
The Protein Encoded by the UL3.5 Gene of the Duck Plague Virus Affects Viral Secondary Envelopment, Release, and Cell-to-Cell Spread


 and
and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells, Viruses, and Primers
2.2. Antibodies
2.3. Construction of Plasmid and Recombinant BAC Virus Plasmid
2.4. Conventional Polymerase Chain Reaction (PCR)
2.5. Rescue and Identification of Recombinant Virus
2.6. Fluorescent Protein Labeling
2.7. Real-Time Quantitative PCR
2.8. Cell Viability Assessment by CCK-8
2.9. GCV/CHX Drug Inhibition Assay
2.10. Western Blotting Analysis
2.11. Determination of Multi-Step Growth Curve In Vitro
2.12. Virus Adsorption, Invasion, Replication, and Release Assays
2.12.1. Adsorption
2.12.2. Invasion
2.12.3. Replication
2.12.4. Release
2.13. Electron Microscopy
2.14. Analysis of Virus Cell-to-Cell Spread
2.15. Data Analysis
3. Results
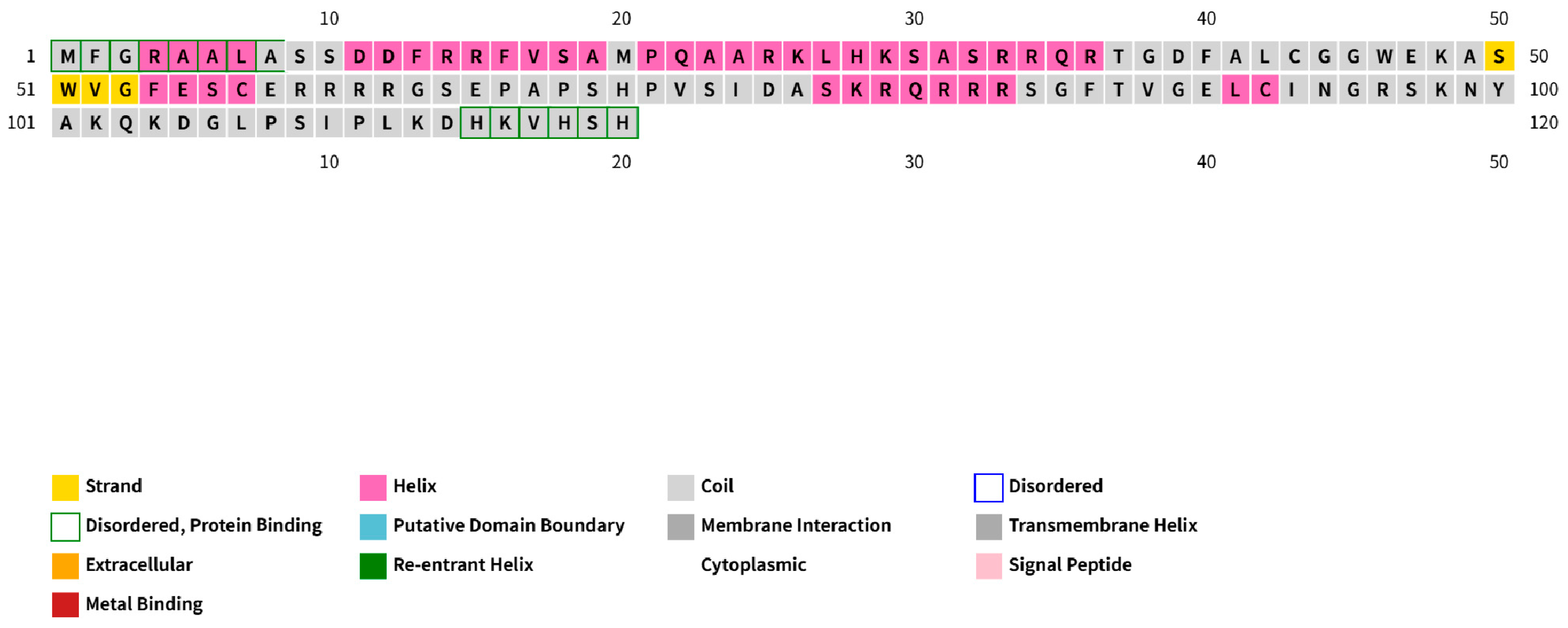
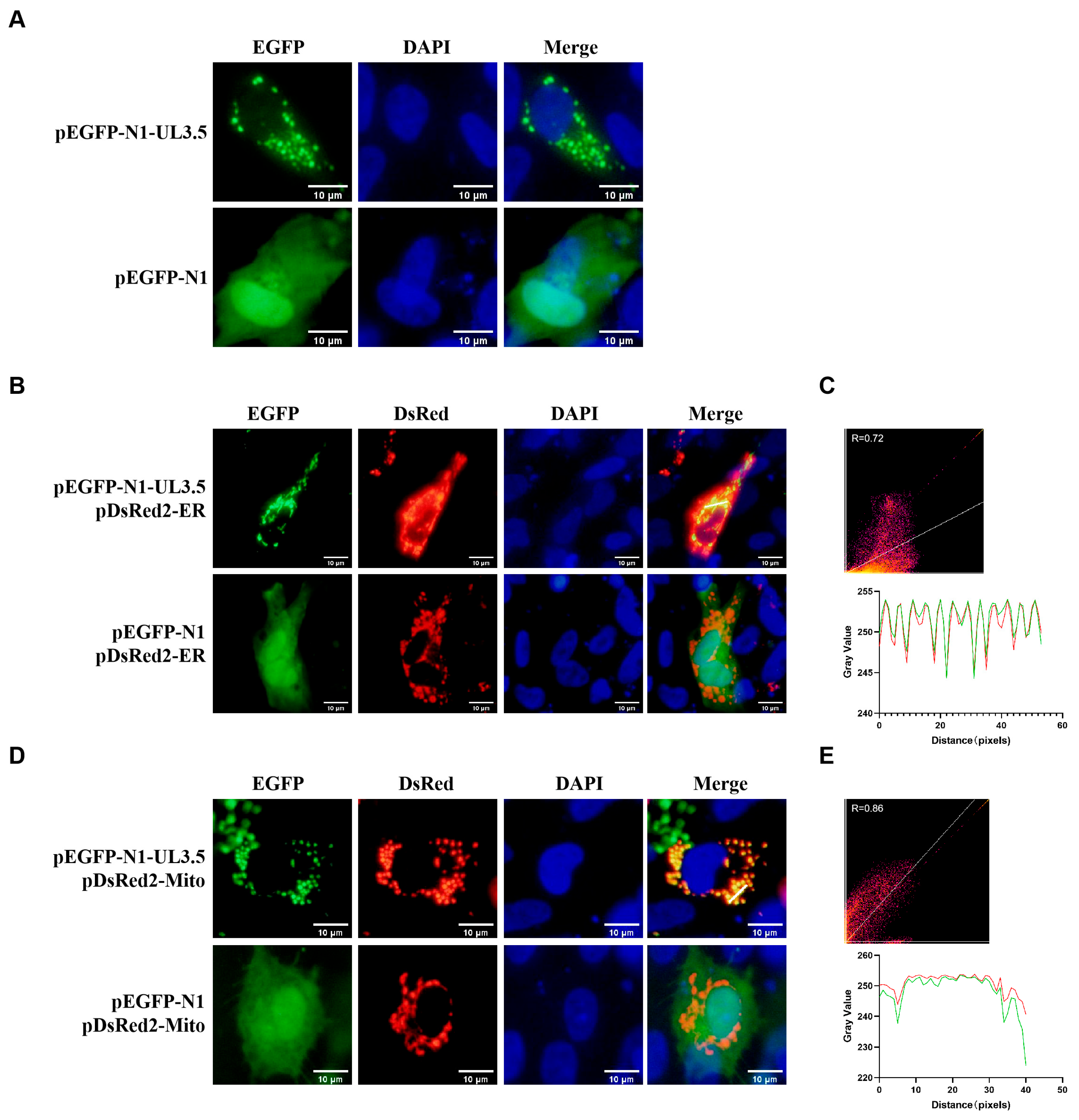
3.1. DPV pUL3.5 Is a Multicellular Localization Protein
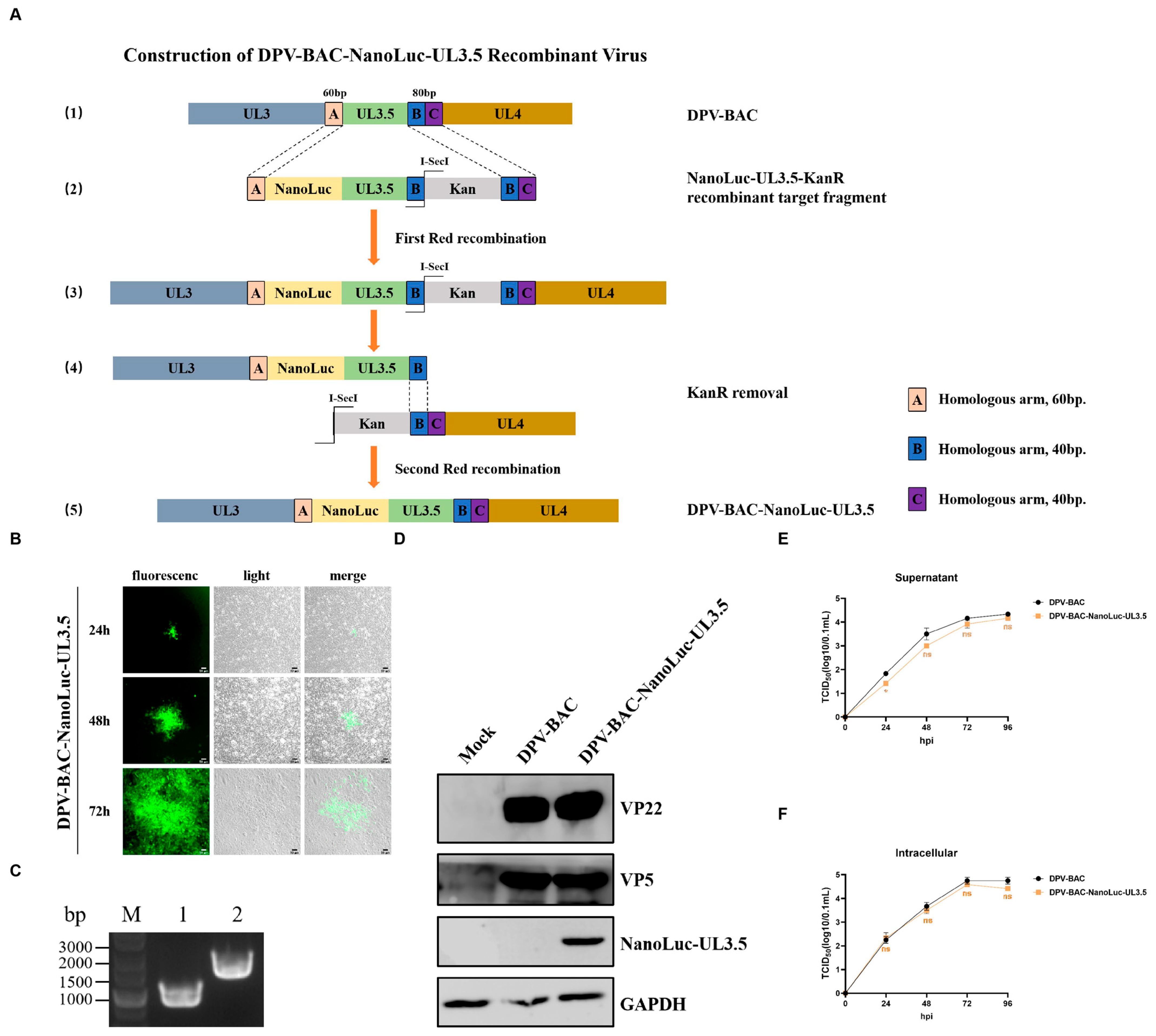
3.2. DPV UL3.5 Encodes an Early Viral Protein
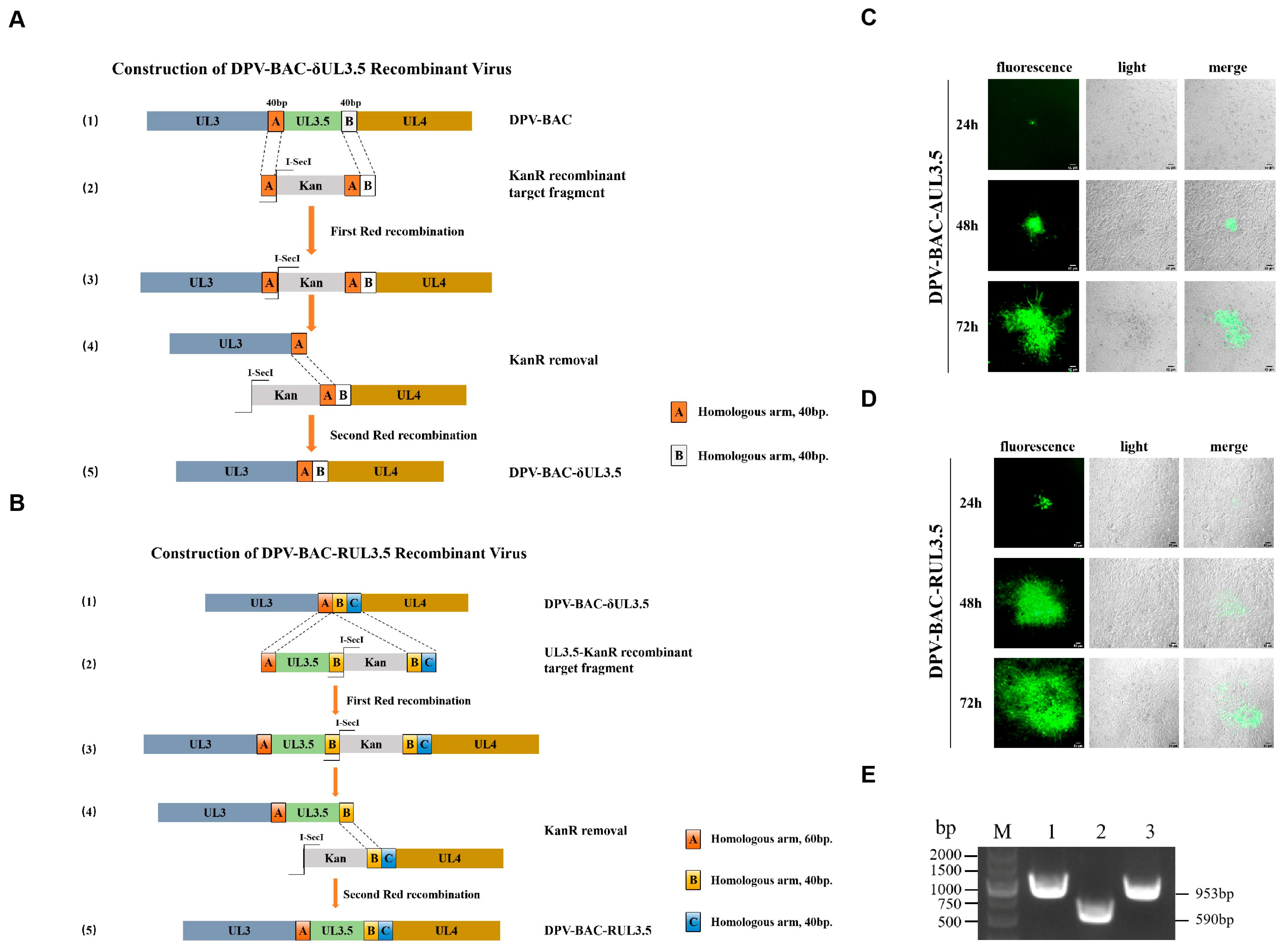
3.3. Construction and Rescue of DPV-BAC-δUL3.5 and DPV-BAC-RUL3.5 Recombinant Virus Strains
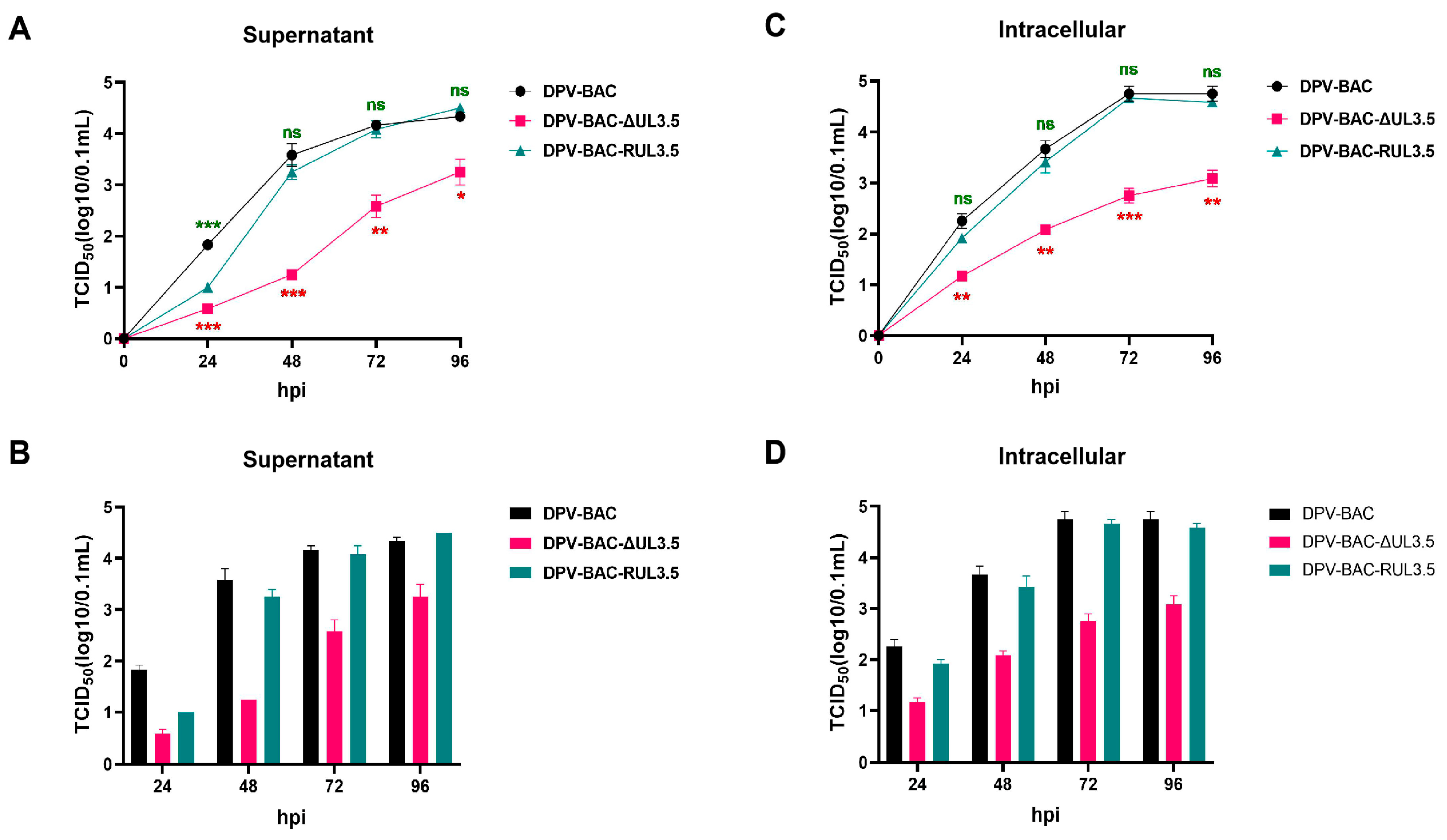
3.4. The Deletion of UL3.5 Leads to the Slow Growth Rate In Vitro
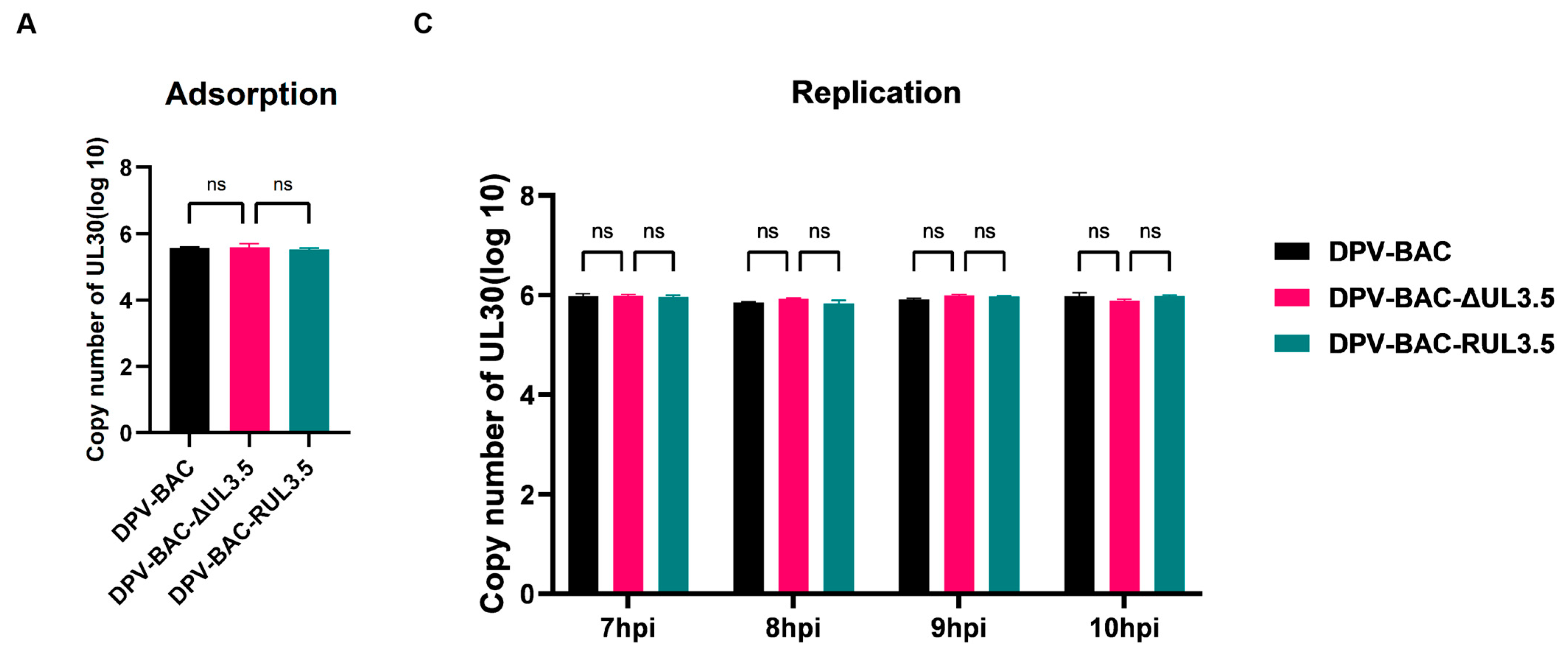
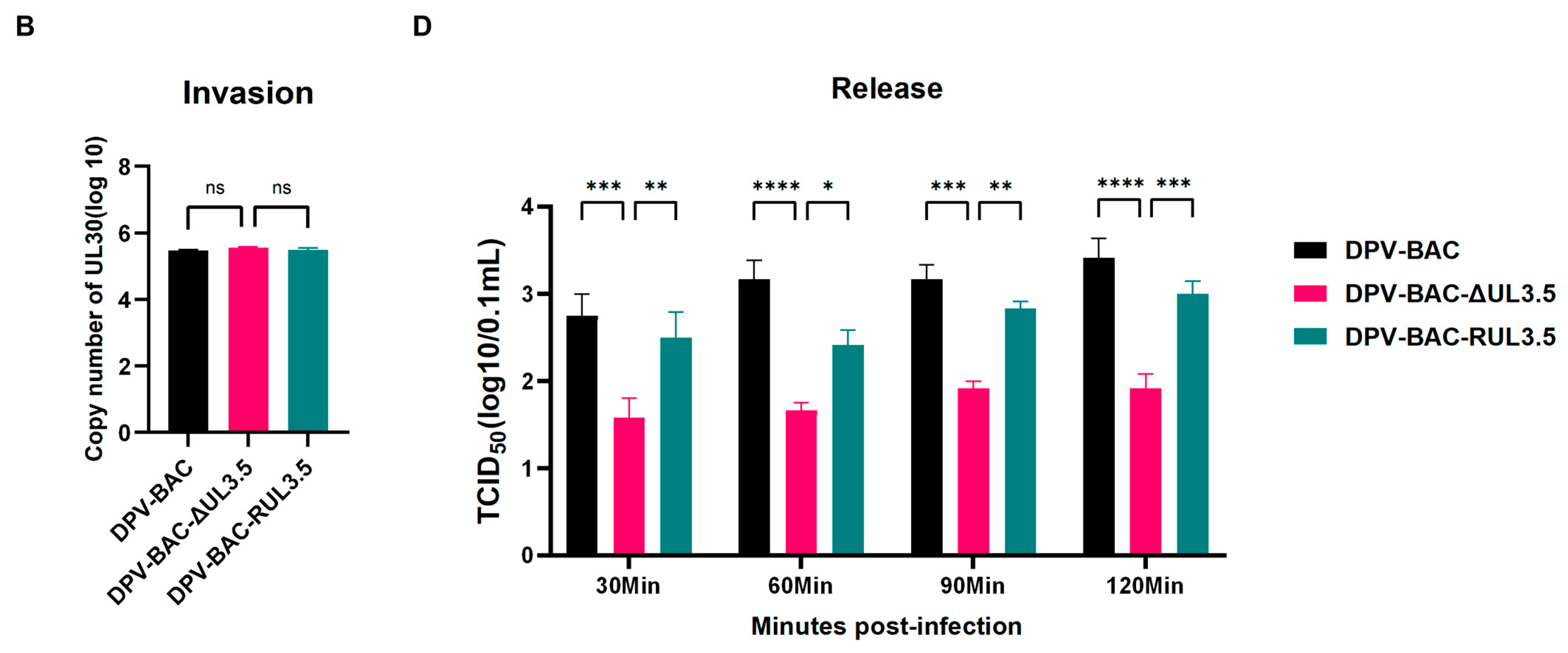
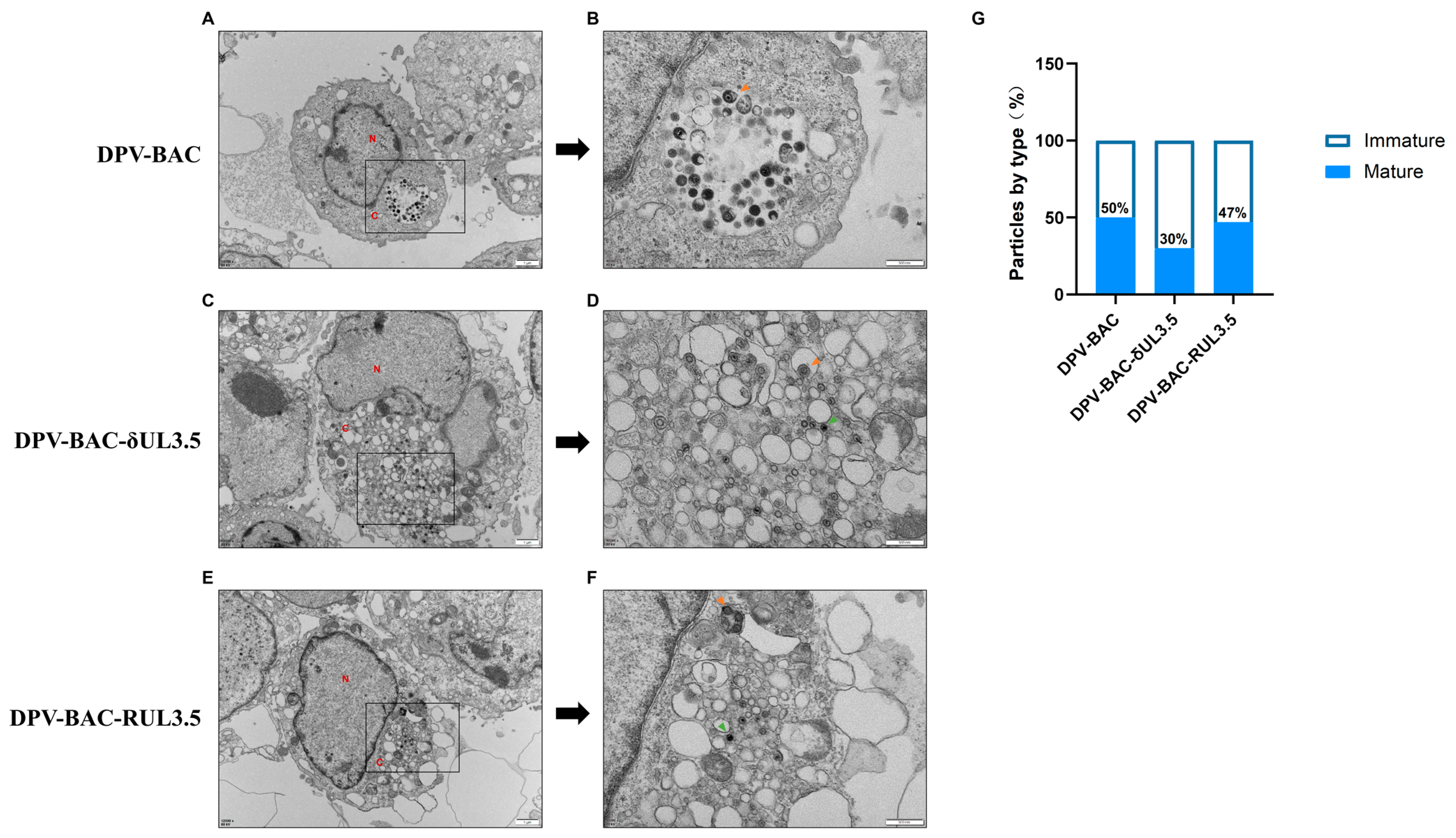
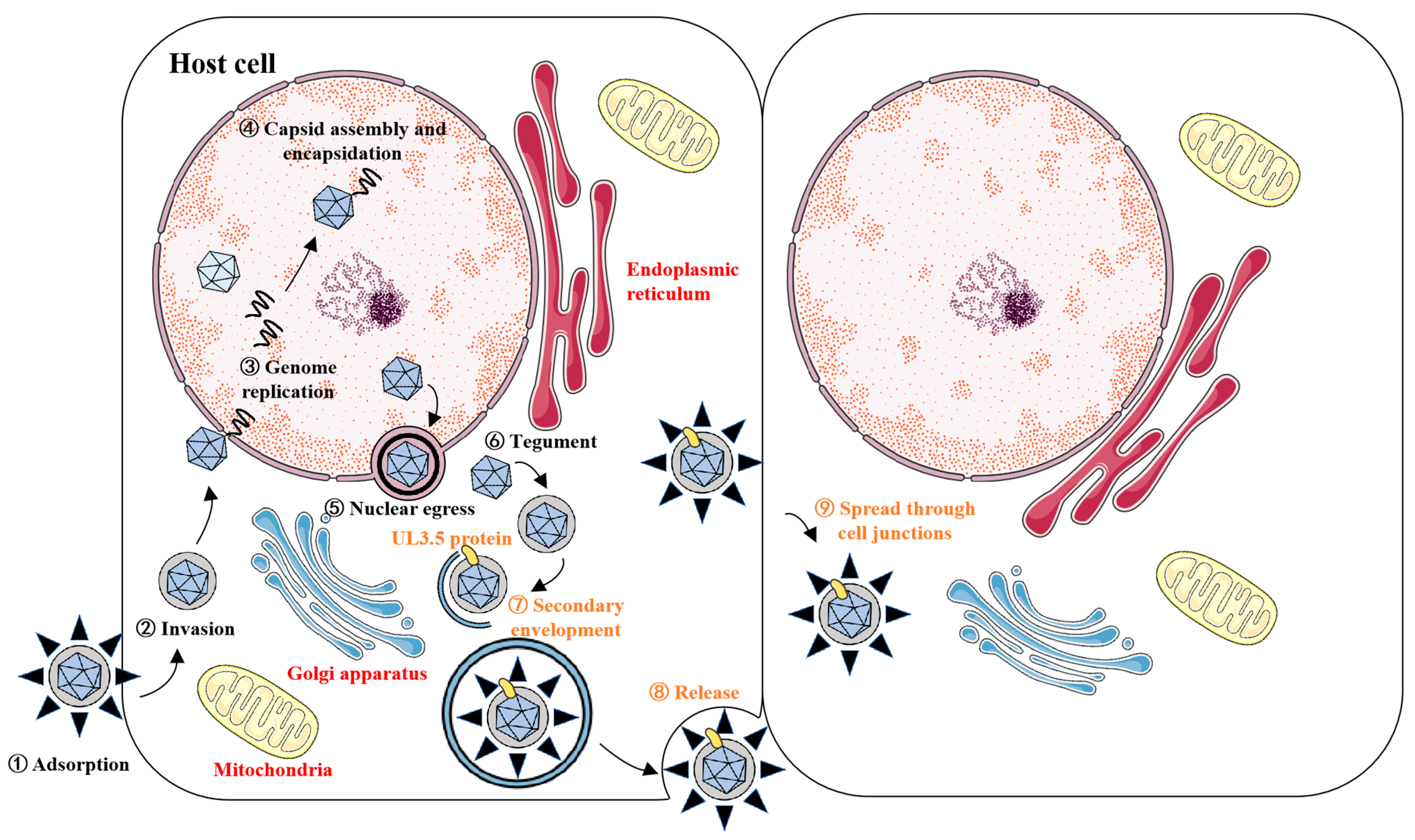
3.5. UL3.5 Protein Participated in the Viral Secondary Envelopment and Virion Release Process
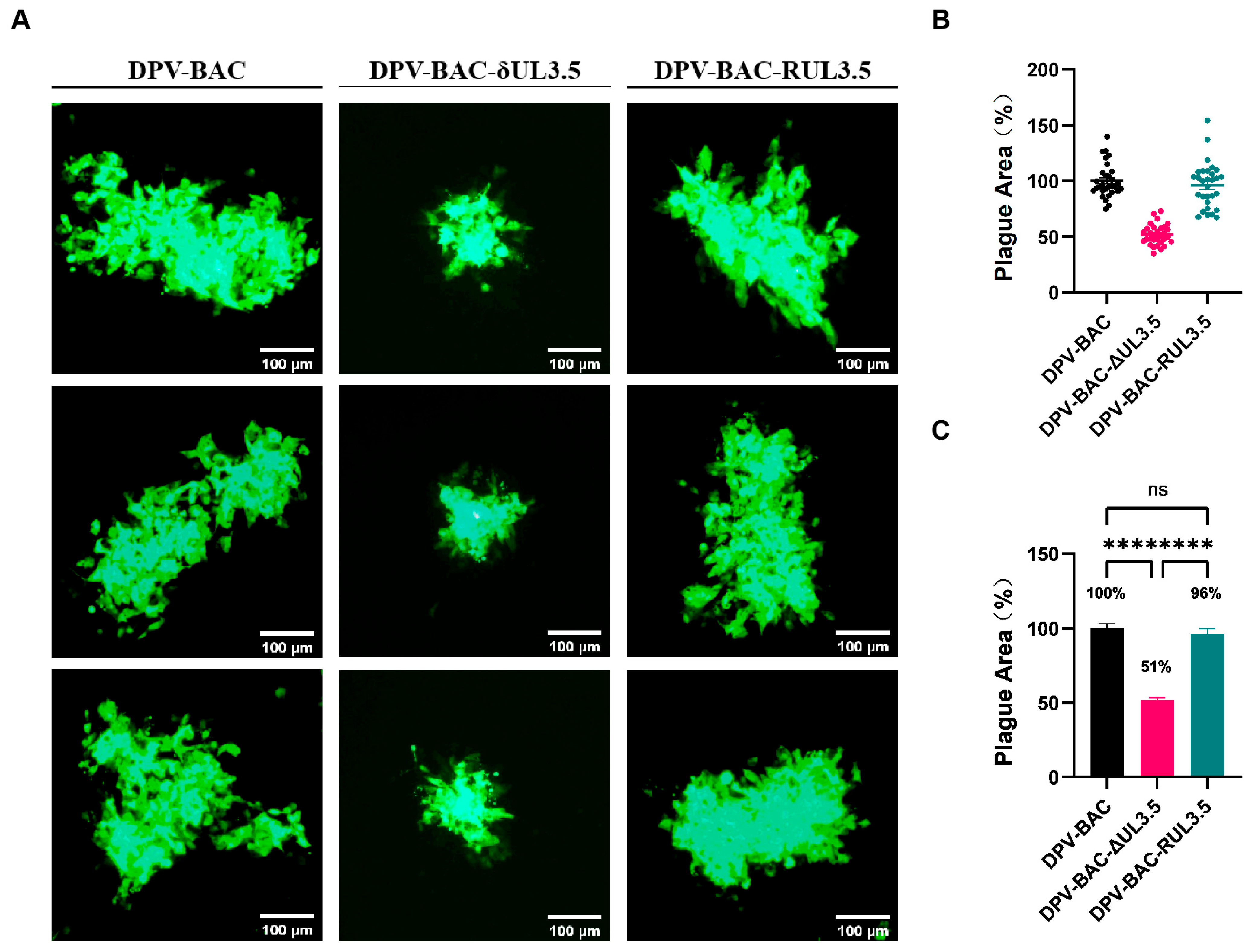
3.6. UL3.5 Exerts a Significant Impact on Cell-to-Cell Spread
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| aa | Amino acid |
| AnHV-1 | Anas herpesviruses 1 |
| BAC | Bacterial artificial chromosome |
| BHV-1 | Bovine herpesvirus 1 |
| cDNA | Complementary DNA |
| CHX | Cycloheximide |
| DEFs | Duck embryo fibroblast |
| DEV | Duck enteritis virus |
| DMEM | Dulbecco Modified Eagle Medium |
| DNA | Deoxyribonucleotide acid |
| DP | Duck plague |
| DPV | Duck plague virus |
