
Transcriptomic Profiling of Thermotolerant Sarcomyxa edulis PQ650759 Reveals the Key Genes and Pathways During Fruiting Body Formation

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Mycelial and Fruiting Body Cultures
2.2. Transcriptome Sequencing
2.2.1. Sample RNA Extraction, cDNA Library Construction, and Sequencing
2.2.2. Data Processing and Differential Gene Expression Analysis
2.2.3. Quantitative RT-PCR Validation
3. Results
3.1. Quality Control of Sequencing Data and FPKM
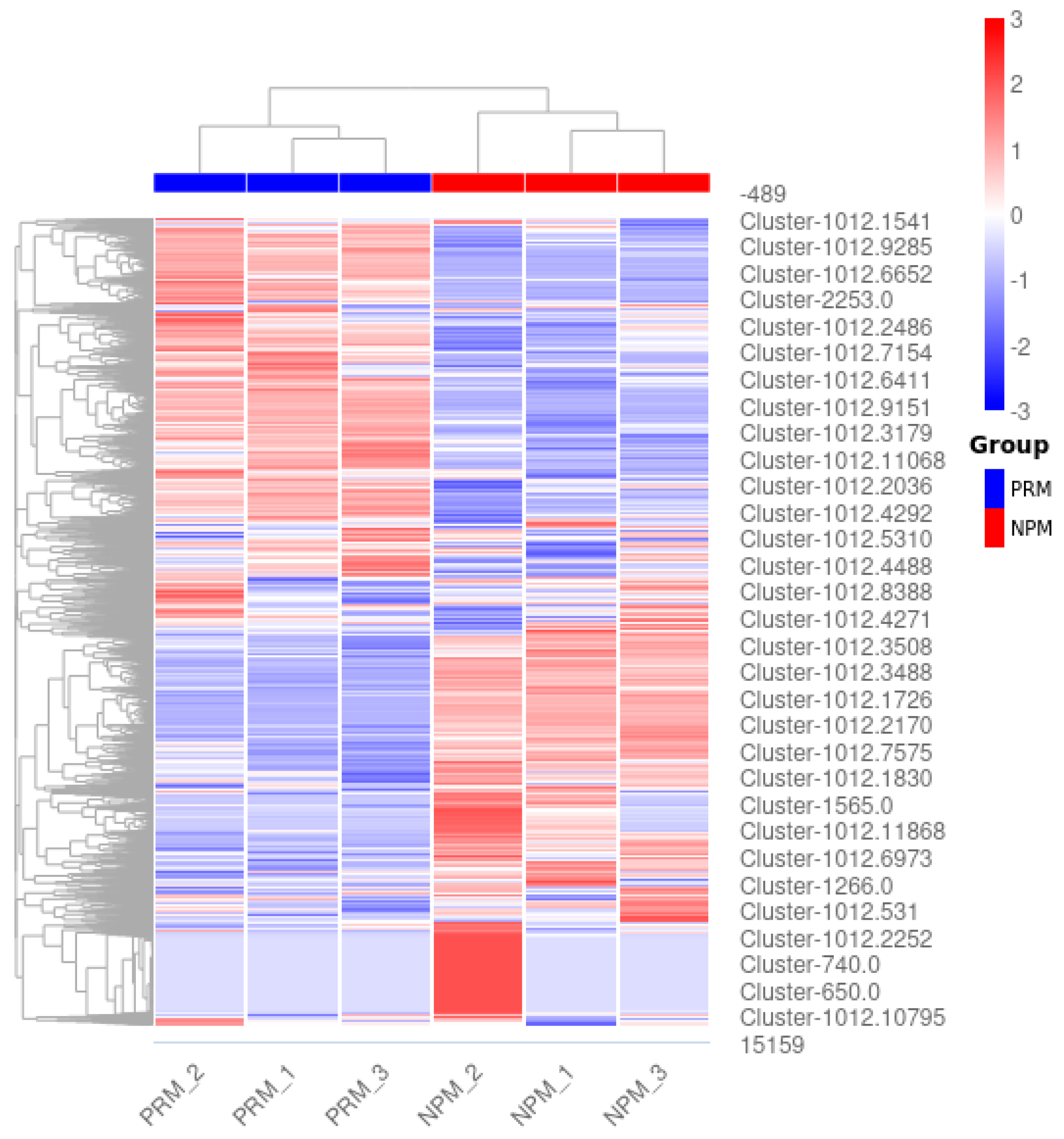
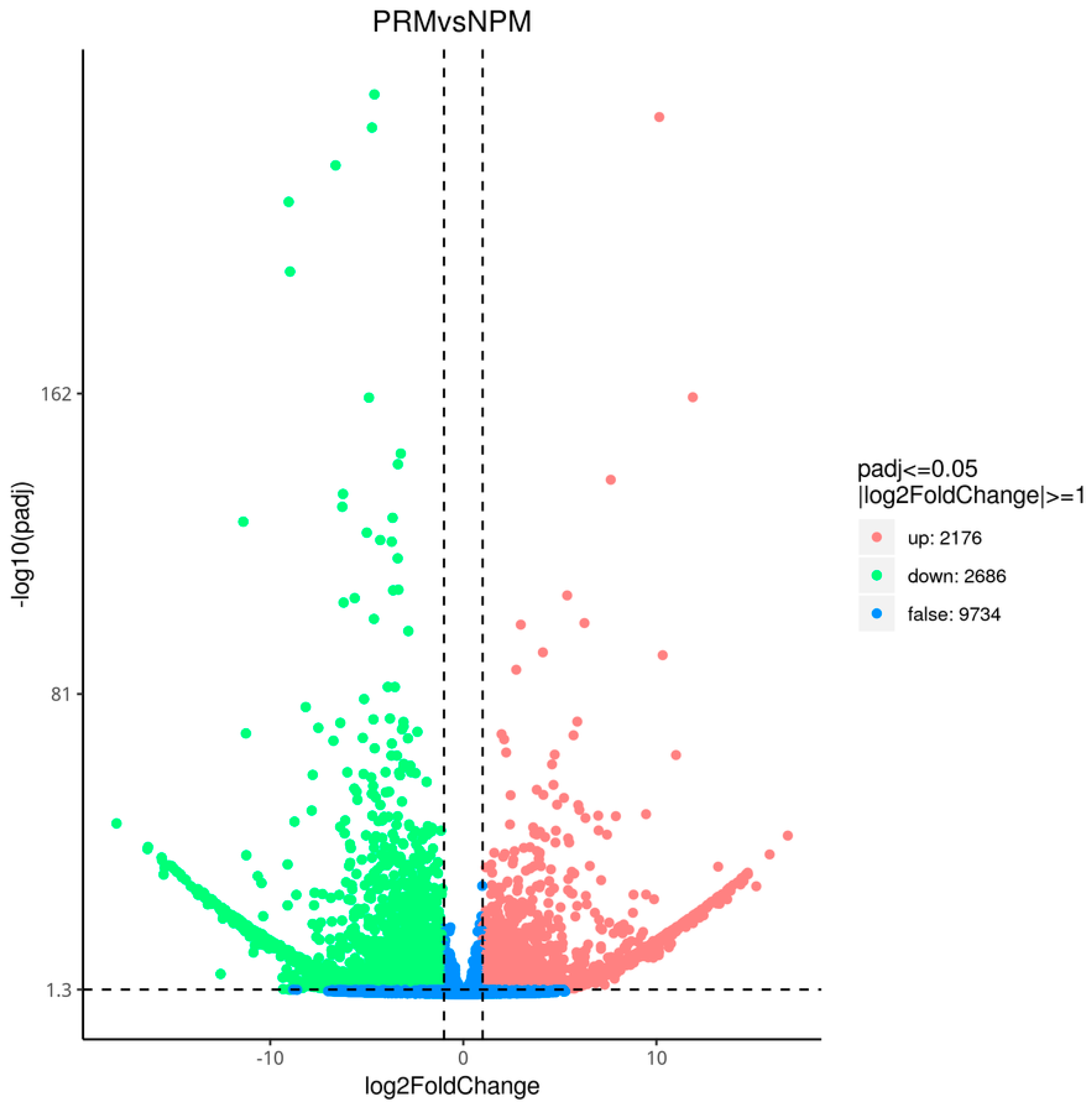
3.2. Screening of DEGs
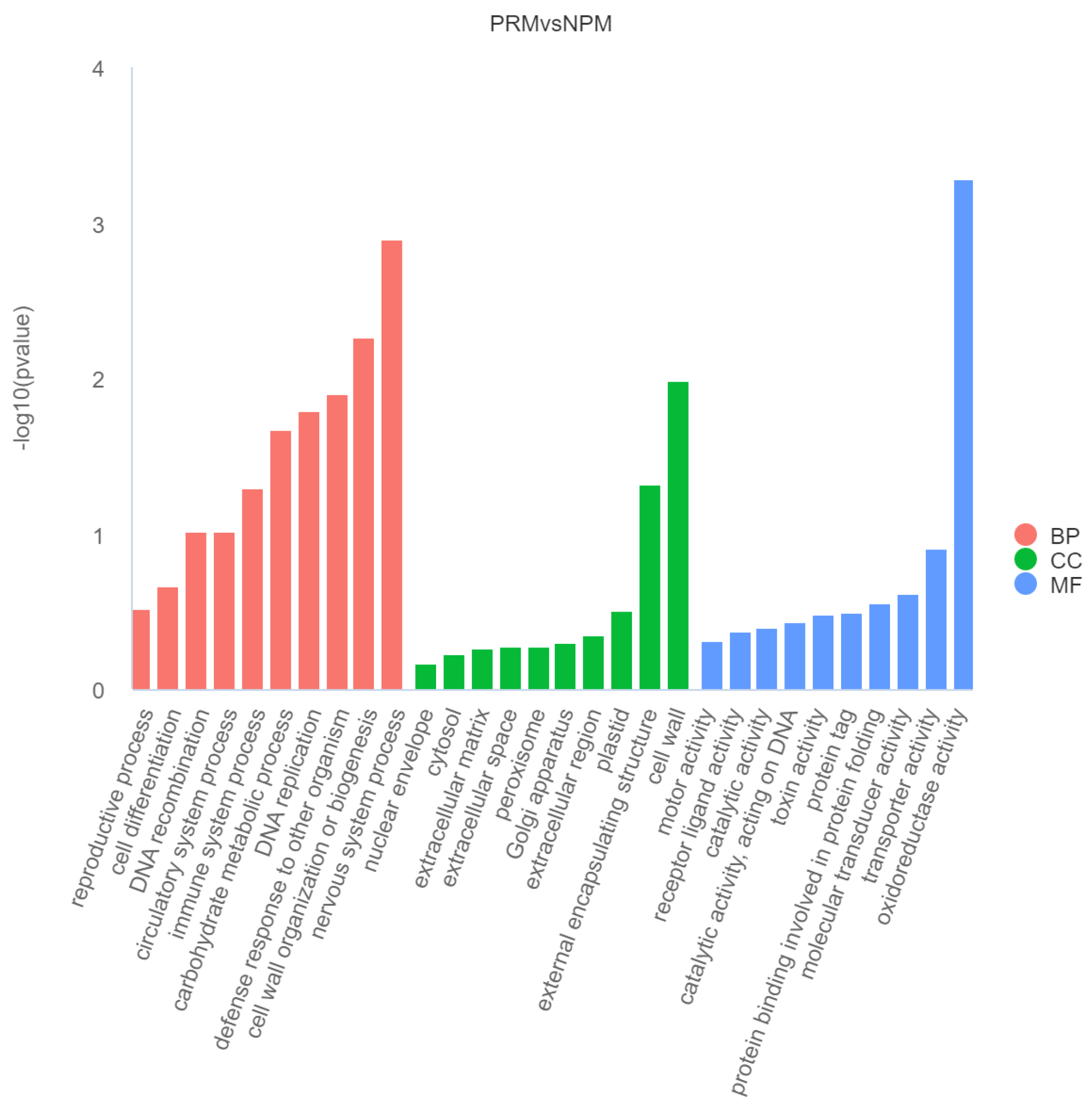
3.3. GO Functional Enrichment Analysis of DEGs
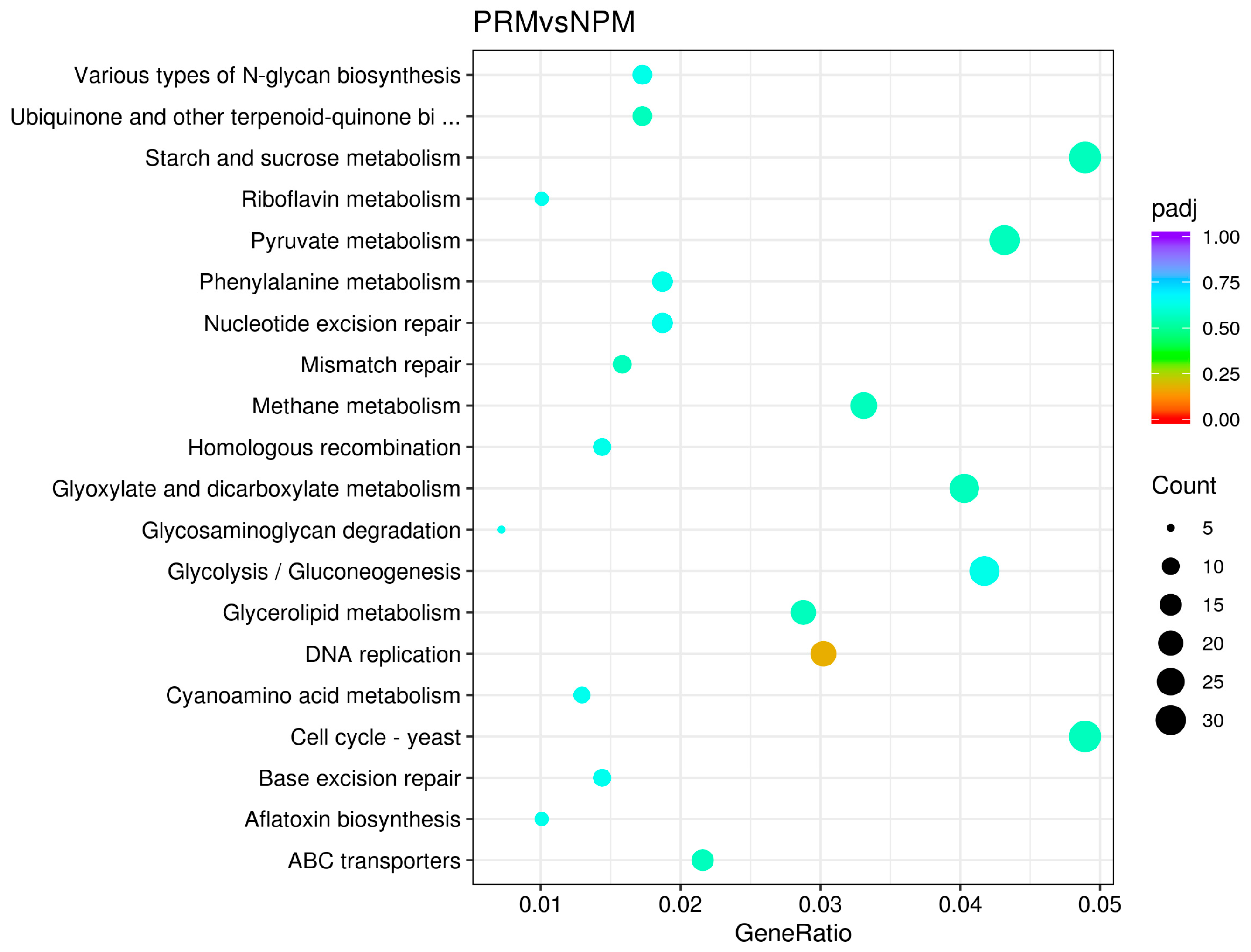
3.4. KEGG Metabolic Pathway Enrichment Analysis of DEGs
3.5. Analysis of Key Genes for Fruiting Body Occurrence of S. edulis
3.5.1. Genetic Information Processing and Its Key Genes
3.5.2. Carbohydrate Metabolism and Its Key Genes
3.5.3. Biosynthetic Metabolism and Its Key Genes
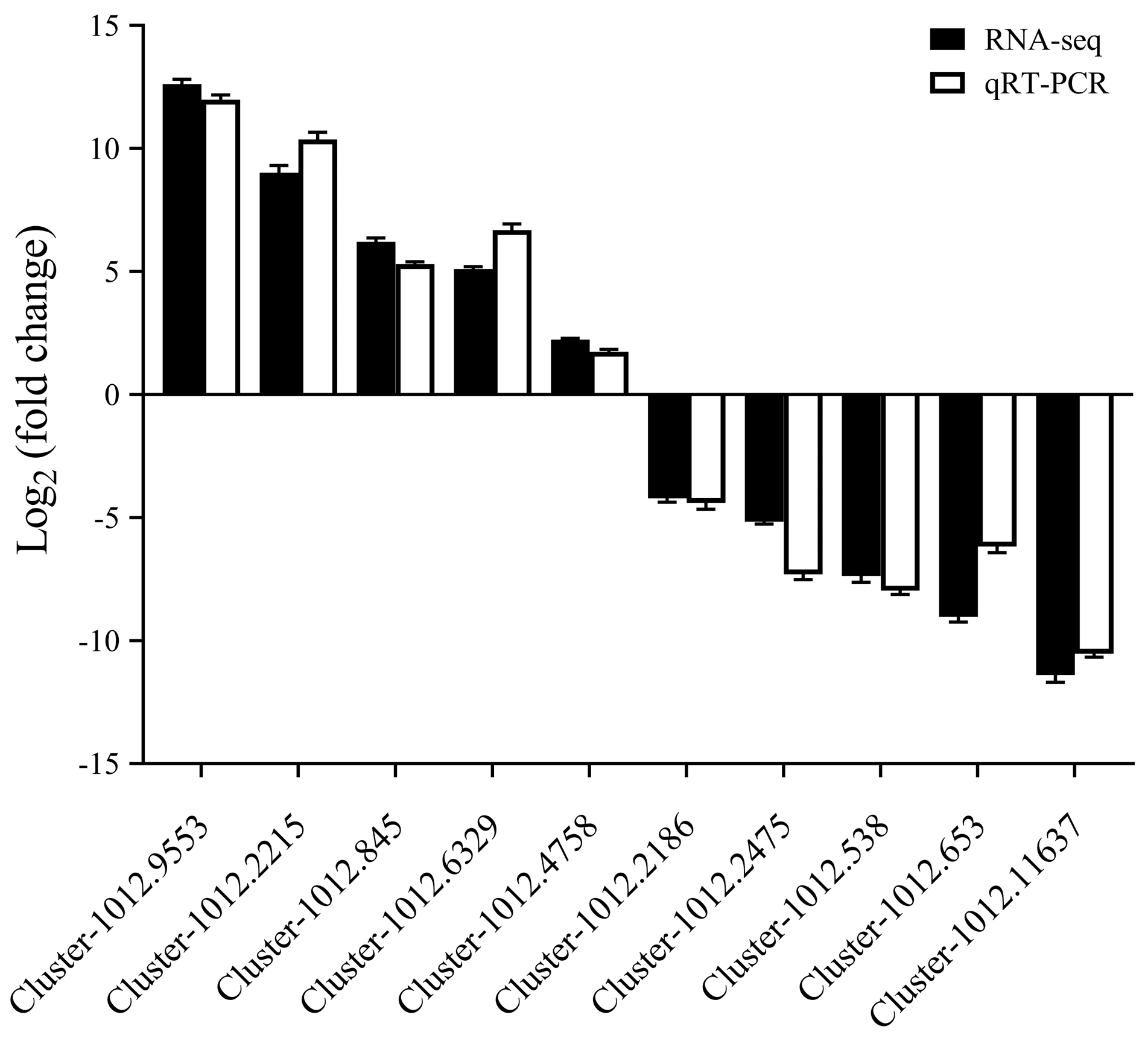
3.6. qRT-PCR
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tian, F.; Li, C.; Li, Y. Genomic Analysis of Sarcomyxa edulis Reveals the Basis of Its Medicinal Properties and Evolutionary Relationships. Front. Microbiol. 2021, 12, 652324. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, Y.; Sheng, C.; Wang, F.; Zhang, P.; Qi, Y.; Wang, J.; Shi, L.; Yu, H.; Zhao, J. Screening and Characterization of Wild Sarcomyxa edulis Strains from Heilongjiang, China, for Strain Development. Horticulturae 2024, 10, 1061. [Google Scholar] [CrossRef]
- Qaswaa, Y.J.; Nameer, K.M. Protective rules of natural antioxidants against gamma-induced damage—A review. Food Sci. Nutr. 2021, 9, 5263–5278. [Google Scholar]
- Duan, C.; Tian, T.; Yao, L.; Lv, J.-H.; Tian, F.-H.; Jia, C.-W.; Li, C.-T. Artificial culvivation and evaluation of two late fall oyster strains (Sarcomyxa edulis) from jilin, China. Mycol. Prog. 2023, 22, 47. [Google Scholar] [CrossRef]
- Duan, C.; Yao, L.; Lv, J.-H.; Jia, C.-W.; Tian, F.-H.; Li, C.-T. Systematic analysis of changes across different developmental stages of the mushroom Sarcomyxa eduli. Gene 2022, 824, 146450. [Google Scholar] [CrossRef]
- Pan, X.R. Macrofungi of the Xiaoxingan Mts; Northeastern Forestry University Press: Harbin, China, 1995; pp. 10–15. [Google Scholar]
- Ma, Y.; Mizino, T.; Ito, H. Antitumor activity of some polysaccharides isolated from a Chinese mushroom, “Huangmo” the fruiting body of Hohenbuehelia serotina. Agric. Biol. Chem. 1991, 55, 2701–2710. [Google Scholar] [CrossRef]
