

Mining Porcine Blood Whole-DNA Sequencing Datasets to Uncover Pig Viromes: An Exploratory Application to Identify Potential Infecting Agents of an Undefined Disease Outbreak

 ,
,  ,
,  ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. General Disease Description and Standard Laboratory Diagnostic Analyses
2.2. Sampled Pigs and Whole-Genome Sequencing
2.3. Mining Whole-Genome Sequencing Datasets for Virus Sequences
- The development of a comprehensive virus database was based on the NCBI Virus resource (https://www.ncbi.nlm.nih.gov/genome/viruses; accessed on 5 February 2025), which contains DNA sequences of reference viral genomes and their strains. Processing and curating this resource are essential to create a reliable database.
- Host-related reads were removed by aligning sequenced reads to the Sus scrofa genome, which was assembled using Sscrofa11.1. Unmapped reads, which represent non-host DNA, likely originating from microorganisms or viruses, were then extracted.
- Screening for viral sequences involved aligning the extracted unmapped reads to the curated virus database to identify viral sequences. This process included a quick alignment using the BWA v.0.7.17 tool, followed by confirming alignments with a more sensitive approach, like the BLAST+ v.2.7.1 tool. Only viral genomes with a minimum of three aligned read pairs (six reads) were considered representative of the sample.
2.4. Statistical Analyses
3. Results
3.1. Description of the Disease Outbreak at the Farm Level and Standard Laboratory Analyses
3.2. Overview of Virus Sequences in Whole-DNA Sequencing Datasets
3.3. Viral DNA Profiles in the Pigs Studied
3.4. Putative Association of Virus Profiles with a Disease Condition
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ELISA | Enzyme-linked immunosorbent assay |
| ENA | European Nucleotide Archive |
| Gb | Giga base pairs |
| PCV2 | Porcine circovirus 2 |
| PCV3 | Porcine circovirus 3 |
| PCVAD | Porcine circovirus-associated disease |
| PCVM | Porcine cytomegalovirus |
| PLHV1 | Porcine lymphotropic herpesvirus 1 |
| PLHV2 | Porcine lymphotropic herpesvirus 2 |
| PMWS | Post-weaning Multisystemic Wasting Syndrome |
| PPV2 | Porcine parvovirus 2 |
| PPV3 | Porcine parvovirus 3 |
| PPV4 | Porcine parvovirus 4 |
| PPV5 | Porcine parvovirus 5 |
| PPV6 | Porcine parvovirus 6 |
| PRRS | Porcine reproductive and respiratory syndrome |
| PRRSV | Porcine reproductive and respiratory syndrome virus |
| SIV | Swine influenza virus A |
| SuBHV2 | Suid betaherpesvirus 2 |
| SuGHV3 | Suid gammaherpesvirus 3 |
| SuGHV4 | Suid gammaherpesvirus 4 |
References
- Cassedy, A.; Parle-McDermott, A.; O’Kennedy, R. Virus Detection: A Review of the Current and Emerging Molecular and Immunological Methods. Front. Mol. Biosci. 2021, 8, 637559. [Google Scholar] [CrossRef] [PubMed]
- Neujahr, A.C.; Loy, D.S.; Loy, J.D.; Brodersen, B.W.; Fernando, S.C. Rapid detection of high consequence and emerging viral pathogens in pigs. Front. Vet. Sci. 2024, 11, 1341783. [Google Scholar] [CrossRef] [PubMed]
- McLean, R.K.; Graham, S.P. The pig as an amplifying host for new and emerging zoonotic viruses. One Health 2022, 14, 100384. [Google Scholar] [CrossRef]
- Alarcón, L.V.; Allepuz, A.; Mateu, E. Biosecurity in pig farms: A review. Porcine Health Manag. 2021, 7, 5. [Google Scholar] [CrossRef]
- Yoon, K.-J. Overview of Viruses. In Diseases of Swine, 10th ed.; Zimmerman, J.J., Karriker, L.A., Ramirez, A., Schwartz, K.J., Stevenson, G.W., Eds.; Wiley-Blackwell: West Sussex, UK, 2012; pp. 383–391. [Google Scholar]
- Blomström, A.-L.; Fossum, C.; Wallgren, P.; Berg, M. Viral metagenomic analysis displays the co-infection situation in healthy and PMWS affected pigs. PLoS ONE 2016, 11, e0166863. [Google Scholar] [CrossRef]
- Shan, T.; Li, L.; Simmonds, P.; Wang, C.; Moeser, A.; Delwart, E. The fecal virome of pigs on a high-density farm. J. Virol. 2011, 85, 11697–11708. [Google Scholar] [CrossRef]