| ER | Endoplasmic reticulum |
| EGFP | Enhanced green fluorescent protein |
| FBS | Fetal bovine serum |
| GCV | Ganciclovir |
| HSV-1/2 | Herpes simplex virus 1/2 |
| ILTV | Infectious laryngotracheitis virus |
| MOI | Multiplicity of infection |
| NanoLuc | NanoLuc® luciferase |
| ORF | Open reading frame |
| PBS | Phosphate-buffered saline solution |
| PBST | Phosphate-buffered saline with Tween |
| PCR | Polymerase chain reaction |
| RNA | Ribonucleic Acid |
| RT-qPCR | Quantitative real-time PCR |
| TBST | Tris-buffered saline with Tween |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid/Gene | Primer | Primer Sequence (5′ → 3′) |
|---|---|---|
| pEGFP-N1-UL3.5 | F | AGATCTCGAGCTCAAGCTTCGAATTCGCCACCATGTTCCGGGCGCGGCATT |
| R | GACCGGTGGATCCCGGGCCCGCGGTACCGTAATGAGAATGAACCTTATGAT | |
| DPV-BAC-NanoLuc-UL3.5 | F | TTTACATCGGCGACAACGTAATAGAGGTCAGTACGCCTCAGTCCTGAAGTGATCGCGAACTATGGATTACAAGGACGACGATGACAAGAACTCCTTCTCC |
| R | GCGATTGCTGGTCTATGAGTCGATAACGGCCGGTGTTGATGATTACCTGGTTCAGGCCGCTCGTTTCCGCCGAGTGATCCAGCCAGTGTTACAACCAAT | |
| DPV-BAC-δUL3.5 | F | TAGAGGTCAGTACGCCTCAGTCCTGAAGTGATCGCGAACTTAGGGATAACAGGGTAATCGATTT |
| R | ATTACCTGGTTCAGGCCGCTCGTTTCCGCCGAGTGATCCAAGTTCGCGATCACTTCAGGACTGAGGCGTACTGACCTCTAGCCAGTGTTACAACCAAT | |
| DPV-BAC-RUL3.5 | F | TTACATCGCCGACAACGTAATAGAGGTCAGTACGCCTCAGTCCTGAAGTGATCGCGAACTATGTTCCGGGCGCGGCATTA |
| R | GCGATTGCTGGTCTATGAGTCGATAACGGCCGGTGTTGATGATTACCTGGTTCAGGCCGCTCGTTTCCGCCGAGTGATCCAGCCAGTGTTACAACCAAT | |
| Identification of UL3.5 | F | GTGGCCACGGCAATAAAAGGC |
| R | GTGAGTCGTCATCTAGAACGG | |
| ICP27 gene [55] | F | TTGCTCGACCTAAGACAGGA |
| R | TCATTAGCAGTAGTTCCGCG | |
| UL28 gene [55] | F | TTAAGCTAAGCCGTGCCGAT |
| R | TGGAGCCAGTTGTTGAAGCA | |
| UL47 gene [55] | F | AACGGAGTTGCTTGGAGAACA |
| R | TGGGCGATGAAACAGAGTAGG | |
| β-actin [40] | F | GATCACAGCCCTGGCACC |
| R | CGGATTCATCATACTCCTGCTT | |
| UL30 gene [55] | F | TTTCCTCCTCCTCGCTGAGTG |
| R | GTGAGTCGTCATCTAGAACG |
| Drug Treatment | Concentration | OD450 Values (mean ± SD) | Cell Viability |
|---|---|---|---|
| DMSO | 0.2% | 2.91 ± 0.08 | 100.00% |
| GCV | 300 μg/mL | 2.89 ± 0.01 | 99.33% |