- Inafuku, M.; Nagao, K.; Nomura, S.; Shirouchi, B.; Inoue, N.; Nagamori, N.; Nakayama, H.; Toda, T.; Yanagita, T. Protective effects of fractional extracts from Panellus serotinus on nonalcoholic fatty liver disease in obese, diabetic db/db mice. Br. J. Nutr. 2012, 107, 639–646. [Google Scholar] [CrossRef]
- Zhang, R.; Zhao, L.Y.; Wang, H.X.; Ng, T.B. A novel ribonuclease with antiproliferative activity toward leukemia and lymphoma cells and HIV-1 reverse transcriptase inhibitory activity from the mushroom. Hohenbuehelia serotina. Int. J. Mol. Med. 2014, 33, 209–214. [Google Scholar] [CrossRef]
- Dai, Y.C.; Niemela, T.; Qin, G.F. Changbai wood-rotting fungi. a new pleurotoid species Panellus edulis. Ann. Bot. Fennici 2003, 40, 107–112. [Google Scholar]
- Saito, T.; Tonouchi, A.; Harada, Y. Biological characteristics and molecular phylogeny of Sarcomyxa edulis comb. nov. and S. serotina. Jpn. J. Mycol. 2014, 55, 19–28. [Google Scholar]
- Nagy, L.G.; Vonk, P.J.; Künzler, M.; Földi, C.; Virágh, M.; Ohm, R.A.; Hennicke, F.; Bálint, B.; Csernetics, Á.; Hegedüs, B.; et al. Lessons on fruiting body morphogenesis from genomes and transcriptomes of Agaricomycetes. Stud. Mycol. 2023, 104, 1–85. [Google Scholar] [CrossRef] [PubMed]
- Royse, D.J.; Baars, J.; Tan, Q. Current overview of mushroom production in the world. In Edible and Medicinal Mushrooms: Technology and Applications; Wiley: Hoboken, NJ, USA, 2017. [Google Scholar] [CrossRef]
- Ma, G.; Yang, W.; Zhao, L.; Pei, F.; Fang, D.; Hu, Q. A critical review on the health promoting effects of mushrooms nutraceuticals. Food Sci. Human Wellness 2018, 7, 125–133. [Google Scholar] [CrossRef]
- Whiteford, J.R.; Thurston, C.F. The molecular genetics of cultivated mushrooms. Adv. Microbiol. Physiol. 2020, 42, 1–23. [Google Scholar] [CrossRef]
- Kües, U.; Liu, Y. Fruiting body production in basidiomycetes. Appl. Microbiol. Biotechnol. 2000, 54, 141–152. [Google Scholar] [CrossRef]
- Ando, A.; Harada, A.; Miura, K.; Tamai, Y. A gene encoding a hydrophobin, fvh1, is specifically expressed after the induction of fruiting in the edible mushroom Flammulina velutipes. Curr. Genet. 2001, 39, 190–197. [Google Scholar] [CrossRef]
- Joh, J.H.; Kim, K.Y.; Lim, J.H.; Son, E.S.; Park, H.R.; Park, Y.J.; Kong, W.S.; Yoo, Y.B.; Lee, C.S. Comparative analysis of expressed sequence tags from Flammulina velutipes at different developmental stages. J. Microbiol. Biotechnol. 2009, 19, 774–780. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, A.; Chen, Y.; Wang, Q.; Wang, W. Combined analyses of transcriptome and proteome during fruiting body development of Flammulina filiformis. Mycosystema 2024, 43, 240109-1. [Google Scholar] [CrossRef]