- Karlsson, O.E.; Larsson, J.; Hayer, J.; Berg, M.; Jacobson, M. The intestinal eukaryotic virome in healthy and diarrhoeic neonatal piglets. PLoS ONE 2016, 11, e0151481. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, D.; Ji, Z.; Zhang, Y.; Wang, Y.; Chen, X.; He, Y.; Lu, X.; Li, R.; Guo, Y.; et al. Viral Metagenomics Reveals Diverse Viruses in Tissue Samples of Diseased Pigs. Viruses 2022, 14, 2048. [Google Scholar] [CrossRef]
- He, B.; Gong, W.; Yan, X.; Zhao, Z.; Yang, L.; Tan, Z.; Xu, L.; Zhu, A.; Zhang, J.; Rao, J.; et al. Viral Metagenome-Based Precision Surveillance of Pig Population at Large Scale Reveals Viromic Signatures of Sample Types and Influence of Farming Management on Pig Virome. mSystems 2021, 6, e0042021. [Google Scholar] [CrossRef]
- Bhatta, T.R.; Chamings, A.; Alexandersen, S. Exploring the cause of diarrhoea and poor growth in 8–11-week-old pigs from an Australian pig herd using metagenomic sequencing. Viruses 2021, 13, 1608. [Google Scholar] [CrossRef]
- Schuele, L.; Lizarazo-Forero, E.; Strutzberg-Minder, K.; Schütze, S.; Löbert, S.; Lambrecht, C.; Harlizius, J.; Friedrich, A.W.; Peter, S.; Rossen, J.W.A.; et al. Application of shotgun metagenomics sequencing and targeted sequence capture to detect circulating porcine viruses in the Dutch-German border region. Transbound. Emerg. Dis. 2022, 69, 2306–2319. [Google Scholar] [CrossRef]
- Zhou, L.; Zhou, H.; Fan, Y.; Wang, J.; Zhang, R.; Guo, Z.; Li, Y.; Kang, R.; Zhang, Z.; Yang, D.; et al. Metagenomics to Identify Viral Communities Associated with Porcine Respiratory Disease Complex in Tibetan Pigs in the Tibetan Plateau, China. Pathogens 2024, 13, 404. [Google Scholar] [CrossRef]
- Klenner, J.; Kohl, C.; Dabrowski, P.W.; Nitsche, A. Comparing viral metagenomic extraction methods. Curr. Issues Mol. Biol. 2017, 24, 59–70. [Google Scholar] [CrossRef]
- Kohl, C.; Brinkmann, A.; Dabrowski, P.W.; Radonić, A.; Nitsche, A.; Kurth, A. Protocol for metagenomic virus detection in clinical specimens. Emerg. Infect. Dis. 2015, 21, 48. [Google Scholar] [CrossRef]
- Zhang, D.; Lou, X.; Yan, H.; Pan, J.; Mao, H.; Tang, H.; Shu, Y.; Zhao, Y.; Liu, L.; Li, J.; et al. Metagenomic analysis of viral nucleic acid extraction methods in respiratory clinical samples. BMC Genom. 2018, 19, 773. [Google Scholar] [CrossRef]
- Thurber, R.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate Viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef]
- Mokili, J.L.; Rohwer, F.; Dutilh, B.E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2012, 2, 63–77. [Google Scholar] [CrossRef]
- Conceição-Neto, N.; Zeller, M.; Lefrère, H.; De Bruyn, P.; Beller, L.; Deboutte, W.; Yinda, C.K.; Lavigne, R.; Mae, P.; Van Ranst, M.; et al. Modular approach to customise sample preparation procedures for viral metagenomics: A reproducible protocol for virome analysis. Sci. Rep. 2015, 5, 16532. [Google Scholar] [CrossRef]
- Kieft, K.; Anantharaman, K. Virus genomics: What is being overlooked? Curr. Opin. Virol. 2022, 53, 101200. [Google Scholar] [CrossRef]
- Parras-Moltó, M.; Rodríguez-Galet, A.; Suárez-Rodríguez, P.; López-Bueno, A. Evaluation of bias induced by viral enrichment and random amplification protocols in metagenomic surveys of saliva DNA viruses. Microbiome 2018, 6, 119. [Google Scholar] [CrossRef]
- Callanan, J.; Stockdale, S.R.; Shkoporov, A.; Draper, L.A.; Ross, R.P.; Hill, C. Biases in viral metagenomics-based detection, cataloguing and quantification of bacteriophage genomes in human faeces, a review. Microorganisms 2021, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- Haagmans, R.; Charity, O.J.; Baker, D.; Telatin, A.; Savva, G.M.; Adriaenssens, E.M.; Powell, P.P.; Carding, S.R. Assessing Bias and Reproducibility of Viral Metagenomics Methods for the Combined Detection of Faecal RNA and DNA Viruses. Viruses 2025, 17, 155. [Google Scholar] [CrossRef] [PubMed]
- Aulicino, P.C.; Kimata, J.T. Beyond surveillance: Leveraging the potential of next generation sequencing in clinical virology. Front. Trop. Dis. 2024, 5, 1512606. [Google Scholar] [CrossRef]