| CHX | 200 μg/mL | 2.89 ± 0.06 | 99.64% |
| GCV + CHX | 300 μg/mL + 200 μg/mL | 2.88 ± 0.01 | 99.15% |
References
- Zerbini, F.M.; Siddell, S.G.; Mushegian, A.R.; Walker, P.J.; Lefkowitz, E.J.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dutilh, B.E.; García, M.L.; Junglen, S.; et al. Differentiating between viruses and virus species by writing their names correctly. Arch. Virol. 2022, 167, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Zerbini, F.M.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; Hendrickson, R.C.; et al. Changes to virus taxonomy and the ICTV Statutes ratified by the International Committee on Taxonomy of Viruses (2023). Arch. Virol. 2023, 168, 175. [Google Scholar] [CrossRef]
- ICTV. The new scope of virus taxonomy: Partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol. 2020, 5, 668–674. [Google Scholar] [CrossRef]
- Gatherer, D.; Depledge, D.P.; Hartley, C.A.; Szpara, M.L.; Vaz, P.K.; Benkő, M.; Brandt, C.R.; Bryant, N.A.; Dastjerdi, A.; Doszpoly, A.; et al. ICTV Virus Taxonomy Profile: Herpesviridae 2021. J. Gen. Virol. 2021, 102, 001673. [Google Scholar] [CrossRef]
- Li, Y.; Huang, B.; Ma, X.; Wu, J.; Li, F.; Ai, W.; Song, M.; Yang, H. Molecular characterization of the genome of duck enteritis virus. Virology 2009, 391, 151–161. [Google Scholar] [CrossRef]
- Chen, L.; Yu, B.; Hua, J.; Ye, W.; Ni, Z.; Yun, T.; Deng, X.; Zhang, C. Construction of a full-length infectious bacterial artificial chromosome clone of duck enteritis virus vaccine strain. Virol. J. 2013, 10, 328. [Google Scholar] [CrossRef]
- Zhang, B.; Huang, X.; Yang, Y.; Zhang, M.; Song, Y.; Yang, C. Complete genome sequence of an isolate of duck enteritis virus from China. Arch. Virol. 2020, 165, 1687–1689. [Google Scholar] [CrossRef]
- Kong, J.; Feng, K.; Zhao, Q.; Chen, Y.; Wang, J.; Chen, S.; Shao, G.; Liao, L.; Li, Y.; Xie, Z.; et al. Pathogenicity and transmissibility studies on live attenuated duck enteritis virus vaccine in non-target species. Front. Microbiol. 2022, 13, 979368. [Google Scholar] [CrossRef]
