Innate Immune Responses to Avian Influenza Viruses in Ducks and Chickens


Abstract
1. Introduction
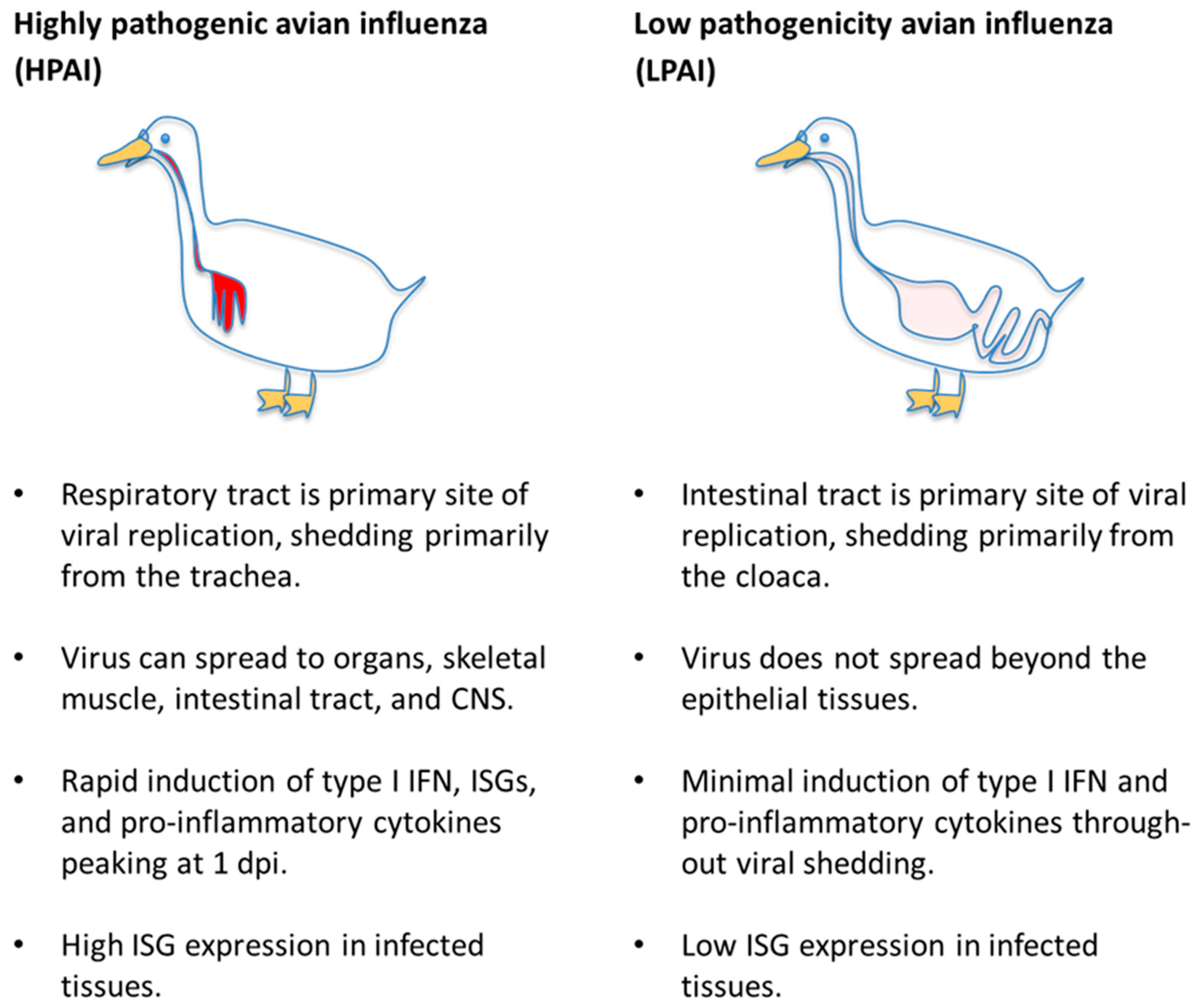
2. Sialic Acid Receptor Distribution and Influenza Virus Tissue Tropism in Ducks and Chickens
3. Factors Affecting Susceptibility to Disease in Mallards
3.1. Susceptibility of Different Anseriform Species
3.2. Age of Birds
3.3. Viral Strain-Dependent Differences in Infection Outcome
4. Innate Immune Signaling—Pro-Inflammatory and Interferon Responses
4.1. Inflammation
4.2. Type I and III Interferon Responses
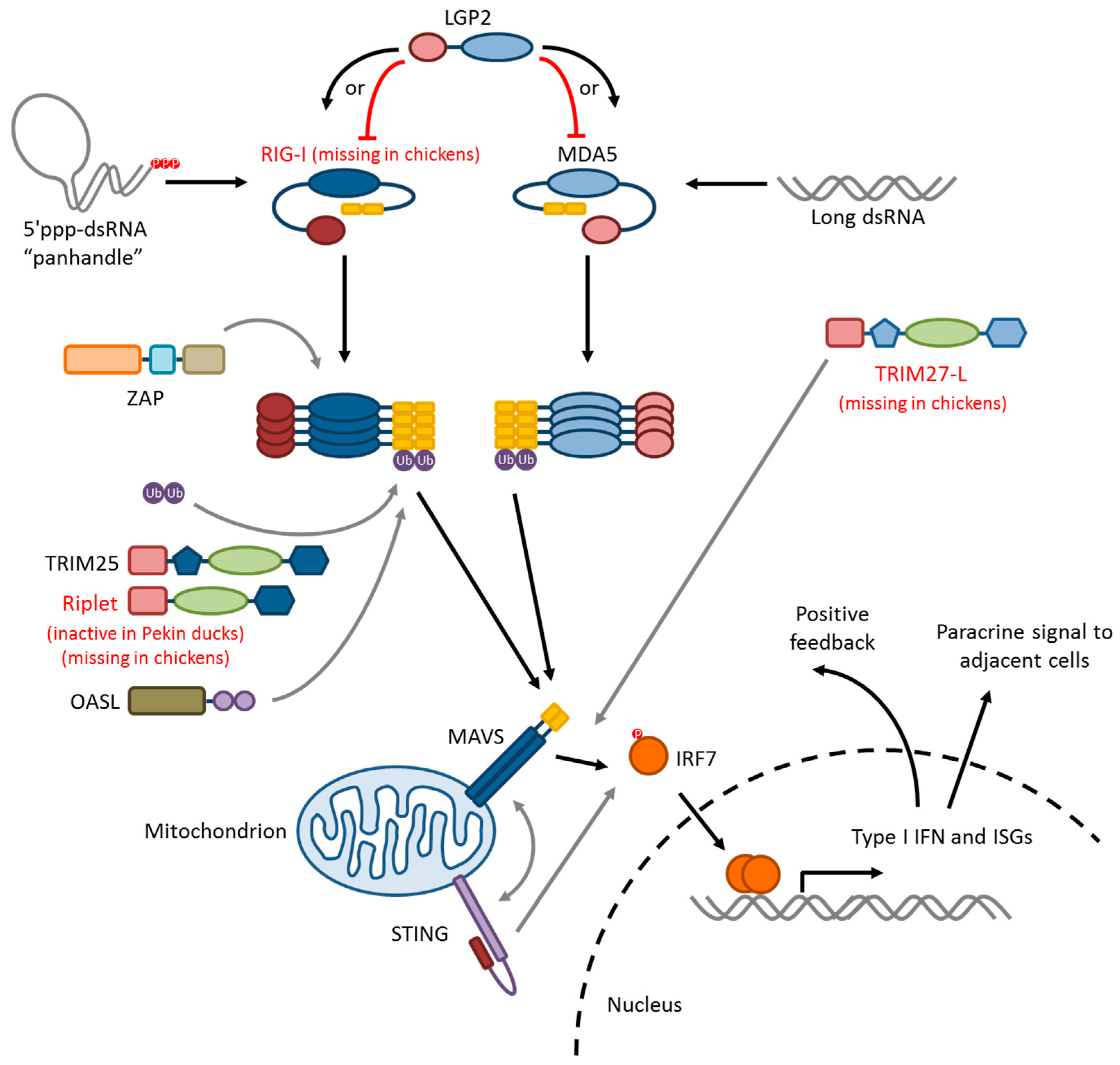
5. Influenza Virus Pattern Recognition—RIG-I-Like Receptors
5.1. RIG-I
5.2. MDA5
5.3. LGP2
5.4. RLR-Signaling Pathway
6. Modifiers of RIG-I/MAVS Signaling
6.1. Ubiquitin Ligases: TRIM25, Riplet, and RNF125
6.2. TRIM27-L and TRIM27.1
6.3. ZAP
6.4. STING
7. Influenza Virus Pattern Recognition—Duck Toll-Like Receptors
7.1. TLR7 and TLR 8
7.2. TLR3 and TRIF
8. Other Pattern Recognition Receptors Relevant to Influenza Infection
8.1. LSm14A—A Possible Viral RNA Sensor in Ducks
8.2. NLRP3
9. Interferon-Stimulated Genes (ISGs)
9.1. Viperin
9.2. IFIT5
9.3. PKR
9.4. CCL19
9.5. Mx
9.6. OASL
9.7. IFITM3
10. Viral Inhibition of Type I IFN Signaling—Influenza A Virus NS1
10.1. NS1 in Ducks
10.2. NS1 in Chickens
10.3. Species-Specificity of the C-Terminal PDZ-Binding Motif
11. Other Potential Mechanisms of Influenza Disease Resistance in Mallards
11.1. Rapid Apoptotic Response
11.2. Limiting Inflammation with USP18
11.3. Humoral Adaptive Immunity
11.4. Cellular Adaptive Immunity
11.5. MicroRNA Regulation
12. Final Note on Comparing Duck and Chicken Innate Immune Responses to Influenza Infection
13. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Taubenberger, J.K.; Kash, J.C. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [PubMed]
- Krauss, S.; Webster, R.G. Avian Influenza Virus Surveillance and Wild Birds: Past and Present. Avian Dis. 2010, 54, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Marchenko, V.Y.; Alekseev, A.Y.; Sharshov, K.A.; Petrov, V.N.; Silko, N.Y.; Susloparov, I.M.; Tserennorov, D.; Otgonbaatar, D.; Savchenko, I.A.; Shestopalov, A.M. Ecology of Influenza Virus in Wild Bird Populations in Central Asia. Avian Dis. 2012, 56, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Jourdain, E.; Gunnarsson, G.; Wahlgren, J.; Latorre-Margalef, N.; Bröjer, C.; Sahlin, S.; Svensson, L.; Waldenström, J.; Lundkvist, Å.; Olsen, B. Influenza Virus in a Natural Host, the Mallard: Experimental Infection Data. PLoS ONE 2010, 5, e8935. [Google Scholar] [CrossRef] [PubMed]
- Kida, H.; Yanagawa, R.; Matsuoka, Y. Duck influenza lacking evidence of disease signs and immune response. Infect. Immun. 1980, 30, 547–553. [Google Scholar] [PubMed]
- Olsen, B.; Munster, V.J.; Wallensten, A.; Waldenström, J.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Global Patterns of Influenza A Virus in Wild Birds. Science 2006, 312, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Runstadler, J.A.; Happ, G.M.; Slemons, R.D.; Sheng, Z.M.; Gundlach, N.; Petrula, M.; Senne, D.; Nolting, J.; Evers, D.L.; Modrell, A.; et al. Using RRT-PCR analysis and virus isolation to determine the prevalence of avian influenza virus infections in ducks at Minto Flats State Game Refuge, Alaska, during August 2005. Arch. Virol. 2007, 152, 1901–1910. [Google Scholar] [CrossRef]
- Kim, J.-K.; Negovetich, N.J.; Forrest, H.L.; Webster, R.G. Ducks: The “Trojan horses” of H5N1 influenza. Influenza Other Respir. Viruses 2009, 3, 121–128. [Google Scholar] [CrossRef]
- Swayne, D.E.; Suarez, D.L. Highly pathogenic avian influenza. Rev. Sci. Tech. 2000, 19, 463–475. [Google Scholar] [CrossRef]
- OIE—World Organisation for Animal Health. Health Manual of Diagnostic Tests and Vaccines for Terrestrial Animals 2018. Available online: http://www.oie.int/standard-setting/terrestrial-manual/access-online/ (accessed on 26 October 2018).
- Pantin-Jackwood, M.J.; Costa-Hurtado, M.; Shepherd, E.; DeJesus, E.; Smith, D.; Spackman, E.; Kapczynski, D.R.; Suarez, D.L.; Stallknecht, D.E.; Swayne, D.E. Pathogenicity and Transmission of H5 and H7 Highly Pathogenic Avian Influenza Viruses in Mallards. J. Virol. 2016, 90, 9967–9982. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Veldhuis Kroeze, E.J.B.; Reperant, L.A.; Richard, M.; Kuiken, T. Influenza virus and endothelial cells: A species specific relationship. Front. Microbiol. 2014, 5, 653. [Google Scholar] [CrossRef] [PubMed]
- Bertram, S.; Glowacka, I.; Steffen, I.; Kühl, A.; Pöhlmann, S. Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev. Med. Virol. 2010, 20, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Horimoto, T.; Kawaoka, Y. Reverse genetics provides direct evidence for a correlation of hemagglutinin cleavability and virulence of an avian influenza A virus. J. Virol. 1994, 68, 3120–3128. [Google Scholar] [PubMed]
- Rott, R. The pathogenic determinant of influenza virus. Vet. Microbiol. 1992, 33, 303–310. [Google Scholar] [CrossRef]
- Stieneke-Gröber, A.; Vey, M.; Angliker, H.; Shaw, E.; Thomas, G.; Roberts, C.; Klenk, H.D.; Garten, W. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J. 1992, 11, 2407–2414. [Google Scholar] [CrossRef] [PubMed]
- Matrosovich, M.; Zhou, N.; Kawaoka, Y.; Webster, R. The Surface Glycoproteins of H5 Influenza Viruses Isolated from Humans, Chickens, and Wild Aquatic Birds Have Distinguishable Properties. J. Virol. 1999, 73, 1146–1155. [Google Scholar] [PubMed]
- Yamada, S.; Shinya, K.; Takada, A.; Ito, T.; Suzuki, T.; Suzuki, Y.; Le, Q.M.; Ebina, M.; Kasai, N.; Kida, H.; et al. Adaptation of a Duck Influenza A Virus in Quail. J. Virol. 2012, 86, 1411–1420. [Google Scholar] [CrossRef]
- Alexander, D.J.; Parsons, G.; Manvell, R.J. Experimental assessment of the pathogenicity of eight avian influenza A viruses of H5 subtype for chickens, turkeys, ducks and quail. Avian Pathol. J. WVPA 1986, 15, 647–662. [Google Scholar] [CrossRef]
- Cooley, A.J.; Van Campen, H.; Philpott, M.S.; Easterday, B.C.; Hinshaw, V.S. Pathological lesions in the lungs of ducks infected with influenza A viruses. Vet. Pathol. 1989, 26, 1–5. [Google Scholar] [CrossRef]
- Laudert, E.A.; Sivanandan, V.; Halvorson, D.A. Effect of intravenous inoculation of avian influenza virus on reproduction and growth in mallard ducks. J. Wildl. Dis. 1993, 29, 523–526. [Google Scholar] [CrossRef]
- Chen, H.; Smith, G.J.D.; Zhang, S.Y.; Qin, K.; Wang, J.; Li, K.S.; Webster, R.G.; Peiris, J.S.M.; Guan, Y. Avian flu: H5N1 virus outbreak in migratory waterfowl. Nature 2005, 436, 191–192. [Google Scholar] [CrossRef] [PubMed]
- Kleyheeg, E.; Slaterus, R.; Bodewes, R.; Rijks, J.M.; Spierenburg, M.A.H.; Beerens, N.; Kelder, L.; Poen, M.J.; Stegeman, J.A.; Fouchier, R.A.M.; et al. Deaths among Wild Birds during Highly Pathogenic Avian Influenza A(H5N8) Virus Outbreak, the Netherlands. Emerg. Infect. Dis. 2017, 23, 2050–2054. [Google Scholar] [CrossRef] [PubMed]
- Njoto, E.N.; Scotch, M.; Bui, C.M.; Adam, D.C.; Chughtai, A.A.; MacIntyre, C.R. Phylogeography of H5N1 avian influenza virus in Indonesia. Transbound. Emerg. Dis. 2018, 65, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Food and Agriculture Organization of the United Nations (FAO). Approaches to Controlling, Preventing and Eliminating H5N1 Highly Pathogenic Avian Influenza in Endemic Countries. Available online: http://www.fao.org/docrep/014/i2150e/i2150e00.htm (accessed on 26 October 2018).
- Lee, Y.J.; Kang, H.M.; Lee, E.K.; Song, B.M.; Jeong, J.; Kwon, Y.K.; Kim, H.R.; Lee, K.J.; Hong, M.S.; Jang, I.; et al. Novel Reassortant Influenza A(H5N8) Viruses, South Korea, 2014. Emerg. Infect. Dis. 2014, 20, 1087–1089. [Google Scholar] [CrossRef] [PubMed]
- ProMED-Mail. Avian Influenza (74): South Korea (CN) HPAI H5N8, Duck, Reoccurrence, OIE. ProMED-Mail 2014. 26 September:20140926.2804737. Available online: http://www.promedmail.org (accessed on 26 October 2018).
- Pasick, J.; Berhane, Y.; Joseph, T.; Bowes, V.; Hisanaga, T.; Handel, K.; Alexandersen, S. Reassortant Highly Pathogenic Influenza A H5N2 Virus Containing Gene Segments Related to Eurasian H5N8 in British Columbia, Canada, 2014. Sci. Rep. 2015, 5, 9484. [Google Scholar] [CrossRef]
- Kilpatrick, A.M.; Chmura, A.A.; Gibbons, D.W.; Fleischer, R.C.; Marra, P.P.; Daszak, P. Predicting the global spread of H5N1 avian influenza. Proc. Natl. Acad. Sci. USA 2006, 103, 19368–19373. [Google Scholar] [CrossRef]
- Costa, T.; Chaves, A.J.; Valle, R.; Darji, A.; van Riel, D.; Kuiken, T.; Majó, N.; Ramis, A. Distribution patterns of influenza virus receptors and viral attachment patterns in the respiratory and intestinal tracts of seven avian species. Vet. Res. 2012, 43, 28. [Google Scholar] [CrossRef] [PubMed]
- Kuchipudi, S.V.; Nelli, R.; White, G.A.; Bain, M.; Chang, K.C.; Dunham, S. Differences in influenza virus receptors in chickens and ducks: Implications for interspecies transmission. J. Mol. Genet. Med. Int. J. Biomed. Res. 2009, 3, 143–151. [Google Scholar] [CrossRef]
- Webster, R.G.; Yakhno, M.; Hinshaw, V.S.; Bean, W.J.; Copal Murti, K. Intestinal influenza: Replication and characterization of influenza viruses in ducks. Virology 1978, 84, 268–278. [Google Scholar] [CrossRef]
- Carranza-Flores, J.M.; Padilla-Noriega, L.; Loza-Rubio, E.; García-Espinosa, G. Prolonged excretion of a low-pathogenicity H5N2 avian influenza virus strain in the Pekin duck. J. Vet. Sci. 2013, 14, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Daoust, P.-Y.; Kibenge, F.S.B.; Fouchier, R.A.M.; van de Bildt, M.W.G.; van Riel, D.; Kuiken, T. Replication of low pathogenic avian influenza virus in naturally infected mallard ducks (anas platyrhynchos) causes no morphologic lesions. J. Wildl. Dis. 2011, 47, 401–409. [Google Scholar] [CrossRef] [PubMed]
- França, M.; Stallknecht, D.E.; Poulson, R.; Brown, J.; Howerth, E.W. The Pathogenesis of Low Pathogenic Avian Influenza in Mallards. Avian Dis. 2012, 56, 976–980. [Google Scholar] [CrossRef]
- Vanderven, H.A.; Petkau, K.; Ryan-Jean, K.E.E.; Aldridge, J.R.; Webster, R.G.; Magor, K.E. Avian influenza rapidly induces antiviral genes in duck lung and intestine. Mol. Immunol. 2012, 51, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Daoust, P.-Y.; van de Bildt, M.; van Riel, D.; van Amerongen, G.; Bestebroer, T.; Vanderstichel, R.; Fouchier, R.A.M.; Kuiken, T. Replication of 2 Subtypes of Low-Pathogenicity Avian Influenza Virus of Duck and Gull Origins in Experimentally Infected Mallard Ducks. Vet. Pathol. 2013, 50, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.P.; Brown, J.D.; Howerth, E.W.; Stallknecht, D.E. Variation in viral shedding patterns between different wild bird species infected experimentally with low-pathogenicity avian influenza viruses that originated from wild birds. Avian Pathol. 2011, 40, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Smith, N.; Yu, L.; Paton, I.R.; Gutowska, M.W.; Forrest, H.L.; Danner, A.F.; Seiler, J.P.; Digard, P.; Webster, R.G.; et al. A comparative analysis of host responses to avian influenza infection in ducks and chickens highlights a role for the interferon-induced transmembrane proteins in viral resistance. BMC Genom. 2015, 16, 574. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, J.B.W.J.; Post, J.; Peeters, B.; Vervelde, L.; Rebel, J.M.J. Differential innate responses of chickens and ducks to low-pathogenic avian influenza. Avian Pathol. 2012, 41, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Antarasena, C.; Sirimujalin, R.; Prommuang, P.; Blacksell, S.D.; Promkuntod, N.; Prommuang, P. Tissue tropism of a Thailand strain of high-pathogenicity avian influenza virus (H5N1) in tissues of naturally infected native chickens (Gallus gallus), Japanese quail (Coturnix coturnix japonica) and ducks (Anas spp.). Avian Pathol. 2006, 35, 250–253. [Google Scholar] [CrossRef]
- Bingham, J.; Green, D.J.; Lowther, S.; Klippel, J.; Burggraaf, S.; Anderson, D.E.; Wibawa, H.; Hoa, D.M.; Long, N.T.; Vu, P.P.; et al. Infection studies with two highly pathogenic avian influenza strains (Vietnamese and Indonesian) in Pekin ducks (Anas platyrhynchos), with particular reference to clinical disease, tissue tropism and viral shedding. Avian Pathol. 2009, 38, 267–278. [Google Scholar] [CrossRef]
- Brown, J.D.; Stallknecht, D.E.; Beck, J.R.; Suarez, D.L.; Swayne, D.E. Susceptibility of North American Ducks and Gulls to H5N1 Highly Pathogenic Avian Influenza Viruses. Emerg. Infect. Dis. 2006, 12, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.D.; Stallknecht, D.E.; Swayne, D.E. Experimental Infection of Swans and Geese with Highly Pathogenic Avian Influenza Virus (H5N1) of Asian Lineage. Emerg. Infect. Dis. 2008, 14, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Pantin-Jackwood, M.J.; Swayne, D.E. Pathobiology of Asian Highly Pathogenic Avian Influenza H5N1 Virus Infections in Ducks. Avian Dis. 2007, 51, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Vidaña, B.; Dolz, R.; Busquets, N.; Ramis, A.; Sánchez, R.; Rivas, R.; Valle, R.; Cordón, I.; Solanes, D.; Martínez, J.; et al. Transmission and immunopathology of the avian influenza virus A/Anhui/1/2013 (H7N9) human isolate in three commonly commercialized avian species. Zoonoses Public Health 2018, 65, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.K.; Thomas, C.; Swayne, D.E. Variability in Pathobiology of South Korean H5N1 High-Pathogenicity Avian Influenza Virus Infection for 5 Species of Migratory Waterfowl. Vet. Pathol. 2010, 47, 495–506. [Google Scholar] [CrossRef] [PubMed]
- França, M.; Stallknecht, D.E.; Howerth, E.W. Expression and distribution of sialic acid influenza virus receptors in wild birds. Avian Pathol. 2013, 42, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Cagle, C.; To, T.L.; Nguyen, T.; Wasilenko, J.; Adams, S.C.; Cardona, C.J.; Spackman, E.; Suarez, D.L.; Pantin-Jackwood, M.J. Pekin and Muscovy ducks respond differently to vaccination with a H5N1 highly pathogenic avian influenza (HPAI) commercial inactivated vaccine. Vaccine 2011, 29, 6549–6557. [Google Scholar] [CrossRef] [PubMed]
- Cagle, C.; Wasilenko, J.; Adams, S.C.; Cardona, C.J.; To, T.L.; Nguyen, T.; Spackman, E.; Suarez, D.L.; Smith, D.; Shepherd, E.; et al. Differences in Pathogenicity, Response to Vaccination, and Innate Immune Responses in Different Types of Ducks Infected with a Virulent H5N1 Highly Pathogenic Avian Influenza Virus from Vietnam. Avian Dis. 2012, 56, 479–487. [Google Scholar] [CrossRef]
- Kim, J.-K.; Seiler, P.; Forrest, H.L.; Khalenkov, A.M.; Franks, J.; Kumar, M.; Karesh, W.B.; Gilbert, M.; Sodnomdarjaa, R.; Douangngeun, B.; et al. Pathogenicity and Vaccine Efficacy of Different Clades of Asian H5N1 Avian Influenza A Viruses in Domestic Ducks. J. Virol. 2008, 82, 11374–11382. [Google Scholar] [CrossRef]
- Ryan, G.B.; Majno, G. Acute inflammation. A review. Am. J. Pathol. 1977, 86, 183–276. [Google Scholar]
- Peper, R.L.; Van Campen, H. Tumor necrosis factor as a mediator of inflammation in influenza A viral pneumonia. Microb. Pathog. 1995, 19, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Uyeki, T.M. Human Infection with Highly Pathogenic Avian Influenza A (H5N1) Virus: Review of Clinical Issues. Clin. Infect. Dis. 2009, 49, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Matthay, M.A. The Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S.-M.; Rothwell, N.J.; Gibson, R.M. The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 2006, 147, S232–S240. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.Y.; Poon, L.L.M.; Lau, A.S.; Luk, W.; Lau, Y.L.; Shortridge, K.F.; Gordon, S.; Guan, Y.; Peiris, J.S.M. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: A mechanism for the unusual severity of human disease? Lancet Lond. Engl. 2002, 360, 1831–1837. [Google Scholar] [CrossRef]
- Hagau, N.; Slavcovici, A.; Gonganau, D.N.; Oltean, S.; Dirzu, D.S.; Brezoszki, E.S.; Maxim, M.; Ciuce, C.; Mlesnite, M.; Gavrus, R.L.; et al. Clinical aspects and cytokine response in severe H1N1 influenza A virus infection. Crit. Care. 2010, 14, R203. [Google Scholar] [CrossRef] [PubMed]
- Yuen, K.Y.; Wong, S.S.Y. Human infection by avian influenza A H5N1. Hong Kong Med. J. Xianggang Yi Xue Za Zhi 2005, 11, 189–199. [Google Scholar]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.C.; Wang, H.; Liu, H. Identification of Oxidative Stress and Toll-like Receptor 4 Signaling as a Key Pathway of Acute Lung Injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- Sun, H.; Jiao, P.; Jia, B.; Xu, C.; Wei, L.; Shan, F.; Luo, K.; Xin, C.; Zhang, K.; Liao, M. Pathogenicity in quails and mice of H5N1 highly pathogenic avian influenza viruses isolated from ducks. Vet. Microbiol. 2011, 152, 258–265. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Walsh, K.B.; Cahalan, S.; Fremgen, D.M.; Roberts, E.; Scott, F.; Martinborough, E.; Peach, R.; Oldstone, M.B.A.; Rosen, H. Endothelial Cells Are Central Orchestrators of Cytokine Amplification during Influenza Virus Infection. Cell 2011, 146, 980–991. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Walsh, K.B.; Rice, S.; Rosen, H.; Oldstone, M.B.A. Mapping the innate signaling cascade essential for cytokine storm during influenza virus infection. Proc. Natl. Acad. Sci. USA 2014, 111, 3799–3804. [Google Scholar] [CrossRef] [PubMed]
- Burggraaf, S.; Karpala, A.J.; Bingham, J.; Lowther, S.; Selleck, P.; Kimpton, W.; Bean, A.G.D. H5N1 infection causes rapid mortality and high cytokine levels in chickens compared to ducks. Virus Res. 2014, 185, 23–31. [Google Scholar] [CrossRef]
- Wei, L.; Jiao, P.; Song, Y.; Cao, L.; Yuan, R.; Gong, L.; Cui, J.; Zhang, S.; Qi, W.; Yang, S.; Liao, M. Host immune responses of ducks infected with H5N1 highly pathogenic avian influenza viruses of different pathogenicities. Vet. Microbiol. 2013, 166, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Green, S.J.; Scheller, L.F.; Marletta, M.A.; Seguin, M.C.; Klotz, F.W.; Slayter, M.; Nelson, B.J.; Nacy, C.A. Nitric oxide: Cytokine-regulation of nitric oxide in host resistance to intracellular pathogens. Immunol. Lett. 1994, 43, 87–94. [Google Scholar] [CrossRef]
- Sturm-Ramirez, K.M.; Hulse-Post, D.J.; Govorkova, E.A.; Humberd, J.; Seiler, P.; Puthavathana, P.; Buranathai, C.; Nguyen, T.D.; Chaisingh, A.; Long, H.T.; et al. Are ducks contributing to the endemicity of highly pathogenic H5N1 influenza virus in Asia? J. Virol. 2005, 79, 11269–11279. [Google Scholar] [CrossRef] [PubMed]
- Saito, L.B.; Diaz-Satizabal, L.; Evseev, D.; Fleming-Canepa, X.; Mao, S.; Webster, R.G.; Magor, K.E. IFN and cytokine responses in ducks to genetically similar H5N1 influenza A viruses of varying pathogenicity. J. Gen. Virol. 2018, 99, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.C.; Xing, Z.; Li, J.; Cardona, C.J. Immune-related gene expression in response to H11N9 low pathogenic avian influenza virus infection in chicken and Pekin duck peripheral blood mononuclear cells. Mol. Immunol. 2009, 46, 1744–1749. [Google Scholar] [CrossRef]
- Kuchipudi, S.V.; Tellabati, M.; Sebastian, S.; Londt, B.Z.; Jansen, C.; Vervelde, L.; Brookes, S.M.; Brown, I.H.; Dunham, S.P.; Chang, K.-C. Highly pathogenic avian influenza virus infection in chickens but not ducks is associated with elevated host immune and pro-inflammatory responses. Vet. Res. 2014, 45, 118. [Google Scholar] [CrossRef]
- Cornelissen, J.B.W.J.; Vervelde, L.; Post, J.; Rebel, J.M.J. Differences in highly pathogenic avian influenza viral pathogenesis and associated early inflammatory response in chickens and ducks. Avian Pathol. 2013, 42, 347–364. [Google Scholar] [CrossRef]
- Liang, Q.; Luo, J.; Zhou, K.; Dong, J.; He, H. Immune-related gene expression in response to H5N1 avian influenza virus infection in chicken and duck embryonic fibroblasts. Mol. Immunol. 2011, 48, 924–930. [Google Scholar] [CrossRef]
- Baquero-Perez, B.; Kuchipudi, S.V.; Ho, J.; Sebastian, S.; Puranik, A.; Howard, W.; Brookes, S.M.; Brown, I.H.; Chang, K.-C. Chicken and Duck Myotubes Are Highly Susceptible and Permissive to Influenza Virus Infection. J. Virol. 2014, 89, 2494–2506. [Google Scholar] [CrossRef] [PubMed]
- Burggraaf, S.; Bingham, J.; Payne, J.; Kimpton, W.G.; Lowenthal, J.W.; Bean, A.G.D. Increased Inducible Nitric Oxide Synthase Expression in Organs Is Associated with a Higher Severity of H5N1 Influenza Virus Infection. PLoS ONE 2011, 6, e14561. [Google Scholar] [CrossRef] [PubMed]
- García-Sastre, A. Induction and evasion of type I interferon responses by influenza viruses. Virus Res. 2011, 162, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Alsharifi, M.; Müllbacher, A.; Regner, M. Interferon type I responses in primary and secondary infections. Immunol Cell Biol. 2008, 86, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Yang, H.; Kapczynski, D.R. Chicken interferon alpha pretreatment reduces virus replication of pandemic H1N1 and H5N9 avian influenza viruses in lung cell cultures from different avian species. Virol. J. 2011, 8, 447. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.K.; Chen, G.; Zheng, D.; Tang, H.; Cheng, G. The host type I interferon response to viral and bacterial infections. Cell Res. 2005, 15, 407–422. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef]
- Schultz, U.; Kaspers, B.; Staeheli, P. The interferon system of non-mammalian vertebrates. Dev. Comp. Immunol. 2004, 28, 499–508. [Google Scholar] [CrossRef]
- Jørgensen, S.E.; Christiansen, M.; Ryø, L.B.; Gad, H.H.; Gjedsted, J.; Staeheli, P.; Mikkelsen, J.G.; Storgaard, M.; Hartmann, R.; Mogensen, T.H. Defective RNA sensing by RIG-I in severe influenza virus infection. Clin. Exp. Immunol. 2018, 192, 366–376. [Google Scholar] [CrossRef]
- Koerner, I.; Kochs, G.; Kalinke, U.; Weiss, S.; Staeheli, P. Protective role of beta interferon in host defense against influenza A virus. J. Virol. 2007, 81, 2025–2030. [Google Scholar] [CrossRef]
- Krishna, V.D.; Roach, E.; Zaidman, N.A.; Panoskaltsis-Mortari, A.; Rotschafer, J.H.; O’Grady, S.M.; Cheeran, M.C.-J. Differential Induction of Type I and Type III Interferons by Swine and Human Origin H1N1 Influenza A Viruses in Porcine Airway Epithelial Cells. PLoS ONE 2015, 10, e0138704. [Google Scholar] [CrossRef] [PubMed]
- Tumpey, T.M.; Szretter, K.J.; Van Hoeven, N.; Katz, J.M.; Kochs, G.; Haller, O.; García-Sastre, A.; Staeheli, P. The Mx1 gene protects mice against the pandemic 1918 and highly lethal human H5N1 influenza viruses. J. Virol. 2007, 81, 10818–10821. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Goldsmith, C.; Thawatsupha, P.; Chittaganpitch, M.; Waicharoen, S.; Zaki, S.; Tumpey, T.M.; Katz, J.M. Highly Pathogenic Avian Influenza H5N1 Viruses Elicit an Attenuated Type I Interferon Response in Polarized Human Bronchial Epithelial Cells. J. Virol. 2007, 81, 12439–12449. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Wang, Y.-H.; Liu, Y.-J. Plasmacytoid dendritic cell precursors/type I interferon-producing cells sense viral infection by Toll-like receptor (TLR) 7 and TLR9. Springer Semin. Immunopathol. 2005, 26, 221–229. [Google Scholar] [CrossRef]
- Liu, Y.-J. IPC: Professional Type 1 Interferon-Producing Cells and Plasmacytoid Dendritic Cell Precursors. Annu. Rev. Immunol. 2005, 23, 275–306. [Google Scholar] [CrossRef]
- Colonna, M.; Trinchieri, G.; Liu, Y.-J. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 2004, 5, 1219–1226. [Google Scholar] [CrossRef]
- Billiau, A. Anti-inflammatory properties of Type I interferons. Antivir. Res. 2006, 71, 108–116. [Google Scholar] [CrossRef]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Förster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type I Interferon Inhibits Interleukin-1 Production and Inflammasome Activation. Immunity 2011, 34, 213–223. [Google Scholar] [CrossRef]
- Trinchieri, G. Type I interferon: Friend or foe? J. Exp. Med. 2010, 207, 2053–2063. [Google Scholar] [CrossRef]
- Arimori, Y.; Nakamura, R.; Yamada, H.; Shibata, K.; Maeda, N.; Kase, T.; Yoshikai, Y. Type I interferon limits influenza virus-induced acute lung injury by regulation of excessive inflammation in mice. Antivir. Res. 2013, 99, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Shahangian, A.; Chow, E.K.; Tian, X.; Kang, J.R.; Ghaffari, A.; Liu, S.Y.; Belperio, J.A.; Cheng, G.; Deng, J.C. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Investig. 2009, 119, 1910–1920. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Vijayakumar, P.; Gandhale, P.N.; Ranaware, P.B.; Kumar, H.; Kulkarni, D.D.; Raut, A.A.; Mishra, A. Genome-wide gene expression pattern underlying differential host response to high or low pathogenic H5N1 avian influenza virus in ducks. Acta Virol. 2017, 61, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.; Nagarajan, S.; Kumar, M.; Murugkar, H.V.; Kalaiyarasu, S.; Venkatesh, G.; Tosh, C. Antigenic characterization of H5N1 highly pathogenic avian influenza viruses isolated from poultry in India, 2006–2015. Arch. Virol. 2017, 162, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, S.; Tosh, C.; Smith, D.K.; Peiris, J.S.M.; Murugkar, H.V.; Sridevi, R.; Kumar, M.; Katare, M.; Jain, R.; Syed, Z.; et al. Avian Influenza (H5N1) Virus of Clade 2.3.2 in Domestic Poultry in India. PLoS ONE 2012, 7, e31844. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Xiang, B.; Li, Y.; Li, Y.; Sun, M.; Kang, Y.; Xie, P.; Chen, L.; Lin, Q.; Liao, M.; Ren, T. Therapeutic Effect of Duck Interferon-Alpha Against H5N1 Highly Pathogenic Avian Influenza Virus Infection in Peking Ducks. J. Interferon Cytokine Res. 2018, 38, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.P.; Kotenko, S.V. Interferon-Lambda: A New Addition to an Old Family. J. Interferon Cytokine Res. 2010, 30, 555–564. [Google Scholar] [CrossRef]
- Hamming, O.J.; Gad, H.H.; Paludan, S.; Hartmann, R. Lambda Interferons: New Cytokines with Old Functions. Pharmaceuticals 2010, 3, 795–809. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, W.; Chen, S.; Wang, A.; Sun, L.; Wang, M.; Jia, R.; Zhu, D.; Liu, M.; Sun, K.; et al. Identification of Type III Interferon (IFN-λ) in Chinese Goose: Gene Structure, Age-Dependent Expression Profile, and Antiviral Immune Characteristics In Vivo and In Vitro. J. Interferon Cytokine Res. 2017, 37, 269–277. [Google Scholar] [CrossRef]
- Soubies, S.M.; Volmer, C.; Guérin, J.-L.; Volmer, R. Truncation of the NS1 Protein Converts a Low Pathogenic Avian Influenza Virus into a Strong Interferon Inducer in Duck Cells. Avian Dis. 2010, 54, 527–531. [Google Scholar] [CrossRef]
- Volmer, C.; Soubies, S.M.; Grenier, B.; Guérin, J.-L.; Volmer, R. Immune response in the duck intestine following infection with low-pathogenic avian influenza viruses or stimulation with a Toll-like receptor 7 agonist administered orally. J. Gen. Virol. 2011, 92, 534–543. [Google Scholar] [CrossRef]
- Cao, Y.; Huang, Y.; Xu, K.; Liu, Y.; Li, X.; Xu, Y.; Zhong, W.; Hao, P. Differential responses of innate immunity triggered by different subtypes of influenza a viruses in human and avian hosts. BMC Med. Genom. 2017, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Moulin, H.R.; Liniger, M.; Python, S.; Guzylack-Piriou, L.; Ocaña-Macchi, M.; Ruggli, N.; Summerfield, A. High interferon type I responses in the lung, plasma and spleen during highly pathogenic H5N1 infection of chicken. Vet. Res. 2011, 42, 6. [Google Scholar] [CrossRef] [PubMed]
- Penski, N.; Härtle, S.; Rubbenstroth, D.; Krohmann, C.; Ruggli, N.; Schusser, B.; Pfann, M.; Reuter, A.; Gohrbandt, S.; Hundt, J.; et al. Highly Pathogenic Avian Influenza Viruses Do Not Inhibit Interferon Synthesis in Infected Chickens but Can Override the Interferon-Induced Antiviral State. J. Virol. 2011, 85, 7730–7741. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Cardona, C.J.; Li, J.; Dao, N.; Tran, T.; Andrada, J. Modulation of the immune responses in chickens by low-pathogenicity avian influenza virus H9N2. J. Gen. Virol. 2008, 89, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.R.W.; Aldridge, J.R.; Webster, R.G.; Magor, K.E. Association of RIG-I with innate immunity of ducks to influenza. Proc. Natl. Acad. Sci. USA 2010, 107, 5913–5918. [Google Scholar] [CrossRef]
- Chen, S.N.; Zou, P.F.; Nie, P. Retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) in fish: Current knowledge and future perspectives. Immunology 2017, 151, 16–25. [Google Scholar] [CrossRef]
- Cui, X.-F.; Imaizumi, T.; Yoshida, H.; Borden, E.C.; Satoh, K. Retinoic acid-inducible gene-I is induced by interferon-gamma and regulates the expression of interferon-gamma stimulated gene 15 in MCF-7 cells. Biochem. Cell Biol. Biochim. Biol. Cell 2004, 82, 401–405. [Google Scholar] [CrossRef]
- Imaizumi, T.; Aratani, S.; Nakajima, T.; Carlson, M.; Matsumiya, T.; Tanji, K.; Ookawa, K.; Yoshida, H.; Tsuchida, S.; McIntyre, T.M.; et al. Retinoic acid-inducible gene-I is induced in endothelial cells by LPS and regulates expression of COX-2. Biochem. Biophys. Res. Commun. 2002, 292, 274–279. [Google Scholar] [CrossRef]
- Imaizumi, T.; Hatakeyama, M.; Yamashita, K.; Yoshida, H.; Ishikawa, A.; Taima, K.; Satoh, K.; Mori, F.; Wakabayashi, K. Interferon-gamma induces retinoic acid-inducible gene-I in endothelial cells. Endothel. J. Endothel. Cell Res. 2004, 11, 169–173. [Google Scholar]
- Li, K.; Chen, Z.; Kato, N.; Gale, M.; Lemon, S.M. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J. Biol. Chem. 2005, 280, 16739–16747. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, H.; Imaizumi, T.; Matsumiya, T.; Kusumi, A.; Nakagawa, H.; Kubota, K.; Nishi, N.; Nakamura, T.; Hirashima, M.; Satoh, K.; et al. Retinoic acid-inducible gene-I is induced by interleukin-1beta in cultured human gingival fibroblasts. Oral Microbiol. Immunol. 2005, 20, 47–50. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.-M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; García-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 Signaling by RNA Viruses in Innate Immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Opitz, B.; Rejaibi, A.; Dauber, B.; Eckhard, J.; Vinzing, M.; Schmeck, B.; Hippenstiel, S.; Suttorp, N.; Wolff, T. IFNβ induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell Microbiol. 2007, 9, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhang, W.; Booth, J.L.; Metcalf, J.P. Influenza A(H1N1)pdm09 Virus Suppresses RIG-I Initiated Innate Antiviral Responses in the Human Lung. PLoS ONE 2012, 7, e49856. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Park, H.-S.; Pyo, H.-M.; Liu, Q.; Zhou, Y. Influenza A Virus Panhandle Structure is Directly Involved in RIG-I Activation and IFN Induction. J. Virol. 2015, JVI.00232-15. [Google Scholar] [CrossRef]
- Weber, M.; Gawanbacht, A.; Habjan, M.; Rang, A.; Borner, C.; Schmidt, A.M.; Veitinger, S.; Jacob, R.; Devignot, S.; Kochs, G.; et al. Incoming RNA Virus Nucleocapsids Containing a 5′-Triphosphorylated Genome Activate RIG-I and Antiviral Signaling. Cell Host Microbe 2013, 13, 336–346. [Google Scholar] [CrossRef]
- te Velthuis, A.J.; Long, J.C.; Bauer, D.L.; Fan, R.L.; Yen, H.L.; Sharps, J.; Siegers, J.Y.; Killip, M.J.; French, H.; Oliva-Martín, M.J.; et al. Mini viral RNAs act as innate immune agonists during influenza virus infection. Nat. Microbiol. 2018, 3, 1234. [Google Scholar] [CrossRef]
- Baum, A.; Sachidanandam, R.; García-Sastre, A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 16303–16308. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M.; Hartmann, G. The Chase for the RIG-I Ligand—Recent Advances. Mol. Ther. 2010, 18, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Lazzarini, R.A.; Keene, J.D.; Schubert, M. The origins of defective interfering particles of the negative-strand RNA viruses. Cell 1981, 26, 145–154. [Google Scholar] [CrossRef]
- Liu, G.; Lu, Y.; Raman, S.N.T.; Xu, F.; Wu, Q.; Li, Z.; Brownlie, R.; Liu, Q.; Zhou, Y. Nuclear-resident RIG-I senses viral replication inducing antiviral immunity. Nat. Commun. 2018, 9, 3199. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Sediri, H.; Felgenhauer, U.; Binzen, I.; Bänfer, S.; Jacob, R.; Brunotte, L.; García-Sastre, A.; Schmid-Burgk, J.L.; Schmidt, T.; et al. Influenza Virus Adaptation PB2-627K Modulates Nucleocapsid Inhibition by the Pathogen Sensor RIG-I. Cell Host Microbe 2015, 17, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Huang, Q.; Ji, W.; Du, B.; Fu, Q.; An, H.; Li, J.; Wang, H.; Yan, Y.; Ding, C.; Sun, J. Muscovy duck retinoic acid-induced gene I (MdRIG-I) functions in innate immunity against H9N2 avian influenza viruses (AIV) infections. Vet. Immunol. Immunopathol. 2015, 163, 183–193. [Google Scholar] [CrossRef]
- Fleming-Canepa, X.; Aldridge, J.R.; Canniff, L.; Kobewka, M.; Jax, E.; Webster, R.G.; Magor, K.E. Duck innate immune responses to high and low pathogenicity H5 avian influenza viruses. Vet. Microbiol. 2019, 228, 101–111. [Google Scholar] [CrossRef]
- Magor, K.E.; Miranzo Navarro, D.; Barber, M.R.W.; Petkau, K.; Fleming-Canepa, X.; Blyth, G.A.D.; Blaine, A.H. Defense genes missing from the flight division. Dev. Comp. Immunol. 2013, 41, 377–388. [Google Scholar] [CrossRef]
- Kallfass, C.; Lienenklaus, S.; Weiss, S.; Staeheli, P. Visualizing the Beta Interferon Response in Mice during Infection with Influenza A Viruses Expressing or Lacking Nonstructural Protein 1. J. Virol. 2013, 87, 6925–6930. [Google Scholar] [CrossRef]
- Jiang, M.; Österlund, P.; Sarin, L.P.; Poranen, M.M.; Bamford, D.H.; Guo, D.; Julkunen, I. Innate Immune Responses in Human Monocyte-Derived Dendritic Cells Are Highly Dependent on the Size and the 5′ Phosphorylation of RNA Molecules. J. Immunol. 2011, 187, 1713–1721. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.-P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Reis e Sousa, C. Activation of MDA5 Requires Higher-Order RNA Structures Generated during Virus Infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef] [PubMed]
- Benitez, A.A.; Panis, M.; Xue, J.; Varble, A.; Shim, J.V.; Frick, A.L.; López, C.B.; Sachs, D.; ten Oever, B.R. In Vivo RNAi Screening Identifies MDA5 as a Significant Contributor to the Cellular Defense against Influenza A Virus. Cell Rep. 2015, 11, 1714–1726. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Cui, J.; Song, Y.; Zhang, S.; Han, F.; Yuan, R.; Gong, L.; Jiao, P.; Liao, M. Duck MDA5 functions in innate immunity against H5N1 highly pathogenic avian influenza virus infections. Vet. Res. 2014, 45, 66. [Google Scholar] [CrossRef] [PubMed]
- Karpala, A.J.; Stewart, C.; McKay, J.; Lowenthal, J.W.; Bean, A.G.D. Characterization of Chicken Mda5 Activity: Regulation of IFN-β in the Absence of RIG-I Functionality. J Immunol. 2011, 1003712. [Google Scholar] [CrossRef] [PubMed]
- Bamming, D.; Horvath, C.M. Regulation of signal transduction by enzymatically inactive antiviral RNA helicase proteins MDA5, RIG-I, and LGP2. J. Biol. Chem. 2009, 284, 9700–9712. [Google Scholar] [CrossRef]
- Li, X.; Ranjith-Kumar, C.T.; Brooks, M.T.; Dharmaiah, S.; Herr, A.B.; Kao, C.; Li, P. The RIG-I-like receptor LGP2 recognizes the termini of double-stranded RNA. J. Biol. Chem. 2009, 284, 13881–13891. [Google Scholar] [CrossRef] [PubMed]
- Murali, A.; Li, X.; Ranjith-Kumar, C.T.; Bhardwaj, K.; Holzenburg, A.; Li, P.; Kao, C.C. Structure and function of LGP2, a DEX(D/H) helicase that regulates the innate immunity response. J. Biol. Chem. 2008, 283, 15825–15833. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Hirai, R.; Loo, Y.-M.; Owen, D.; Johnson, C.L.; Sinha, S.C.; Akira, S.; Fujita, T.; Gale, M. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 2007, 104, 582–587. [Google Scholar] [CrossRef]
- Takahasi, K.; Kumeta, H.; Tsuduki, N.; Narita, R.; Shigemoto, T.; Hirai, R.; Yoneyama, M.; Horiuchi, M.; Ogura, K.; Fujita, T.; et al. Solution structures of cytosolic RNA sensor MDA5 and LGP2 C-terminal domains: Identification of the RNA recognition loop in RIG-I-like receptors. J. Biol. Chem. 2009, 284, 17465–17474. [Google Scholar] [CrossRef]
- Satoh, T.; Kato, H.; Kumagai, Y.; Yoneyama, M.; Sato, S.; Matsushita, K.; Tsujimura, T.; Fujita, T.; Akira, S.; Takeuchi, O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. USA 2010, 107, 1512–1517. [Google Scholar] [CrossRef]
- Venkataraman, T.; Valdes, M.; Elsby, R.; Kakuta, S.; Caceres, G.; Saijo, S.; Iwakura, Y.; Barber, G.N. Loss of DExD/H Box RNA Helicase LGP2 Manifests Disparate Antiviral Responses. J. Immunol. 2007, 178, 6444–6455. [Google Scholar] [CrossRef] [PubMed]
- Jiao, P.R.; Wei, L.M.; Song, Y.F.; Cui, J.; Zhang, S.; Han, F.; Yuan, R.Y.; Liao, M. Molecular cloning and immune responsive expression of LGP2 gene, a pivotal member of the RLR gene family from Muscovy duck Cairina moschata. Poult. Sci. 2015, 94, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Peisley, A.; Tetrault, D.; Li, Z.; Egelman, E.H.; Magor, K.E.; Walz, T.; Penczek, P.A.; Hur, S. Molecular imprinting as a signal-activation mechanism of the viral RNA sensor RIG-I. Mol. Cell 2014, 55, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Miranzo-Navarro, D.; Magor, K.E. Activation of duck RIG-I by TRIM25 is independent of anchored ubiquitin. PLoS ONE 2014, 9, e86968. [Google Scholar] [CrossRef]
- Barber, M.R.W.; Aldridge, J.R.; Fleming-Canepa, X.; Wang, Y.-D.; Webster, R.G.; Magor, K.E. Identification of avian RIG-I responsive genes during influenza infection. Mol. Immunol. 2013, 54, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, Q.; Li, Y.; Liu, R.; Huang, Z.; Wang, B.; Chen, G. Gene expression profile after activation of RIG-I in 5′ppp-dsRNA challenged DF1. Dev. Comp. Immunol. 2016, 65, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Reeves, M.B.; Caulfield, A.F.; Evseev, D.; Magor, K.E. The core promoter controls basal and inducible expression of duck retinoic acid inducible gene-I (RIG-I). Mol. Immunol. 2018, 103, 156–165. [Google Scholar] [CrossRef]
- Arimoto, K.; Takahashi, H.; Hishiki, T.; Konishi, H.; Fujita, T.; Shimotohno, K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. USA 2007, 104, 7500–7505. [Google Scholar] [CrossRef]
- Gack, M.U.; Shin, Y.C.; Joo, C.-H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Matsumoto, M.; Hatakeyama, S.; Seya, T. Riplet/RNF135, a RING Finger Protein, Ubiquitinates RIG-I to Promote Interferon-β Induction during the Early Phase of Viral Infection. J. Biol. Chem. 2009, 284, 807–817. [Google Scholar] [CrossRef]
- Oshiumi, H.; Miyashita, M.; Inoue, N.; Okabe, M.; Matsumoto, M.; Seya, T. The Ubiquitin Ligase Riplet Is Essential for RIG-I-Dependent Innate Immune Responses to RNA Virus Infection. Cell Host Microbe 2010, 8, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Rajsbaum, R.; Albrecht, R.A.; Wang, M.K.; Maharaj, N.P.; Versteeg, G.A.; Nistal-Villán, E.; García-Sastre, A.; Gack, M.U. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 2012, 8, e1003059. [Google Scholar] [CrossRef] [PubMed]
- Peisley, A.; Wu, B.; Xu, H.; Chen, Z.J.; Hur, S. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 2014, 509, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Sun, L.; Jiang, X.; Chen, X.; Hou, F.; Adhikari, A.; Xu, M.; Chen, Z.J. Reconstitution of the RIG-I Pathway Reveals a Pivotal Role of Unanchored Polyubiquitin Chains in Innate Immunity. Cell 2010, 141, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, N.R.; Zhou, L.; Guo, Y.R.; Zhao, C.; Tao, Y.J.; Krug, R.M.; Sawyer, S.L. Nuclear TRIM25 Specifically Targets Influenza Virus Ribonucleoproteins to Block the Onset of RNA Chain Elongation. Cell Host Microbe 2017, 22, 627–638.e7. [Google Scholar] [CrossRef] [PubMed]
- Blaine, A.H.; Miranzo-Navarro, D.; Campbell, L.K.; Aldridge, J.R.; Webster, R.G.; Magor, K.E. Duck TRIM27-L enhances MAVS signaling and is absent in chickens and turkeys. Mol. Immunol. 2015, 67, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.A.; Gonçalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chen, G.; Lv, F.; Wang, X.; Ji, X.; Xu, Y.; Sun, J.; Wu, L.; Zheng, Y.-T.; Gao, G. Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proc. Natl. Acad. Sci. USA 2011, 108, 15834–15839. [Google Scholar] [CrossRef]
- Hayakawa, S.; Shiratori, S.; Yamato, H.; Kameyama, T.; Kitatsuji, C.; Kashigi, F.; Goto, S.; Kameoka, S.; Fujikura, D.; Yamada, T.; et al. ZAPS is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nat. Immunol. 2011, 12, 37–44. [Google Scholar] [CrossRef]
- Goossens, K.E.; Karpala, A.J.; Ward, A.; Bean, A.G.D. Characterisation of chicken ZAP. Dev. Comp. Immunol. 2014, 46, 373–381. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.-Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; Shu, H.-B. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Zevini, A.; Olagnier, D.; Hiscott, J. Cross-Talk between the Cytoplasmic RIG-I and STING Sensing Pathways. Trends Immunol. 2017, 38, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef]
- Diner, E.J.; Burdette, D.L.; Wilson, S.C.; Monroe, K.M.; Kellenberger, C.A.; Hyodo, M.; Hayakawa, Y.; Hammond, M.C.; Vance, R.E. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 2013, 3, 1355–1361. [Google Scholar] [CrossRef]
- Liu, Y.; Lin, R.; Olagnier, D. RIGulation of STING expression: At the crossroads of viral RNA and DNA sensing pathways. Inflamm. Cell Signal. 2017, 4, e1491. [Google Scholar]
- Cheng, Y.; Sun, Y.; Wang, H.; Yan, Y.; Ding, C.; Sun, J. Chicken STING Mediates Activation of the IFN Gene Independently of the RIG-I Gene. J. Immunol. 2015, 195, 3922–3936. [Google Scholar] [CrossRef]
- Chen, S.; Wu, Z.; Zhang, J.; Wang, M.; Jia, R.; Zhu, D.; Liu, M.; Sun, K.; Yang, Q.; Wu, Y.; Zhao, X.; Cheng, A. Duck stimulator of interferon genes plays an important role in host anti-duck plague virus infection through an IFN-dependent signalling pathway. Cytokine 2018, 102, 191–199. [Google Scholar] [CrossRef]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef]
- Feldman, S.; Stein, D.; Amrute, S.; Denny, T.; Garcia, Z.; Kloser, P.; Sun, Y.; Megjugorac, N.; Fitzgerald-Bocarsly, P. Decreased Interferon-α Production in HIV-Infected Patients Correlates with Numerical and Functional Deficiencies in Circulating Type 2 Dendritic Cell Precursors. Clin. Immunol. 2001, 101, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Feldman, S.B.; Milone, M.C.; Kloser, P.; Fitzgerald-Bocarsly, P. Functional deficiencies in two distinct interferon alpha-producing cell populations in peripheral blood mononuclear cells from human immunodeficiency virus seropositive patients. J. Leukoc. Biol. 1995, 57, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Gobl, A.E.; Funa, K.; Alm, G.V. Different induction patterns of mRNA for IFN-alpha and -beta in human mononuclear leukocytes after in vitro stimulation with herpes simplex virus-infected fibroblasts and Sendai virus. J. Immunol. 1988, 140, 3605–3609. [Google Scholar] [PubMed]
- MacDonald, M.R.W.; Xia, J.; Smith, A.L.; Magor, K.E. The duck toll like receptor 7: Genomic organization, expression and function. Mol. Immunol. 2008, 45, 2055–2061. [Google Scholar] [CrossRef] [PubMed]
- Philbin, V.J.; Iqbal, M.; Boyd, Y.; Goodchild, M.J.; Beal, R.K.; Bumstead, N.; Young, J.; Smith, A.L. Identification and characterization of a functional, alternatively spliced Toll-like receptor 7 (TLR7) and genomic disruption of TLR8 in chickens. Immunology 2005, 114, 507–521. [Google Scholar] [CrossRef]
- Iqbal, M.; Philbin, V.J.; Smith, A.L. Expression patterns of chicken Toll-like receptor mRNA in tissues, immune cell subsets and cell lines. Vet. Immunol. Immunopathol. 2005, 104, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Kogut, M.H.; Swaggerty, C.; He, H.; Pevzner, I.; Kaiser, P. Toll-like receptor agonists stimulate differential functional activation and cytokine and chemokine gene expression in heterophils isolated from chickens with differential innate responses. Microbes Infect. 2006, 8, 1866–1874. [Google Scholar] [CrossRef]
- Stewart, C.R.; Bagnaud-Baule, A.; Karpala, A.J.; Lowther, S.; Mohr, P.G.; Wise, T.G.; Lowenthal, J.W.; Bean, A.G. Toll-Like Receptor 7 Ligands Inhibit Influenza A Infection in Chickens. J. Interferon Cytokine Res. 2012, 32, 46–51. [Google Scholar] [CrossRef]
- Wu, W.; Zhang, W.; Duggan, E.S.; Booth, J.L.; Zou, M.-H.; Metcalf, J.P. RIG-I and TLR3 are both required for maximum interferon induction by influenza virus in human lung alveolar epithelial cells. Virology 2015, 482, 181–188. [Google Scholar] [CrossRef]
- Le Goffic, R.; Balloy, V.; Lagranderie, M.; Alexopoulou, L.; Escriou, N.; Flavell, R.; Chignard, M.; Si-Tahar, M. Detrimental Contribution of the Toll-Like Receptor (TLR)3 to Influenza A Virus–Induced Acute Pneumonia. PLoS Pathog. 2006, 2, e53. [Google Scholar] [CrossRef] [PubMed]
- Karpala, A.J.; Lowenthal, J.W.; Bean, A.G. Activation of the TLR3 pathway regulates IFNβ production in chickens. Dev. Comp. Immunol. 2008, 32, 435–444. [Google Scholar] [CrossRef]
- Jiao, P.R.; Wei, L.M.; Cheng, Y.Q.; Yuan, R.Y.; Han, F.; Liang, J.; Liu, W.L.; Ren, T.; Xin, C.A.; Liao, M. Molecular cloning, characterization, and expression analysis of the Muscovy duck Toll-like receptor 3 (MdTLR3) gene. Poult. Sci. 2012, 91, 2475–2481. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Cheng, A.; Wang, M. Innate sensing of viruses by pattern recognition receptors in birds. Vet. Res. 2013, 44, 82. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Hiromoto, Y.; Chaichoune, K.; Patchimasiri, T.; Chakritbudsabong, W.; Prayoonwong, N.; Chaisilp, N.; Wiriyarat, W.; Parchariyanon, S.; Ratanakorn, P.; Uchida, Y.; Saito, T. Host Cytokine Responses of Pigeons Infected with Highly Pathogenic Thai Avian Influenza Viruses of Subtype H5N1 Isolated from Wild Birds. PLoS ONE 2011, 6, e23103. [Google Scholar] [CrossRef]
- Oshiumi, H.; Matsumoto, M.; Funami, K.; Akazawa, T.; Seya, T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3–mediated interferon-β induction. Nat. Immunol. 2003, 4, 161–167. [Google Scholar] [CrossRef]
- Wei, X.; Qian, W.; Sizhu, S.; Shi, L.; Jin, M.; Zhou, H. Molecular cloning and functional analysis of the duck TIR domain-containing adaptor inducing IFN-β (TRIF) gene. Dev. Comp. Immunol. 2016, 65, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, R.; Zhou, Q.; Xu, Z.; Li, C.; Wang, S.; Mao, A.; Zhang, X.; He, W.; Shu, H.-B. LSm14A is a processing body-associated sensor of viral nucleic acids that initiates cellular antiviral response in the early phase of viral infection. Proc. Natl. Acad. Sci. USA 2012, 109, 11770–11775. [Google Scholar] [CrossRef]
- Hua, K.; Li, H.; Chen, H.; Foda, M.F.; Luo, R.; Jin, H. Functional characterization of duck LSm14A in IFN-β induction. Dev. Comp. Immunol. 2017, 76, 255–261. [Google Scholar] [CrossRef]
- Leemans, J.C.; Cassel, S.L.; Sutterwala, F.S. Sensing damage by the NLRP3 inflammasome. Immunol. Rev. 2011, 243, 152–162. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P.-Y. The NLRP3 Inflammasome Mediates In Vivo Innate Immunity to Influenza A Virus through Recognition of Viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.G.; Dash, P.; Aldridge, J.R.; Ellebedy, A.H.; Reynolds, C.; Funk, A.J.; Martin, W.J.; Lamkanfi, M.; Webby, R.J.; Boyd, K.L.; et al. The Intracellular Sensor NLRP3 Mediates Key Innate and Healing Responses to Influenza A Virus via the Regulation of Caspase-1. Immunity 2009, 30, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Pothlichet, J.; Meunier, I.; Davis, B.K.; Ting, J.P.-Y.; Skamene, E.; von Messling, V.; Vidal, S.M. Type I IFN Triggers RIG-I/TLR3/NLRP3-dependent Inflammasome Activation in Influenza A Virus Infected Cells. PLoS Pathog. 2013, 9, e1003256. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Yu, M.; Zhang, K.; Liu, J.; Wang, Q.; Tao, P.; Jia, K.; Liao, M.; Ning, Z. Tissue-specific expression pattern and histological distribution of NLRP3 in Chinese yellow chicken. Vet. Res. Commun. 2015, 39, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, Y.; Burt, D.W.; Chen, H.; Zhang, Y.; Qian, W.; Kim, H.; Gan, S.; Zhao, Y.; Li, J.; Yi, K. The duck genome and transcriptome provide insight into an avian influenza virus reservoir species. Nat. Genet. 2013, 45, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A. The Interferon Inducible Gene: Viperin. J. Interferon Cytokine Res. 2011, 31, 131–135. [Google Scholar] [CrossRef]
- Helbig, K.J.; Beard, M.R. The Role of Viperin in the Innate Antiviral Response. J. Mol. Biol. 2014, 426, 1210–1219. [Google Scholar] [CrossRef]
- Wang, X.; Hinson, E.R.; Cresswell, P. The Interferon-Inducible Protein Viperin Inhibits Influenza Virus Release by Perturbing Lipid Rafts. Cell Host Microbe 2007, 2, 96–105. [Google Scholar] [CrossRef]
- Zhong, Z.; Ji, Y.; Fu, Y.; Liu, B.; Zhu, Q. Molecular characterization and expression analysis of the duck viperin gene. Gene 2015, 570, 100–107. [Google Scholar] [CrossRef]
- Goossens, K.E.; Karpala, A.J.; Rohringer, A.; Ward, A.; Bean, A.G.D. Characterisation of chicken viperin. Mol. Immunol. 2015, 63, 373–380. [Google Scholar] [CrossRef]
- Abbas, Y.M.; Pichlmair, A.; Górna, M.W.; Superti-Furga, G.; Nagar, B. Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature 2013, 494, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Katibah, G.E.; Lee, H.J.; Huizar, J.P.; Vogan, J.M.; Alber, T.; Collins, K. tRNA binding, structure, and localization of the human interferon-induced protein IFIT5. Mol. Cell 2013, 49, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Lassnig, C.; Eberle, C.A.; Górna, M.W.; Baumann, C.L.; Burkard, T.R.; Bürckstümmer, T.; Stefanovic, A.; Krieger, S.; Bennett, K.L.; et al. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Rong, E.; Hu, J.; Yang, C.; Chen, H.; Wang, Z.; Liu, X.; Liu, W.; Lu, C.; He, P.; Wang, X.; et al. Broad-spectrum antiviral functions of duck interferon-induced protein with tetratricopeptide repeats (AvIFIT). Dev. Comp. Immunol. 2018, 84, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Clemens, M.J.; Elia, A. The Double-Stranded RNA-Dependent Protein Kinase PKR: Structure and Function. J. Interferon Cytokine Res. 1997, 17, 503–524. [Google Scholar] [CrossRef]
- García, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of Protein Kinase PKR in Cell Biology: From Antiviral to Antiproliferative Action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef]
- Gil, J.; Esteban, M. Induction of apoptosis by the dsRNA-dependent protein kinase (PKR): Mechanism of action. Apoptosis 2000, 5, 107–114. [Google Scholar] [CrossRef]
- Nanduri, S.; Carpick, B.W.; Yang, Y.; Williams, B.R.G.; Qin, J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 1998, 17, 5458–5465. [Google Scholar] [CrossRef]
- Balachandran, S.; Roberts, P.C.; Brown, L.E.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential Role for the dsRNA-Dependent Protein Kinase PKR in Innate Immunity to Viral Infection. Immunity 2000, 13, 129–141. [Google Scholar] [CrossRef]
- Takizawa, T.; Ohashi, K.; Nakanishi, Y. Possible involvement of double-stranded RNA-activated protein kinase in cell death by influenza virus infection. J. Virol. 1996, 70, 8128–8132. [Google Scholar] [PubMed]
- Liu, W.; Yang, Y.; Huang, Y.; zhou, D.-R.; Xu, D.; Cao, N.; Jiang, D.; Pan, J.; Tian, Y. Identification of Goose PKR Gene: Structure, Expression Profiling, and Antiviral Activity Against Newcastle Disease Virus. J. Interferon Cytokine Res. 2018, 38, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.-H.; Asano, A.; Kon, Y.; Watanabe, T.; Agui, T. Characterization of the Chicken PKR: Polymorphism of the gene and Antiviral Activity against Vesicular Stomatitis Virus. Jpn. J. Vet. Res. 2004, 51, 123–133. [Google Scholar]
- Förster, R.; Davalos-Misslitz, A.C.; Rot, A. CCR7 and its ligands: Balancing immunity and tolerance. Nat. Rev. Immunol. 2008, 8, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Fleming-Canepa, X.; Brusnyk, C.; Aldridge, J.R.; Ross, K.L.; Moon, D.; Wang, D.; Xia, J.; Barber, M.R.W.; Webster, R.G.; Magor, K.E. Expression of duck CCL19 and CCL21 and CCR7 receptor in lymphoid and influenza-infected tissues. Mol. Immunol. 2011, 48, 1950–1957. [Google Scholar] [CrossRef] [PubMed]
- Ranaware, P.B.; Mishra, A.; Vijayakumar, P.; Gandhale, P.N.; Kumar, H.; Kulkarni, D.D.; Raut, A.A. Genome Wide Host Gene Expression Analysis in Chicken Lungs Infected with Avian Influenza Viruses. PLoS ONE 2016, 11, e0153671. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Moreno, J.; Moyron-Quiroz, J.E.; Hartson, L.; Kusser, K.; Randall, T.D. Pulmonary expression of CXC chemokine ligand 13, CC chemokine ligand 19, and CC chemokine ligand 21 is essential for local immunity to influenza. Proc. Natl. Acad. Sci. USA 2007, 104, 10577–10582. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Acklin, M.; Staeheli, P. Influenza Virus Resistance of Wild Mice: Wild-Type and Mutant Mx Alleles Occur at Comparable Frequencies. J. Interferon Res. 1987, 7, 647–656. [Google Scholar] [CrossRef]
- Haller, O.; Staeheli, P.; Kochs, G. Protective role of interferon-induced Mx GTPases against influenza viruses. Rev. Sci. Tech. 2009, 28, 219–231. [Google Scholar] [CrossRef]
- Manuja, B.K.; Manuja, A.; Dahiya, R.; Singh, S.; Sharma, R.C.; Gahlot, S.K. Diversity of interferon inducible Mx gene in horses and association of variations with susceptibility vis-à-vis resistance against equine influenza infection. Infect. Genet. Evol. 2014, 27, 142–148. [Google Scholar] [CrossRef]
- Numajiri Haruki, A.; Naito, T.; Nishie, T.; Saito, S.; Nagata, K. Interferon-inducible antiviral protein MxA enhances cell death triggered by endoplasmic reticulum stress. J. Interferon Cytokine Res. 2011, 31, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Staeheli, P.; Grob, R.; Meier, E.; Sutcliffe, J.G.; Haller, O. Influenza virus-susceptible mice carry Mx genes with a large deletion or a nonsense mutation. Mol. Cell. Biol. 1988, 8, 4518–4523. [Google Scholar] [CrossRef]
- Haller, O.; Staeheli, P.; Schwemmle, M.; Kochs, G. Mx GTPases: Dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015, 23, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Horisberger, M.A.; Staritzky, K.D. Expression and Stability of the Mx Protein in Different Tissues of Mice, in Response to Interferon Inducers or to Influenza Virus Infection. J. Interferon Res. 1989, 9, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Staeheli, P.; Pitossi, F.; Pavlovic, J. Mx proteins: GTPases with antiviral activity. Trends Cell Biol. 1993, 3, 268–272. [Google Scholar] [CrossRef]
- Verhelst, J.; Hulpiau, P.; Saelens, X. Mx Proteins: Antiviral Gatekeepers That Restrain the Uninvited. Microbiol. Mol. Biol. Rev. MMBR 2013, 77, 551–566. [Google Scholar] [CrossRef]
- Xiao, H.; Killip, M.J.; Staeheli, P.; Randall, R.E.; Jackson, D. The Human Interferon-Induced MxA Protein Inhibits Early Stages of Influenza A Virus Infection by Retaining the Incoming Viral Genome in the Cytoplasm. J. Virol. 2013, 87, 13053–13058. [Google Scholar] [CrossRef]
- Zimmermann, P.; Mänz, B.; Haller, O.; Schwemmle, M.; Kochs, G. The Viral Nucleoprotein Determines Mx Sensitivity of Influenza A Viruses. J. Virol. 2011, 85, 8133–8140. [Google Scholar] [CrossRef] [PubMed]
- Turan, K.; Mibayashi, M.; Sugiyama, K.; Saito, S.; Numajiri, A.; Nagata, K. Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic Acids Res. 2004, 32, 643–652. [Google Scholar] [CrossRef]
- Pavlovic, J.; Haller, O.; Staeheli, P. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J. Virol. 1992, 66, 2564–2569. [Google Scholar]
- Bazzigher, L.; Schwarz, A.; Staeheli, P. No Enhanced Influenza Virus Resistance of Murine and Avian Cells Expressing Cloned Duck Mx Protein. Virology 1993, 195, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.-H.; Jin, H.-K.; Asano, A.; Takada, A.; Ninomiya, A.; Kida, H.; Hokiyama, H.; Ohara, M.; Tsuzuki, M.; Nishibori, M.; et al. Polymorphisms and the Differential Antiviral Activity of the Chicken Mx Gene. Genome Res. 2002, 12, 595–601. [Google Scholar] [CrossRef]
- Ko, J.H.; Takada, A.; Mitsuhashi, T.; Agui, T.; Watanabe, T. Native antiviral specificity of chicken Mx protein depends on amino acid variation at position 631. Anim. Genet. 2004, 35, 119–122. [Google Scholar] [CrossRef]
- Dillon, D.; Runstadler, J. Mx gene diversity and influenza association among five wild dabbling duck species (Anas spp.) in Alaska. Infect. Genet. Evol. 2010, 10, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Patzina, C.; Haller, O.; Kochs, G. Structural Requirements for the Antiviral Activity of the Human MxA Protein against Thogoto and Influenza A Virus. J. Biol. Chem. 2014, 289, 6020–6027. [Google Scholar] [CrossRef]
- Götz, V.; Magar, L.; Dornfeld, D.; Giese, S.; Pohlmann, A.; Höper, D.; Kong, B.-W.; Jans, D.A.; Beer, M.; Haller, O.; et al. Influenza A viruses escape from MxA restriction at the expense of efficient nuclear vRNP import. Sci. Rep. 2016, 6, 23138. [Google Scholar] [CrossRef]
- Mänz, B.; Dornfeld, D.; Götz, V.; Zell, R.; Zimmermann, P.; Haller, O.; Kochs, G.; Schwemmle, M. Pandemic Influenza A Viruses Escape from Restriction by Human MxA through Adaptive Mutations in the Nucleoprotein. PLoS Pathog. 2013, 9, e1003279. [Google Scholar] [CrossRef] [PubMed]
- Riegger, D.; Hai, R.; Dornfeld, D.; Mänz, B.; Leyva-Grado, V.; Sánchez-Aparicio, M.T.; Albrecht, R.A.; Palese, P.; Haller, O.; Schwemmle, M.; et al. The Nucleoprotein of Newly Emerged H7N9 Influenza A Virus Harbors a Unique Motif Conferring Resistance to Antiviral Human MxA. J. Virol. 2014, 89, 2241–2252. [Google Scholar] [CrossRef] [PubMed]
- Benfield, C.T.O.; Lyall, J.W.; Kochs, G.; Tiley, L.S. Asparagine 631 Variants of the Chicken Mx Protein Do Not Inhibit Influenza Virus Replication in Primary Chicken Embryo Fibroblasts or In Vitro Surrogate Assays. J. Virol. 2008, 82, 7533–7539. [Google Scholar] [CrossRef]
- Bernasconi, D.; Schultz, U.; Staeheli, P. The Interferon-Induced Mx Protein of Chickens Lacks Antiviral Activity. J. Interferon Cytokine Res. 1995, 15, 47–53. [Google Scholar] [CrossRef]
- Fulton, J.E.; Arango, J.; Ali, R.A.; Bohorquez, E.B.; Lund, A.R.; Ashwell, C.M.; Settar, P.; O’Sullivan, N.P.; Koci, M.D. Genetic Variation within the Mx Gene of Commercially Selected Chicken Lines Reveals Multiple Haplotypes, Recombination and a Protein under Selection Pressure. PLoS ONE 2014, 9, e108054. [Google Scholar] [CrossRef]
- Schusser, B.; Reuter, A.; Malsburg A von der Penski, N.; Weigend, S.; Kaspers, B.; Staeheli, P.; Härtle, S. Mx Is Dispensable for Interferon-Mediated Resistance of Chicken Cells against Influenza A Virus. J. Virol. 2011, 85, 8307–8315. [Google Scholar] [CrossRef]
- Gao, S.; von der Malsburg, A.; Dick, A.; Faelber, K.; Schröder, G.F.; Haller, O.; Kochs, G.; Daumke, O. Structure of Myxovirus Resistance Protein A Reveals Intra- and Intermolecular Domain Interactions Required for the Antiviral Function. Immunity 2011, 35, 514–525. [Google Scholar] [CrossRef]
- Sarkar, S.N.; Bandyopadhyay, S.; Ghosh, A.; Sen, G.C. Enzymatic Characteristics of Recombinant Medium Isozyme of 2′-5′ Oligoadenylate Synthetase. J. Biol. Chem. 1999, 274, 1848–1855. [Google Scholar] [CrossRef]
- Sarkar, S.N.; Ghosh, A.; Wang, H.-W.; Sung, S.-S.; Sen, G.C. The Nature of the Catalytic Domain of 2′-5′-Oligoadenylate Synthetases. J. Biol. Chem. 1999, 274, 25535–25542. [Google Scholar] [CrossRef]
- Ishibashi, M.; Wakita, T.; Esumi, M. 2′,5′-Oligoadenylate synthetase-like gene highly induced by hepatitis C virus infection in human liver is inhibitory to viral replication in vitro. Biochem. Biophys. Res. Commun. 2010, 392, 397–402. [Google Scholar] [CrossRef]
- Marques, J.; Anwar, J.; Eskildsen-Larsen, S.; Rebouillat, D.; Paludan, S.R.; Sen, G.; Williams, B.R.G.; Hartmann, R. The p59 oligoadenylate synthetase-like protein possesses antiviral activity that requires the C-terminal ubiquitin-like domain. J. Gen. Virol. 2008, 89, 2767–2772. [Google Scholar] [CrossRef] [PubMed]
- Rebouillat, D.; Marié, I.; Hovanessian, A.G. Molecular cloning and characterization of two related and interferon-induced 56-kDa and 30-kDa proteins highly similar to 2′-5′ oligoadenylate synthetase. Eur. J. Biochem. 1998, 257, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Rong, E.; Wang, X.; Chen, H.; Yang, C.; Hu, J.; Liu, W.; Wang, Z.; Chen, X.; Zheng, H.; Pu, J.; et al. Molecular Mechanisms for the Adaptive Switching Between the OAS/RNase L and OASL/RIG-I Pathways in Birds and Mammals. Front. Immunol. 2018, 9, 1398. [Google Scholar] [CrossRef]
- Sokawa, J.; Shimizu, N.; Sokawa, Y. Presence of (2′-5′) Oligoadenylate Synthetase in Avian Erythrocytes. J. Biochem. 1984, 96, 215–222. [Google Scholar] [CrossRef]
- Tag-EL-Din-Hassan, H.T.; Morimatsu, M.; Agui, T. Functional analysis of duck, goose, and ostrich 2′-5′-oligoadenylate synthetase. Infect. Genet. Evol. 2018, 62, 220–232. [Google Scholar] [CrossRef]
- Qian, W.; Wei, X.; Zhou, H.; Jin, M. Molecular cloning and functional analysis of duck ubiquitin-specific protease 18 (USP18) gene. Dev. Comp. Immunol. 2016, 62, 39–47. [Google Scholar] [CrossRef]
- Huang, I.C.; Bailey, C.C.; Weyer, J.L.; Radoshitzky, S.R.; Becker, M.M.; Chiang, J.J.; Brass, A.L.; Ahmed, A.A.; Chi, X.; Dong, L.; et al. Distinct Patterns of IFITM-Mediated Restriction of Filoviruses, SARS Coronavirus, and Influenza A Virus. PLoS Pathog. 2011, 7, e1001258. [Google Scholar] [CrossRef]
- Bailey, C.C.; Huang, I.-C.; Kam, C.; Farzan, M. Ifitm3 Limits the Severity of Acute Influenza in Mice. PLoS Pathog. 2012, 8, e1002909. [Google Scholar] [CrossRef]
- Everitt, A.R.; Clare, S.; Pertel, T.; John, S.P.; Wash, R.S.; Smith, S.E.; Chin, C.R.; Feeley, E.M.; Sims, J.S.; Adams, D.J.; et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 2012, 484, 519–523. [Google Scholar] [CrossRef]
- John, S.P.; Chin, C.R.; Perreira, J.M.; Feeley, E.M.; Aker, A.M.; Savidis, G.; Smith, S.E.; Elia, A.E.H.; Everitt, A.R.; Vora, M.; et al. The CD225 Domain of IFITM3 Is Required for both IFITM Protein Association and Inhibition of Influenza A Virus and Dengue Virus Replication. J. Virol. 2013, 87, 7837–7852. [Google Scholar] [CrossRef]
- Blyth, G.A.D.; Chan, W.F.; Webster, R.G.; Magor, K.E. Duck Interferon-Inducible Transmembrane Protein 3 Mediates Restriction of Influenza Viruses. J. Virol. 2015, 90, 103–116. [Google Scholar] [CrossRef]
- Smith, S.E.; Gibson, M.S.; Wash, R.S.; Ferrara, F.; Wright, E.; Temperton, N.; Kellam, P.; Fife, M. Chicken Interferon-Inducible Transmembrane Protein 3 Restricts Influenza Viruses and Lyssaviruses In Vitro. J. Virol. 2013, 87, 12957–12966. [Google Scholar] [CrossRef]
- Ayllon, J.; García-Sastre, A. The NS1 Protein: A Multitasking Virulence Factor. In Influenza Pathogenesis and Control—Volume II; Oldstone, M.B.A., Compans, R.W., Eds.; Current Topics in Microbiology and Immunology; Springer International Publishing: Cham, Switzerland, 2015; pp. 73–107. [Google Scholar]
- Min, J.-Y.; Li, S.; Sen, G.C.; Krug, R.M. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology 2007, 363, 236–243. [Google Scholar] [CrossRef]
- Nemeroff, M.E.; Barabino, S.M.L.; Li, Y.; Keller, W.; Krug, R.M. Influenza Virus NS1 Protein Interacts with the Cellular 30 kDa Subunit of CPSF and Inhibits 3′ End Formation of Cellular Pre-mRNAs. Mol. Cell 1998, 1, 991–1000. [Google Scholar] [CrossRef]
- Hale, B.G.; Randall, R.E.; Ortín, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef]
- García-Sastre, A.; Egorov, A.; Matassov, D.; Brandt, S.; Levy, D.E.; Durbin, J.E.; Palese, P.; Muster, T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 1998, 252, 324–330. [Google Scholar] [CrossRef]
- Kochs, G.; Koerner, I.; Thiel, L.; Kothlow, S.; Kaspers, B.; Ruggli, N.; Summerfield, A.; Pavlovic, J.; Stech, J.; Staeheli, P. Properties of H7N7 influenza A virus strain SC35M lacking interferon antagonist NS1 in mice and chickens. J. Gen. Virol. 2007, 88, 1403–1409. [Google Scholar] [CrossRef]
- Zhu, Q.; Yang, H.; Chen, W.; Cao, W.; Zhong, G.; Jiao, P.; Deng, G.; Yu, K.; Yang, C.; Bu, Z.; et al. A Naturally Occurring Deletion in Its NS Gene Contributes to the Attenuation of an H5N1 Swine Influenza Virus in Chickens. J. Virol. 2008, 82, 220–228. [Google Scholar] [CrossRef]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.-S.; Huang, I.-C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef]
- Koliopoulos, M.G.; Lethier, M.; van der Veen, A.G.; Haubrich, K.; Hennig, J.; Kowalinski, E.; Stevens, R.V.; Martin, S.R.; Reis e Sousa, C.; Cusack, S.; et al. Molecular mechanism of influenza A NS1-mediated TRIM25 recognition and inhibition. Nat. Commun. 2018, 9, 1820. [Google Scholar] [CrossRef]
- Mok, B.W.-Y.; Song, W.; Wang, P.; Tai, H.; Chen, Y.; Zheng, M.; Wen, X.; Lau, S.-Y.; Wu, W.L.; Matsumoto, K.; et al. The NS1 Protein of Influenza A Virus Interacts with Cellular Processing Bodies and Stress Granules through RNA-Associated Protein 55 (RAP55) during Virus Infection. J. Virol. 2012, 86, 12695–12707. [Google Scholar] [CrossRef]
- Cauthen, A.N.; Swayne, D.E.; Sekellick, M.J.; Marcus, P.I.; Suarez, D.L. Amelioration of Influenza Virus Pathogenesis in Chickens Attributed to the Enhanced Interferon-Inducing Capacity of a Virus with a Truncated NS1 Gene. J. Virol. 2007, 81, 1838–1847. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, Y.; Jiao, P.; Wang, A.; Zhao, F.; Tian, G.; Wang, X.; Yu, K.; Bu, Z.; Chen, H. The NS1 Gene Contributes to the Virulence of H5N1 Avian Influenza Viruses. J. Virol. 2006, 80, 11115–11123. [Google Scholar] [CrossRef]
- Obenauer, J.C.; Denson, J.; Mehta, P.K.; Su, X.; Mukatira, S.; Finkelstein, D.B.; Xu, X.; Wang, J.; Ma, J.; Fan, Y.; et al. Large-Scale Sequence Analysis of Avian Influenza Isolates. Science 2006, 311, 1576–1580. [Google Scholar] [CrossRef]
- Thomas, M.; Kranjec, C.; Nagasaka, K.; Matlashewski, G.; Banks, L. Analysis of the PDZ binding specificities of Influenza A Virus NS1 proteins. Virol. J. 2011, 8, 25. [Google Scholar] [CrossRef]
- Golebiewski, L.; Liu, H.; Javier, R.T.; Rice, A.P. The Avian Influenza Virus NS1 ESEV PDZ Binding Motif Associates with Dlg1 and Scribble to Disrupt Cellular Tight Junctions. J. Virol. 2011, 85, 10639–10648. [Google Scholar] [CrossRef]
- Liu, H.; Golebiewski, L.; Dow, E.C.; Krug, R.M.; Javier, R.T.; Rice, A.P. The ESEV PDZ-Binding Motif of the Avian Influenza A Virus NS1 Protein Protects Infected Cells from Apoptosis by Directly Targeting Scribble. J. Virol. 2010, 84, 11164–11174. [Google Scholar] [CrossRef]
- Jackson, D.; Hossain, M.J.; Hickman, D.; Perez, D.R.; Lamb, R.A. A new influenza virus virulence determinant: The NS1 protein four C-terminal residues modulate pathogenicity. Proc. Natl. Acad. Sci. USA 2008, 105, 4381–4386. [Google Scholar] [CrossRef]
- Soubies, S.M.; Hoffmann, T.W.; Croville, G.; Larcher, T.; Ledevin, M.; Soubieux, D.; Quéré, P.; Guérin, J.-L.; Marc, D.; Volmer, R. Deletion of the C-terminal ESEV domain of NS1 does not affect the replication of a low-pathogenic avian influenza virus H7N1 in ducks and chickens. J. Gen. Virol. 2013, 94, 50–58. [Google Scholar] [CrossRef]
- Soubies, S.M.; Volmer, C.; Croville, G.; Loupias, J.; Peralta, B.; Costes, P.; Lacroux, C.; Guérin, J.-L.; Volmer, R. Species-Specific Contribution of the Four C-Terminal Amino Acids of Influenza A Virus NS1 Protein to Virulence. J. Virol. 2010, 84, 6733–6747. [Google Scholar] [CrossRef]
- Zielecki, F.; Semmler, I.; Kalthoff, D.; Voss, D.; Mauel, S.; Gruber, A.D.; Beer, M.; Wolff, T. Virulence determinants of avian H5N1 influenza A virus in mammalian and avian hosts: Role of the C-terminal ESEV motif in the viral NS1 protein. J. Virol. 2010, 84, 10708–10718. [Google Scholar] [CrossRef]
- Kuchipudi, S.V.; Dunham, S.P.; Nelli, R.; White, G.A.; Coward, V.J.; Slomka, M.J.; Brown, I.H.; Chang, K.C. Rapid death of duck cells infected with influenza: A potential mechanism for host resistance to H5N1. Immunol. Cell Biol. 2012, 90, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Rosebeck, S.; Leaman, D.W. Mitochondrial localization and pro-apoptotic effects of the interferon-inducible protein ISG12a. Apoptosis 2008, 13, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Daidoji, T.; Du, A.; Yang, C.-S.; Ibrahim, M.S.; Ikuta, K.; Nakaya, T. Highly Pathogenic H5N1 Avian Influenza Virus Induces Extracellular Ca2+ Influx, Leading to Apoptosis in Avian Cells. J. Virol. 2010, 84, 3068–3078. [Google Scholar] [CrossRef]
- Basters, A.; Geurink, P.P.; Oualid, F.E.; Ketscher, L.; Casutt, M.S.; Krause, E.; Ovaa, H.; Knobeloch, K.-P.; Fritz, G. Molecular characterization of ubiquitin-specific protease 18 reveals substrate specificity for interferon-stimulated gene 15. FEBS J. 2014, 281, 1918–1928. [Google Scholar] [CrossRef] [PubMed]
- Malakhov, M.P.; Malakhova, O.A.; Kim, K.I.; Ritchie, K.J.; Zhang, D.-E. UBP43 (USP18) Specifically Removes ISG15 from Conjugated Proteins. J. Biol. Chem. 2002, 277, 9976–9981. [Google Scholar] [CrossRef]
- Yang, Z.; Xian, H.; Hu, J.; Tian, S.; Qin, Y.; Wang, R.-F.; Cui, J. USP18 negatively regulates NF-κB signaling by targeting TAK1 and NEMO for deubiquitination through distinct mechanisms. Sci. Rep. 2015, 5, 12738. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, T.; Uchida, Y.; Saito, T. Replication of a low-pathogenic avian influenza virus is enhanced by chicken ubiquitin-specific protease 18. J. Gen. Virol. 2017, 98, 2235–2247. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, M.L.; Middleton, D.L.; Radford, C.; Warr, G.W.; Magor, K.E. Immunoglobulins of the non-galliform birds: Antibody expression and repertoire in the duck. Dev. Comp. Immunol. 2006, 30, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Magor, K.E. Immunoglobulin genetics and antibody responses to influenza in ducks. Dev. Comp. Immunol. 2011, 35, 1008–1016. [Google Scholar] [CrossRef]
- Chaise, C.; Lalmanach, A.-C.; Marty, H.; Soubies, S.M.; Croville, G.; Loupias, J.; Marc, D.; Quéré, P.; Guérin, J.-L. Protection Patterns in Duck and Chicken after Homo- or Hetero-Subtypic Reinfections with H5 and H7 Low Pathogenicity Avian Influenza Viruses: A Comparative Study. PLoS ONE 2014, 9, e105189. [Google Scholar] [CrossRef]
- Dannemiller, N.G.; Webb, C.T.; Wilson, K.R.; Bentler, K.T.; Mooers, N.L.; Ellis, J.W.; Root, J.J.; Franklin, A.B.; Shriner, S.A. Impact of body condition on influenza A virus infection dynamics in mallards following a secondary exposure. PLoS ONE 2017, 12, e0175757. [Google Scholar] [CrossRef]
- Segovia, K.M.; França, M.S.; Leyson, C.L.; Kapczynski, D.R.; Chrzastek, K.; Bahnson, C.S.; Stallknecht, D.E. Heterosubtypic immunity increases infectious dose required to infect Mallard ducks with Influenza A virus. PLoS ONE 2018, 13, e0196394. [Google Scholar] [CrossRef]
- Latorre-Margalef, N.; Brown, J.D.; Fojtik, A.; Poulson, R.L.; Carter, D.; Franca, M.; Stallknecht, D.E. Competition between influenza A virus subtypes through heterosubtypic immunity modulates re-infection and antibody dynamics in the mallard duck. PLoS Pathog. 2017, 13, e1006419. [Google Scholar] [CrossRef]
- Kim, C.H.; Pelus, L.M.; White, J.R.; Applebaum, E.; Johanson, K.; Broxmeyer, H.E. CKβ-11/Macrophage Inflammatory Protein-3β/EBI1-Ligand Chemokine Is an Efficacious Chemoattractant for T and B Cells. J. Immunol. 1998, 160, 2418–2424. [Google Scholar] [PubMed]
- Fleming-Canepa, X.; Jensen, S.M.; Mesa, C.M.; Diaz-Satizabal, L.; Roth, A.J.; Parks-Dely, J.A.; Moon, D.A.; Wong, J.P.; Evseev, D.; Gossen, D.A.; et al. Extensive Allelic Diversity of MHC Class I in Wild Mallard Ducks. J. Immunol. 2016, 197, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.A.; Veniamin, S.M.; Parks-Dely, J.A.; Magor, K.E. The MHC of the Duck (Anas platyrhynchos) Contains Five Differentially Expressed Class I Genes. J. Immunol. 2005, 175, 6702–6712. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, J.; Fan, S.; Chen, R.; Liu, Y.; Zhang, J.; Yuan, H.; Liang, R.; Zhang, N.; Xia, C. Structural Definition of Duck MHC class I Molecules that might explain efficient CTL Immunity against Influenza A Virus. J. Virol. 2017, JVI.02511-16. [Google Scholar] [CrossRef] [PubMed]
- Heimberg, A.M.; Sempere, L.F.; Moy, V.N.; Donoghue, P.C.J.; Peterson, K.J. MicroRNAs and the advent of vertebrate morphological complexity. Proc. Natl. Acad. Sci. USA 2008, 105, 2946–2950. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, J.; Su, J.; Liu, Y.; Guo, J.; Zhang, Y.; Lu, C.; Xing, S.; Guan, Y.; Li, Y.; Sun, B.; Zhao, Z. MicroRNAs in the immune organs of chickens and ducks indicate divergence of immunity against H5N1 avian influenza. FEBS Lett. 2014, 589, 419–425. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene Name | Ensembl ID |
|---|---|
| RSAD2 (Viperin) | ENSAPLT00000006241 |
| IFIT5 | ENSAPLT00000001602 |
| CCL4 | ENSAPLT00000010196 |
| EIF2AK2 (PKR) | ENSAPLT00000010258 |
| ISG15 | ENSAPLT00000016054 |
| IL4I1 | ENSAPLT00000016724 |
| USP18 | ENSAPLT00000007808 |
| EPSTI1 | ENSAPLT00000005561 |
| CMPK2 | ENSAPLT00000006212 |
| CCL19 | ENSAPLT00000005578 |
| MX | ENSAPLT00000016707 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evseev, D.; Magor, K.E. Innate Immune Responses to Avian Influenza Viruses in Ducks and Chickens. Vet. Sci. 2019, 6, 5. https://doi.org/10.3390/vetsci6010005
Evseev D, Magor KE. Innate Immune Responses to Avian Influenza Viruses in Ducks and Chickens. Veterinary Sciences. 2019; 6(1):5. https://doi.org/10.3390/vetsci6010005
Chicago/Turabian StyleEvseev, Danyel, and Katharine E. Magor. 2019. "Innate Immune Responses to Avian Influenza Viruses in Ducks and Chickens" Veterinary Sciences 6, no. 1: 5. https://doi.org/10.3390/vetsci6010005
APA StyleEvseev, D., & Magor, K. E. (2019). Innate Immune Responses to Avian Influenza Viruses in Ducks and Chickens. Veterinary Sciences, 6(1), 5. https://doi.org/10.3390/vetsci6010005