- Moustafa, A.; Xie, C.; Kirkness, E.; Biggs, W.; Wong, E.; Turpaz, Y.; Bloom, K.; Delwart, E.; Nelson, K.E.; Venter, J.C.; et al. The blood DNA virome in 8000 humans. PLoS Pathog. 2017, 13, e1006292. [Google Scholar] [CrossRef]
- Liu, S.; Huang, S.; Chen, F.; Zhao, L.; Yuan, Y.; Francis, S.S.; Fang, L.; Li, Z.; Lin, L.; Liu, R.; et al. Genomic Analyses from Non-invasive Prenatal Testing Reveal Genetic Associations, Patterns of Viral Infections, and Chinese Population History. Cell 2018, 175, 347–359.e14. [Google Scholar] [CrossRef]
- Guo, J.; Huang, X.; Zhang, C.; Huang, P.; Li, Y.; Wen, F.; Wang, X.; Yang, N.; Xu, M.; Bi, Y.; et al. The blood virome of 10,585 individuals from the ChinaMAP. Cell Discov. 2022, 8, 113. [Google Scholar] [CrossRef]
- Bovo, S.; Mazzoni, G.; Ribani, A.; Utzeri, V.J.; Bertolini, F.; Schiavo, G.; Fontanesi, L. A viral metagenomic approach on a non-metagenomic experiment: Mining next generation sequencing datasets from pig DNA identified several porcine parvoviruses for a retrospective evaluation of viral infections. PLoS ONE 2017, 12, e0179462. [Google Scholar] [CrossRef]
- Bovo, S.; Schiavo, G.; Bolner, M.; Ballan, M.; Fontanesi, L. Mining livestock genome datasets for an unconventional characterization of animal DNA viromes. Genomics 2022, 114, 110312. [Google Scholar] [CrossRef]
- Segalés, J.; Domingo, M. Postweaning multisystemic wasting syndrome (PMWS) in pigs. A review. Vet. Q. 2002, 24, 109–124. [Google Scholar] [CrossRef]
- Baekbo, P.; Kristensen, C.S.; Larsen, L.E. Porcine circovirus diseases: A review of PMWS. Trans. Emerg. Dis. 2021, 59, 60–67. [Google Scholar] [CrossRef]
- Arruda, B.; Piñeyro, P.; Derscheid, R.; Hause, B.; Byers, E.; Dion, K.; Long, D.; Sievers, C.; Tangen, J.; Williams, T. PCV3-associated disease in the United States swine herd. Emerg. Microbes Infect. 2019, 8, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Dinh, P.X.; Nguyen, M.N.; Nguyen, H.T.; Tran, V.H.; Tran, Q.D.; Dang, K.H.; Vo, D.T.; Le, H.T.; Nguyen, N.T.T.; Nguyen, T.T. Porcine circovirus genotypes and their copathogens in pigs with respiratory disease in southern provinces of Vietnam. Arch. Virol. 2021, 166, 403–411. [Google Scholar] [CrossRef]
- Guo, Z.; Ruan, H.; Qiao, S.; Deng, R.; Zhang, G. Co-infection status of porcine circoviruses (PCV2 and PCV3) and porcine epidemic diarrhea virus (PEDV) in pigs with watery diarrhea in Henan province, central China. Microb. Pathog. 2020, 142, 104047. [Google Scholar] [CrossRef]
- Segalés, J.; Calsamiglia, M.; Rosell, C.; Soler, M.; Maldonado, J.; Martín, M.; Domingo, M. Porcine reproductive and respiratory syndrome virus (PRRSV) infection status in pigs naturally affected with post-weaning multisystemic wasting syndrome (PMWS) in Spain. Vet. Microbiol. 2002, 85, 23–30. [Google Scholar] [CrossRef]
- Ohlinger, V. Porcine cytomegalovirus (PCMV). In Herpesvirus Diseases of Cattle, Horses, and Pigs; Wittmann, G., Ed.; Kluwer Academic Publishers: Boston, MA, USA, 1989; pp. 326–333. [Google Scholar]
- Yoon, K.J.; Edington, N. Porcine cytomegalovirus. In Diseases of Swine, 9th ed.; Straw, B.E., Zimmerman, J., D’Allaire, S., Taylor, D.J., Eds.; Blackwell Publishing Company: Ames, IW, USA, 2006; pp. 323–329. [Google Scholar]
- Ehlers, B.; Ulrich, S.; Goltz, M. Detection of two novel porcine herpesviruses with high similarity to gammaherpesviruses. J. Gen. Virol. 1999, 80, 971–978. [Google Scholar] [CrossRef]
- Ni, J.; Qiao, C.; Han, X.; Han, T.; Kang, W.; Zi, Z.; Cao, Z.; Zhai, X.; Cai, X. Identification and genomic characterization of a novel porcine parvovirus (PPV6) in China. Virol. J. 2014, 11, 203. [Google Scholar] [CrossRef]
- Vargas-Bermudez, D.S.; Mogollon, J.D.; Franco-Rodriguez, C.; Jaime, J. The novel porcine parvoviruses: Current state of knowledge and their possible implications in clinical syndromes in pigs. Viruses 2023, 15, 2398. [Google Scholar] [CrossRef]
- Segalés, J. Porcine circovirus type 2 (PCV2) infections: Clinical signs, pathology and laboratory diagnosis. Virus Res. 2012, 164, 10–19. [Google Scholar] [CrossRef]
- Hijikata, M.; Abe, K.; Win, K.M.; Shimizu, Y.K.; