support for the splitting of genus Pythium. Mycologia 2022, 114, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Arvind, K.; Rahul, C.; Babu, M.; Grace, T.; Rajesh, M. Comparative secretome prediction and analysis of two Phytophthora spp. Res. J. Biotech 2017, 12, 10–16. [Google Scholar]
- Lee, J.-H.; Siddique, M.I.; Kwon, J.-K.; Kang, B.-C. Comparative genomic analysis reveals genetic variation and adaptive evolution in the pathogenicity-related genes of Phytophthora capsici. Front. Microbiol. 2021, 12, 694136. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Kim, D.; Gilchrist, C.L.M.; Chun, J.; Steinegger, M. UFCG: Database of universal fungal core genes and pipeline for genome-wide phylogenetic analysis of fungi. Nucleic Acids Res. 2022, 51, D777–D784. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Frith, M.C. A new repeat-masking method enables specific detection of homologous sequences. Nucleic Acids Res. 2011, 39, e23. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Slater, G.S.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef]
- Bateman, A. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef]
- Johnson, A.D.; Handsaker, R.E.; Pulit, S.L.; Nizzari, M.M.; O’Donnell, C.J.; de Bakker, P.I.W. SNAP: A web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 2008, 24, 2938–2939. [Google Scholar] [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, X.; Govers, F. The mysterious route of sterols in oomycetes. PLoS Pathog. 2021, 17, e1009591. [Google Scholar] [CrossRef]
- Latijnhouwers, M.; de Wit, P.J.G.M.; Govers, F. Oomycetes and fungi: Similar weaponry to attack plants. Trends Microbiol. 2003, 11, 462–469. [Google Scholar] [CrossRef]
- Zerillo, M.M.; Adhikari, B.N.; Hamilton, J.P.; Buell, C.R.; Lévesque, C.A.; Tisserat, N. Carbohydrate-Active Enzymes in Pythium and Their Role in Plant Cell Wall and Storage Polysaccharide Degradation. PLoS ONE 2013, 8, e72572. [Google Scholar] [CrossRef] [PubMed]
- Breton, C.; Šnajdrová, L.; Jeanneau, C.; Koča, J.; Imberty, A. Structures and mechanisms of glycosyltransferases. Glycobiology 2005, 16, 29R–37R. [Google Scholar] [CrossRef] [PubMed]
- Gavande, P.V.; Goyal, A.; Fontes, C.M.G.A. Carbohydrates and Carbohydrate-Active enZymes (CAZyme): An overview. In Glycoside Hydrolases; Goyal, A., Sharma, K., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 1–23. [Google Scholar]
- Levasseur, A.; Drula, E.; Lombard, V.; Coutinho, P.M.; Henrissat, B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 2013, 6, 41. [Google Scholar] [CrossRef]
- Villarreal, F.; Stocchi, N.; Ten Have, A. Functional classification and characterization of the fungal glycoside hydrolase 28 protein family. J. Fungi 2022, 8, 217. [Google Scholar] [CrossRef]
- Blackman, L.M.; Cullerne, D.P.; Hardham, A.R. Bioinformatic characterisation of genes encoding cell wall degrading enzymes in the Phytophthora parasitica genome. BMC Genom. 2014, 15, 785. [Google Scholar] [CrossRef]
- Chakraborty, S.; Rani, A.; Dhillon, A.; Goyal, A. Polysaccharide Lyases. In Current Developments in Biotechnology and Bioengineering; Pandey, A., Negi, S., Soccol, C.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 527–539. [Google Scholar]
- Lin, S.-C.; Lin, I.P.; Chou, W.-I.; Hsieh, C.-A.; Liu, S.-H.; Huang, R.-Y.; Sheu, C.-C.; Chang, M.D.-T. CBM21 starch-binding domain: A new purification tag for recombinant protein engineering. Protein Expr. Purif. 2009, 65, 261–266. [Google Scholar] [CrossRef]
- Lamour, K.H.; Mudge, J.; Gobena, D.; Hurtado-Gonzales, O.P.; Schmutz, J.; Kuo, A.; Miller, N.A.; Rice, B.J.; Raffaele, S.; Cano, L.M.; et al. Genome sequencing and mapping reveal loss of heterozygosity as a mechanism for rapid adaptation in the vegetable pathogen Phytophthora capsici. Mol. Plant Microb. Interact. 2012, 25, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Stam, R.; Jupe, J.; Howden, A.J.; Morris, J.A.; Boevink, P.C.; Hedley, P.E.; Huitema, E. Identification and characterisation CRN effectors in Phytophthora capsici shows modularity and functional diversity. PLoS ONE 2013, 8, e59517. [Google Scholar] [CrossRef]