- Islam, M.M.; Islam, J.; Islam, M.S.; Ahamed, T.; Islam, M.R.; Khatun, M.M.; Islam, M.A. Duck virus enteritis (duck plague) outbreak in an Australian black swan (Cygnus atratus) flock at safari park in Bangladesh: A case report. J. Adv. Vet. Anim. Res. 2021, 8, 557–562. [Google Scholar] [CrossRef]
- Mellman, I. Endocytosis and molecular sorting. Annu. Rev. Cell Dev. Biol. 1996, 12, 575–625. [Google Scholar] [CrossRef]
- Casasnovas, J.M. Virus-receptor interactions and receptor-mediated virus entry into host cells. Subcell. Biochem. 2013, 68, 441–466. [Google Scholar] [PubMed]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: An update. Virus Res. 2009, 143, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011, 9, 382–394. [Google Scholar] [CrossRef]
- Zeev-Ben-Mordehai, T.; Hagen, C.; Grünewald, K. A cool hybrid approach to the herpesvirus ‘life’ cycle. Curr. Opin. Virol. 2014, 5, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.J.; Crump, C.M.; Graham, S.C. Tegument Assembly and Secondary Envelopment of Alphaherpesviruses. Viruses 2015, 7, 5084–5114. [Google Scholar] [CrossRef]
- Jana, A.K.; May, E.R. Atomistic dynamics of a viral infection process: Release of membrane lytic peptides from a non-enveloped virus. Sci. Adv. 2021, 7, eabe1761. [Google Scholar] [CrossRef]
- Mesters, J.R.; Tan, J.; Hilgenfeld, R. Viral enzymes. Curr. Opin. Struct. Biol. 2006, 16, 776–786. [Google Scholar] [CrossRef]
- Sun, S.; Rao, V.B.; Rossmann, M.G. Genome packaging in viruses. Curr. Opin. Struct. Biol. 2010, 20, 114–120. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Dalrymple, M.A.; Davison, A.J.; Dolan, A.; Frame, M.C.; McNab, D.; Perry, L.J.; Scott, J.E.; Taylor, P. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 1988, 69 Pt 7, 1531–1574. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Cunningham, C.; McIntyre, G.; Dolan, A. Comparative sequence analysis of the long repeat regions and adjoining parts of the long unique regions in the genomes of herpes simplex viruses types 1 and 2. J. Gen. Virol. 1991, 72 Pt 12, 3057–3075. [Google Scholar] [CrossRef]