- Tong, X.; Zhang, H.; Wang, F.; Xue, Z.; Cao, J.; Peng, C.; Guo, J. Comparative transcriptome analysis revealed genes involved in the fruiting body development of Ophiocordyceps sinensis. PeerJ 2020, 1, e8379. [Google Scholar] [CrossRef]
- Zhou, Z.X.; Dong, X.L.; Li, C.R. Comparative transcriptome analysis of different developmental stage of Bactrocera minax (Diptera: Tephritidae): Implication of the molecular basis of its obligatory diapause induction. Comp. Biochem. Physiol. D Genom. Proteom. 2021, 38, 100818. [Google Scholar] [CrossRef]
- Davidson, R.M.; Gowda, M.; Moghe, G.; Lin, H.; Vaillancourt, B.; Shiu, S.-H.; Jiang, N.; Robin Buell, C. Comparative transcriptomics of three Poaceae species reveals patterns of gene expression evolution. Plant J. 2012, 71, 492–502. [Google Scholar] [CrossRef]
- Duan, C.; Tian, F.; Yao, L.; Lv, J.; Jia, C.; Li, C. Comparative transcriptome and WGCNA reveal key genes involved in lignocellulose degradation in Sarcomyxa edulis. Sci. Rep. 2022, 12, 18379. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Kim, D.; Kim, J. Comparative transcriptome analysis of dikaryotic mycelia and mature fruiting bodies in the edible mushroom Lentinula edodes. Sci. Rep. 2018, 8, 8983. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Dai, Y.; Yang, C.; Wei, P.; Song, B.; Yang, Y.; Sun, L.; Zhang, Z.; Li, Y. Comparative Transcriptome Analysis Identified Candidate Genes Related to Bailinggu Mushroom Formation and Genetic Markers for Genetic Analyses and Breeding. Sci. Rep. 2017, 7, 9266. [Google Scholar] [CrossRef]
- Son, S.; Park, Y.; Jung, E.; Singh, D.; Lee, Y.; Kim, J.; Lee, C. Integrated Metabolomics and Transcriptomics Unravel the Metabolic Pathway Variations for Different Sized Beech Mushrooms. Int. J. Mol. Sci. 2019, 20, 6007. [Google Scholar] [CrossRef]
- Heidary, M.; Pahlevan, K.M. TRIzol-based RNA Extraction: A Reliable Method for Gene Expression Studies. J. Sci. 2014, 25, 13–17. [Google Scholar]
- Ulrike, S.; Neil, A.S.; Wang, M.B. A fast and efficient method for preparation of high-quality RNA from fungal mycelia. BMC Res. Notes 2013, 6, 7. [Google Scholar]
- Parkhomchuk, D.; Borodina, T.; Amstislavskiy, V.; Banaru, M.; Hallen, L.; Krobitsch, S.; Lehrach, H.; Soldatov, A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009, 37, e123. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 23, 3150–3152. [Google Scholar] [CrossRef]