- Haas, B.J.; Kamoun, S.; Zody, M.C.; Jiang, R.H.; Handsaker, R.E.; Cano, L.M.; Grabherr, M.; Kodira, C.D.; Raffaele, S.; Torto-Alalibo, T.; et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 2009, 461, 393–398. [Google Scholar] [CrossRef]
- Dale, A.L.; Feau, N.; Everhart, S.E.; Dhillon, B.; Wong, B.; Sheppard, J.; Bilodeau, G.J.; Brar, A.; Tabima, J.F.; Shen, D.; et al. Mitotic recombination and rapid genome evolution in the invasive forest pathogen Phytophthora ramorum. Mbio 2019, 10, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Raffaele, S.; Kamoun, S. The two-speed genomes of filamentous pathogens: Waltz with plants. Curr. Opin. Genet. Dev. 2015, 35, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Petre, B.; Contreras, M.P.; Bozkurt, T.O.; Schattat, M.H.; Sklenar, J.; Schornack, S.; Abd-El-Haliem, A.; Castells-Graells, R.; Lozano-Durán, R.; Dagdas, Y.F.; et al. Host-interactor screens of Phytophthora infestans RXLR proteins reveal vesicle trafficking as a major effector-targeted process. Plant Cell 2021, 33, 1447–1471. [Google Scholar] [CrossRef]
- Ai, G.; Yang, K.; Ye, W.; Tian, Y.; Du, Y.; Zhu, H.; Li, T.; Xia, Q.; Shen, D.; Peng, H.; et al. Prediction and characterization of RXLR effectors in Pythium species. Mol. Plant-Microbe Interact. 2020, 33, 1046–1058. [Google Scholar] [CrossRef]
- Meijer, H.J.G.; Mancuso, F.M.; Espadas, G.; Seidl, M.F.; Chiva, C.; Govers, F.; Sabidó, E. Profiling the secretome and extracellular proteome of the potato late blight pathogen Phytophthora infestans. Mol. Cell. Proteom. 2014, 13, 2101–2113. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Assembly | SS21 | CBS 119.80 | HF1 |
|---|---|---|---|
| Sequencing technology | Illumina HiSeq | Illumina NextSeq | Illumina HiSeq and PacBio RS |
| Accession (NCBI) | JAKLCF000000000.2 | JAADXV000000000.1 | QLOC00000000.1 |
| Genome size (Mbp) | 35.3 | 33.85 | 41.73 |
| Contigs number | 2843 | 3685 | 44 |
| Largest contig (bp) | 124,174 | 185,880 | 3,173,098 |
| Contig N50 (bp) | 31,500 | 29,240 | 1,512,378 |
| Contig N90 (bp) | 5364 | 4392 | 634,848 |
| GC content | 59% | 58.85% | 58.17% |
| BUSCO genome completeness | 99.0% | 90.60% | 92.20% |
| Genome coverage | 412× | 78× | 87× |
| Predicted genes number | 12,619 | 12,693 | 14,537 |
| BUSCO protein completeness | 99% | 99% | 99% |
| Category | Family | Number of Proteins Per Strain | ||
|---|---|---|---|---|
| SS21 | CBS 119.80 | HF1 | ||
| MAMP | Sterol binding proteins | 42 | 52 | 57 |
| Transglutaminase proteins | 4 | 4 | 4 | |
| Apoplastic effectors | CAZymes ** | 89 | 107 | 146 |
| Glucanase inhibitors | 6 | 6 | 12 | |
| Phytotoxins | 0 | 0 | 0 | |
| Necrosis inducing proteins | 28 | 28 | 52 | |
| Cutinases | 0 | 0 | 0 | |
| Protease inhibitors | 27 | 26 | 28 | |
| Cysteine protease inhibitor | 3 | 2 | 2 | |
| Cytoplasmic effectors | CRN * | 85 | 100 | 112 |
| RXLR * | 32 | 62 | 36 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villanueva, O.; Nguyen, H.D.T.; Ellouze, W. Profiling the Genomes and Secreted Effector Proteins in Phytopythium vexans Global Strains. J. Fungi 2025, 11, 477. https://doi.org/10.3390/jof11070477
Villanueva O, Nguyen HDT, Ellouze W. Profiling the Genomes and Secreted Effector Proteins in Phytopythium vexans Global Strains. Journal of Fungi. 2025; 11(7):477. https://doi.org/10.3390/jof11070477
Chicago/Turabian StyleVillanueva, Oscar, Hai D. T. Nguyen, and Walid Ellouze. 2025. "Profiling the Genomes and Secreted Effector Proteins in Phytopythium vexans Global Strains" Journal of Fungi 11, no. 7: 477. https://doi.org/10.3390/jof11070477
APA StyleVillanueva, O., Nguyen, H. D. T., & Ellouze, W. (2025). Profiling the Genomes and Secreted Effector Proteins in Phytopythium vexans Global Strains. Journal of Fungi, 11(7), 477. https://doi.org/10.3390/jof11070477