- Davison, A.J.; Scott, J.E. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 1986, 67 Pt 9, 1759–1816. [Google Scholar] [CrossRef] [PubMed]
- Telford, E.A.; Watson, M.S.; McBride, K.; Davison, A.J. The DNA sequence of equine herpesvirus-1. Virology 1992, 189, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Dean, H.J.; Cheung, A.K. A 3′ coterminal gene cluster in pseudorabies virus contains herpes simplex virus UL1, UL2, and UL3 gene homologs and a unique UL3.5 open reading frame. J. Virol. 1993, 67, 5955–5961. [Google Scholar] [CrossRef]
- Khattar, S.K.; van Drunen Littel-van den Hurk, S.; Babiuk, L.A.; Tikoo, S.K. Identification and transcriptional analysis of a 3′-coterminal gene cluster containing UL1, UL2, UL3, and UL3.5 open reading frames of bovine herpesvirus-1. Virology 1995, 213, 28–37. [Google Scholar] [CrossRef]
- Fuchs, W.; Mettenleiter, T.C. DNA sequence and transcriptional analysis of the UL1 to UL5 gene cluster of infectious laryngotracheitis virus. J. Gen. Virol. 1996, 77 Pt 9, 2221–2229. [Google Scholar] [CrossRef]
- Dean, H.J.; Cheung, A.K. Identification of the pseudorabies virus UL4 and UL5 (helicase) genes. Virology 1994, 202, 962–967. [Google Scholar] [CrossRef]
- Fuchs, W.; Klupp, B.G.; Granzow, H.; Rziha, H.J.; Mettenleiter, T.C. Identification and characterization of the pseudorabies virus UL3.5 protein, which is involved in virus egress. J. Virol. 1996, 70, 3517–3527. [Google Scholar] [CrossRef]
- Fuchs, W.; Granzow, H.; Mettenleiter, T.C. Functional complementation of UL3.5-negative pseudorabies virus by the bovine herpesvirus 1 UL3.5 homolog. J. Virol. 1997, 71, 8886–8892. [Google Scholar] [CrossRef] [PubMed]
- Lam, N.; Letchworth, G.J. Bovine herpesvirus 1 U(L)3.5 interacts with bovine herpesvirus 1 alpha-transinducing factor. J. Virol. 2000, 74, 2876–2884. [Google Scholar] [CrossRef]
- Lam, N.; Letchworth, G. A derivative of bovine herpesvirus 1 (BoHV-1) UL3.5 lacking the last forty amino acids inhibits replication of BoHV-1. Arch. Virol. 2004, 149, 2295–2306. [Google Scholar] [CrossRef]
- Fuchs, W.; Granzow, H.; Klupp, B.G.; Karger, A.; Michael, K.; Maresch, C.; Klopfleisch, R.; Mettenleiter, T.C. Relevance of the interaction between alphaherpesvirus UL3.5 and UL48 proteins for virion maturation and neuroinvasion. J. Virol. 2007, 81, 9307–9318. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Courtney, R.J. Chemical cross-linking of virion envelope and tegument proteins of herpes simplex virus type 1. Virology 1994, 204, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.T.; Harley, C.A.; Wilson, D.W. The cytoplasmic tail of Herpes simplex virus glycoprotein H binds to the tegument protein VP16 in vitro and in vivo. Virology 2003, 317, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cox, E.; Reddy, S.; Iofin, I.; Cohen, J.I. Varicella-zoster virus ORF57, unlike its pseudorabies virus UL3.5 homolog, is dispensable for viral replication in cell culture. Virology 1998, 250, 205–209. [Google Scholar] [CrossRef]
- Messerle, M.; Crnkovic, I.; Hammerschmidt, W.; Ziegler, H.; Koszinowski, U.H. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA 1997, 94, 14759–14763. [Google Scholar] [CrossRef]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. BioTechniques 2006, 40, 191–197. [Google Scholar]
- Mahmoudian, A.; Markham, P.F.; Noormohammadi, A.H.; Browning, G.F. Kinetics of transcription of infectious laryngotracheitis virus genes. Comp. Immunol. Microbiol. Infect. Dis. 2012, 35, 103–115. [Google Scholar] [CrossRef]
- Nygard, A.-B.; Jørgensen, C.B.; Cirera, S.; Fredholm, M. Selection of reference genes for gene expression studies in pig tissues using SYBR green qPCR. BMC Mol. Biol. 2007, 8, 67. [Google Scholar] [CrossRef]
- Falahzadeh, K.; Banaei-Esfahani, A.; Shahhoseini, M. The potential roles of actin in the nucleus. Cell J. 2015, 17, 7–14. [Google Scholar]
- Shen, B.; Ruan, P.; Cheng, A.; Wang, M.; Zhang, W.; Wu, Y.; Yang, Q.; Tian, B.; Ou, X.; Mao, S.; et al. Characterization of a Unique Novel LORF3 Protein of Duck Plague Virus and Its Potential Pathogenesis. J. Virol. 2023, 97, e0157722. [Google Scholar] [CrossRef]
- Hogue, I.B. Tegument Assembly, Secondary Envelopment and Exocytosis. Curr. Issues Mol. Biol. 2021, 42, 551–604. [Google Scholar] [PubMed]