- Smith-Unna, R.; Boursnell, C.; Patro, R.; Hibberd, J.M.; Kelly, S. TransRate: Reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 2016, 26, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Zhang, Q. The Effect and Mechanism of CO2 on Growth of Fruiting Body of Pleurotus giganteus. Master’s Thesis, Ludong University, Yantai, China, 2023. [Google Scholar]
- Sen, K.; Kinoshita, H.; Tazuke, K.; Maki, Y.; Yoshiura, Y.; Yakushi, T.; Shibai, H.; Kurosawa, S.-I. Analysis of the sexual development-promoting region of Schizophyllum commune trp1 gene. Biosci. Biotechnol. Biochem. 2016, 80, 2033–2044. [Google Scholar] [CrossRef]
- Baars, J.J.P.; Scholtmeijer, K.; Sonnenberg, A.S.M.; van Peer, A. Critical Factors Involved in Primordia Building in Agaricus bisporus: A Review. Molecules 2020, 25, 2984. [Google Scholar] [CrossRef]
- Chen, T.; Yuan, B.; Lian, Y.; Ke, L.; Wu, Z.; Deng, Y. Transcriptome analysis of Auricularia cornea fruiting bodies at different developmental stages. Mycosystema 2024, 43, 240082. [Google Scholar] [CrossRef]
- Courty, P.E.; Hoegger, P.J.; Kilaru, S.; Kohler, A.; Buée, M.; Garbaye, J.; Martin, F.; Kües, U. Phylogenetic analysis, genomic organization, and expression analysis of multi-copper oxidases in the ectomycorrhizal basidiomycete Laccaria bicolor. New Phytol. 2009, 182, 736–750. [Google Scholar] [CrossRef]
- Wang, W.; Liu, F.; Jiang, Y.; Wu, G.; Guo, L.; Chen, R.; Chen, B.; Lu, Y.; Dai, Y.; Xie, B. The multigene family of fungal laccases and their expression in the white rot basidiomycete Flammulina velutipes. Gene 2015, 563, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, M.; Xu, Y.; van der Wel, H.; Walden, P.; Hartson, S.D.; West, C.M. Glycosylation of Skp1 Promotes Formation of Skp1–Cullin-1–F-box Protein Complexes in Dictyostelium. Mol. Cell. Proteom. 2014, 14, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Gstaiger, M.; Marti, A.; Krek, W. Association of human SCFSKP2 subunit p19SKP1 with interphase centrosomes and mitotic spindle poles. Exp. Cell Res. 1999, 247, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H. Research on LeSKP1 Gene Function and Auxin Rapid Response Genes of Lentinula edodes. Master’s Thesis, Huazhong Agricultural University, Wuhan, China, 2023. [Google Scholar]
- Bressan, D.A.; Baxter, B.K.; Petrini, J.H.J. The Mre11-Rad50-Xrs2 Protein Complex Facilitates Homologous Recombination-Based Double-Strand Break Repair in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999, 19, 7681–7687. [Google Scholar] [CrossRef]
- Cosma, M.A.; Curtis, N.L.; Pain, C.; Kriechbaumer, V.; Bolanos-Garcia, V.M. Biochemical, biophysical, andfunctional characterisation of the E3 ubiquitin ligase APC/C regulator CDC20 from Arabidopsis thaliana. Front. Physiol. 2022, 13, 938688. [Google Scholar] [CrossRef]
- Ding, D.; Muthuswamy, S.; Meier, I. Functional interaction between the Arabidopsis orthologs of spindle assembly checkpoint proteins MAD1 and MAD2 and the nucleoporin NUA. Plant Mol. Biol. 2012, 79, 203–216. [Google Scholar] [CrossRef]
- Schulman, B.A.; Carrano, A.C.; Jeffrey, P.D.; Bowen, Z.; Kinnucan, E.R.; Finnin, M.S.; Elledge, S.J.; Harper, J.W.; Pagano, M.; Pavletich, N.P. Insights into SCF ubiquitin ligases from the structure of the Skp1-Skp2 complex. Nature 2000, 408, 381–386. [Google Scholar] [CrossRef]
- Devoto, A.; Nieto-Rostro, M.; Xie, D.; Ellis, C.; Harmston, R.; Patrick, E.; Davis, J.; Sherratt, L.; Coleman, M.; Turner, J.G. COI1 links jasmonate signalling and fertility to the SCF ubiquitin-ligase complex in Arabidopsis. Plant J. 2002, 32, 457–466. [Google Scholar] [CrossRef]
- Xu, L.; Liu, F.; Lechner, E.; Genschik, P.; Crosby, W.L.; Ma, H.; Peng, W.; Huang, D.; Xie, D. The SCFCOI1 ubiquitin-ligase complexes are required for jasmonate response in Arabidopsis. Plant Cell 2002, 14, 1919–1935. [Google Scholar] [CrossRef]
- Gomi, K.; Sasaki, A.; Itoh, H.; Ueguchi-Tanaka, M.; Ashikari, M.; Kitano, H.; Matsuoka, M. GID2, an F-box subunit of the SCF E3 complex, specifically interacts with phosphorylated SLR1 protein and regulates the gibberellin-dependent degradation of SLR1 in rice. Plant J. 2004, 37, 626–634. [Google Scholar] [CrossRef]
- Fleet, C.M.; Sun, T.P. A DELLA cate balance: The role of gibberellin in plant morphogenesis. Curr. Opin. Plant Biol. 2005, 8, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Ueguchi-Tanaka, M.; Ashikari, M.; Nakajima, M.; Itoh, H.; Katoh, E.; Kobayashi, M.; Chow, T.Y.; Hsing, Y.I.; Kitano, H.; Yamaguchi, I.; et al. GIBBERELLIN INSENSITIVE DWARF1 encodes a soluble receptor for gibberellin. Nature 2005, 437, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Landherr, L.L.; Frohlich, M.W.; Leebens-Mack, J.; Ma, H.; DePamphilis, C.W. Patterns of gene duplication in the plant SKP1 gene family in angiosperms: Evidence for multiple mechanisms of rapid gene birth. Plant J. 2007, 50, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Miyazaki, Y.; Kaneko, S.; Shishido, K. Developmental regulator Le. CDC5 of the mushroom Lentinula edodes: Analyses of its amount in each of the stages of fruiting-body formation and its distribution in parts of the fruiting bodies. FEMS Microbiol. Lett. 2006, 261, 60–63. [Google Scholar] [CrossRef]
- Nakazawa, T.; Miyazaki, Y.; Kaneko, S.; Shishido, K. Stimulative effects of light and a temperature downshift on transcriptional expressions of developmentally regulated genes in the initial stages of fruiting-body formation of the basidiomycetous mushroom Lentinula edodes. FEMS Microbiol. Lett. 2008, 289, 67–71. [Google Scholar] [CrossRef]
- Markiewicz-Potoczny, M.; Lisby, M.; Lydall, D. A Critical Role for Dna2 at Unwound Telomeres. Genetics 2018, 209, 129–141. [Google Scholar] [CrossRef]
- Lodi, T.; Dallabona, C.; Nolli, C.; Goffrini, P.; Donnini, C.; Baruffini, E. DNA polymerase γ and disease: What we have learned from yeast. Front. Genet. 2015, 6, 106. [Google Scholar] [CrossRef]
- Cerritelli, S.M.; Crouch, R.J. Ribonuclease H: The enzymes in eukaryotes. FEBS J. 2009, 276, 1494–1505. [Google Scholar] [CrossRef]
- Tennen, R.I.; Haye, J.E.; Wijayatilake, H.D.; Arlow, T.; Ponzio, D.; Gammie, A.E. Cell-cycle and DNA damage regulation of the DNA mismatch repair protein Msh2 occurs at the transcriptional and post-transcriptional level. DNA Repair. 2013, 12, 97–109. [Google Scholar] [CrossRef]
- D’Amours, D.; Jackson, S.P. The MRE11 complex: At the crossroads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 317–327. [Google Scholar] [CrossRef]
- Culligan, K.M.; Meyer-Gauen, G.; Lyons-Wilder, J.; Hays, J.B. Evolutionary origin, diversification and specialization of eukaryotic MutS-homolog mismatch-repair proteins. Nucleic Acids Res. 2000, 28, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Grauvogel, C.; Brinkmann, H.; Petersen, J. Evolution of the Glucose-6-Phosphate Isomerase: The Plasticity of Primary Metabolism in Photosynthetic Eukaryotes. Mol. Biol. Evol. 2007, 24, 1611–1621. [Google Scholar] [CrossRef] [PubMed]
- Stover, N.A.; Dixon, T.A.; Cavalcanti, A.R.O.; Moreno, S.N. Multiple Independent Fusions of Glucose-6-Phosphate Dehydrogenase with Enzymes in the Pentose Phosphate Pathway. PLoS ONE 2011, 6, e22269. [Google Scholar] [CrossRef]
- Miclet, E.; Stoven, V.; Michels, P.A.; Opperdoes, F.R.; Lallemand, J.Y.; Duffieux, F. NMR spectroscopic analysis of the first two steps of the pentose-phosphate pathway elucidates the role of 6-phosphogluconolactonase. J. Biol. Chem. 2001, 276, 34840–34846. [Google Scholar] [CrossRef]