- Gruffat, H.; Marchione, R.; Manet, E. Herpesvirus Late Gene Expression: A Viral-Specific Pre-initiation Complex Is Key. Front. Microbiol. 2016, 7, 869. [Google Scholar] [CrossRef] [PubMed]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Spannl, S.; Tereshchenko, M.; Mastromarco, G.J.; Ihn, S.J.; Lee, H.O. Biomolecular condensates in neurodegeneration and cancer. Traffic 2019, 20, 890–911. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Banani, S.F.; Rice, A.M.; Peeples, W.B.; Lin, Y.; Jain, S.; Parker, R.; Rosen, M.K. Compositional Control of Phase-Separated Cellular Bodies. Cell 2016, 166, 651–663. [Google Scholar] [CrossRef]
- Li, P.; Banjade, S.; Cheng, H.-C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Elbaum-Garfinkle, S.; Kim, Y.; Szczepaniak, K.; Chen, C.C.-H.; Eckmann, C.R.; Myong, S.; Brangwynne, C.P. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl. Acad. Sci. USA 2015, 112, 7189–7194. [Google Scholar] [CrossRef]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef]
- Smith, J.; Calidas, D.; Schmidt, H.; Lu, T.; Rasoloson, D.; Seydoux, G. Spatial patterning of P granules by RNA-induced phase separation of the intrinsically-disordered protein MEG-3. eLife 2016, 5, e21337. [Google Scholar] [CrossRef]
- Wang, J.; Choi, J.-M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 2018, 174, 688–699.e16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Peng, F.; He, C.; Liu, Y.; Deng, H.; Fang, X. Large-scale identification of potential phase-separation proteins from plants using a cell-free system. Mol. Plant 2023, 16, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Seyffert, M.; Georgi, F.; Tobler, K.; Bourqui, L.; Anfossi, M.; Michaelsen, K.; Vogt, B.; Greber, U.F.; Fraefel, C. The HSV-1 Transcription Factor ICP4 Confers Liquid-Like Properties to Viral Replication Compartments. Int. J. Mol. Sci. 2021, 22, 4447. [Google Scholar] [CrossRef]