- Khosravi, C.; Battaglia, E.; Kun, R.S.; Dalhuijsen, S.; Visser, J.; Aguilar-Pontes, M.V.; Zhou, M.; Heyman, H.M.; Kim, Y.; Baker, S.E.; et al. Blocking hexose entry into glycolysis activates alternative metabolic conversion of these sugars and upregulates pentose metabolism in Aspergillus nidulans. BMC Genom. 2018, 19, 214. [Google Scholar] [CrossRef]
- Lu, S. Key Regulatory Benchmarks for Fruiting Body Development of Pleurotus ostreatus Shielded by Selection and Functional Assays. Master’s Thesis, Hebei University of Engineering, Handan, China, 2023. [Google Scholar]
- Wang, M.; Gu, B.; Huang, J.; Jiang, S.; Chen, Y.; Yin, Y.; Pan, Y.; Yu, G.; Li, Y.; Wong, B.; et al. Transcriptome and proteome exploration to provide a resource for the study of Agrocybe aegerita. PLoS ONE 2013, 8, e56686. [Google Scholar] [CrossRef]
- Son, H.; Min, K.; Lee, J. Differential roles of pyruvate decarboxylase in aerial and embedded mycelia of the ascomycete Gibberella zeae. FEMS Microbiol. Lett. 2012, 329, 123–130. [Google Scholar] [CrossRef]
- Dong, H.; Beer, S.V. Riboflavin Induces Disease Resistance in Plants by Activating a Novel Signal Transduction Pathway. Phytopathology 2000, 90, 801–811. [Google Scholar] [CrossRef]
- De Carvalho, M.A.A.P.; Popova, T.N.; Santos, T.M.M.D.; Zherebtsov, N.A. Effect of Metabolites of g-Aminobutyric Shunt on Activities of NAD- and NADP-Isocitrate Dehydrogenases and Aconitate Hydratase from Higher Plants. Biol. Bull. 2003, 30, 236–242. [Google Scholar] [CrossRef]
- Kesawat, M.S.; Kherawat, B.S.; Ram, C.; Singh, A.; Dey, P.; Gora, J.S.; Misra, N.; Chung, S.; Kumar, M. Genome-Wide Identification and Expression Profiling of Aconitase Gene Family Members Reveals Their Roles in Plant Development and Adaptation to Diverse Stress in Triticum aestivum L. Plants 2022, 11, 3475. [Google Scholar] [CrossRef]
- Giles, N. Johnson. Physiology of PSI cyclic electron transport in higher plants. Biochim. Biophys. Acta 2011, 1807, 384–389. [Google Scholar] [CrossRef]
- Parul, T.; Deeksha, S.; Shivangi, M.; Ayushi, S.; Rajiv, R. Upcoming progress of transcriptomics studies on plants: An overview. Front. Plant Sci. 2022, 13, 1030890. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Error Rate | Q20 | Q30 | GC pct |
|---|---|---|---|---|---|---|
| PRM1 | 22749365 | 208998714 | 0.01 | 98.31 | 94.99 | 52.10 |
| PRM2 | 24370641 | 22972599 | 0.01 | 98.34 | 95.12 | 51.97 |
| PRM3 | 21640785 | 20346185 | 0.01 | 98.28 | 94.90 | 51.99 |
| NPM1 | 22260169 | 21646551 | 0.01 | 98.36 | 95.20 | 52.35 |
| NPM2 | 23581987 | 21386247 | 0.01 | 98.46 | 95.41 | 52.15 |
| NPM3 | 22721561 | 21143993 | 0.01 | 98.36 | 95.18 | 51.95 |
| Gene Name | Gene ID | log2FoldChange | p Value | padj |
|---|---|---|---|---|
| S-phase kinase-associated protein 1 | Cluster-1012.804 | 5.6367 | 2.54 × 10−3 | 6.89 × 10−3 |
| DNA replication ATP-dependent helicase/nuclease Dna2 | Cluster-1012.804 | 3.6928 | 3.19 × 10−7 | 1.69 × 10−6 |
| Double-strand break Repair protein MRE11 | Cluster-1012.4758 | 2.2675 | 1.07 × 10−16 | 1.65 × 10−15 |
| Glucose-6-phosphate isomerase | Cluster-1222.0 | −7.3875 | 5.33 × 10−3 | 1.34 × 10−2 |
| Pyruvate decarboxylase | Cluster-1012.7332 | −2.0115 | 1.75 × 10−25 | 5.74 × 10−24 |
| 6-Phosphogluconate dehydrogenase | Cluster-1012.2679 | −6.0057 | 5.99 × 10−63 | 1.35 × 10−60 |
| Hexosaminidase | Cluster-1012.10120 | −3.1094 | 1.59 × 10−15 | 2.19 × 10−14 |
| Riboflavin metabolism | Cluster-1012.6329 | 5.1008 | 1.40 × 10−17 | 2.37 × 10−16 |
| Aconitate hydratase | Cluster-1012.319 | −7.9724 | 1.84 × 10−3 | 5.17 × 10−3 |
| NAD(P)H dehydrogenase | Cluster-1012.8236 | −2.4582 | 8.41 × 10−8 | 4.83 × 10−7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Li, M.; Ma, H.; Wang, F.; Shi, L.; Wang, J.; Sheng, C.; Zhang, P.; Yu, H.; Zhao, J.; et al. Transcriptomic Profiling of Thermotolerant Sarcomyxa edulis PQ650759 Reveals the Key Genes and Pathways During Fruiting Body Formation. J. Fungi 2025, 11, 484. https://doi.org/10.3390/jof11070484
Liu Z, Li M, Ma H, Wang F, Shi L, Wang J, Sheng C, Zhang P, Yu H, Zhao J, et al. Transcriptomic Profiling of Thermotolerant Sarcomyxa edulis PQ650759 Reveals the Key Genes and Pathways During Fruiting Body Formation. Journal of Fungi. 2025; 11(7):484. https://doi.org/10.3390/jof11070484
Chicago/Turabian StyleLiu, Zitong, Minglei Li, Hongyu Ma, Fei Wang, Lei Shi, Jinhe Wang, Chunge Sheng, Peng Zhang, Haiyang Yu, Jing Zhao, and et al. 2025. "Transcriptomic Profiling of Thermotolerant Sarcomyxa edulis PQ650759 Reveals the Key Genes and Pathways During Fruiting Body Formation" Journal of Fungi 11, no. 7: 484. https://doi.org/10.3390/jof11070484
APA StyleLiu, Z., Li, M., Ma, H., Wang, F., Shi, L., Wang, J., Sheng, C., Zhang, P., Yu, H., Zhao, J., & Wang, Y. (2025). Transcriptomic Profiling of Thermotolerant Sarcomyxa edulis PQ650759 Reveals the Key Genes and Pathways During Fruiting Body Formation. Journal of Fungi, 11(7), 484. https://doi.org/10.3390/jof11070484