- Li, C.; Wang, M.; Cheng, A.; Tian, B.; Huang, J.; Wu, Y.; Yang, Q.; Gao, Q.; Sun, D.; Zhang, S.; et al. Deleting UL49.5 in duck plague virus causes attachment, entry and spread defects. Vet. Microbiol. 2023, 280, 109707. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wang, M.; Cheng, A.; Tian, B.; Huang, J.; Wu, Y.; Yang, Q.; Ou, X.; Sun, D.; Zhang, S.; et al. Duck plague virus tegument protein vp22 plays a key role in the secondary envelopment and cell-to-cell spread. Vet. Res. 2023, 54, 60. [Google Scholar] [CrossRef]
- Cifuentes-Munoz, N.; El Najjar, F.; Dutch, R.E. Viral cell-to-cell spread: Conventional and non-conventional ways. Adv. Virus Res. 2020, 108, 85–125. [Google Scholar]
- Carmichael, J.C.; Yokota, H.; Craven, R.C.; Schmitt, A.; Wills, J.W. The HSV-1 mechanisms of cell-to-cell spread and fusion are critically dependent on host PTP1B. PLoS Pathog. 2018, 14, e1007054. [Google Scholar] [CrossRef]
- Dingwell, K.S.; Brunetti, C.R.; Hendricks, R.L.; Tang, Q.; Tang, M.; Rainbow, A.J.; Johnson, D.C. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 1994, 68, 834–845. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, H.; Tian, B.; Tian, Y.; Cai, D.; Wang, M.; Jia, R.; Chen, S.; Cheng, A. The Protein Encoded by the UL3.5 Gene of the Duck Plague Virus Affects Viral Secondary Envelopment, Release, and Cell-to-Cell Spread. Vet. Sci. 2025, 12, 510. https://doi.org/10.3390/vetsci12060510
Cao H, Tian B, Tian Y, Cai D, Wang M, Jia R, Chen S, Cheng A. The Protein Encoded by the UL3.5 Gene of the Duck Plague Virus Affects Viral Secondary Envelopment, Release, and Cell-to-Cell Spread. Veterinary Sciences. 2025; 12(6):510. https://doi.org/10.3390/vetsci12060510
Chicago/Turabian StyleCao, Huanhuan, Bin Tian, Yanming Tian, Dongjie Cai, Mingshu Wang, Renyong Jia, Shun Chen, and Anchun Cheng. 2025. "The Protein Encoded by the UL3.5 Gene of the Duck Plague Virus Affects Viral Secondary Envelopment, Release, and Cell-to-Cell Spread" Veterinary Sciences 12, no. 6: 510. https://doi.org/10.3390/vetsci12060510
APA StyleCao, H., Tian, B., Tian, Y., Cai, D., Wang, M., Jia, R., Chen, S., & Cheng, A. (2025). The Protein Encoded by the UL3.5 Gene of the Duck Plague Virus Affects Viral Secondary Envelopment, Release, and Cell-to-Cell Spread. Veterinary Sciences, 12(6), 510. https://doi.org/10.3390/vetsci12060510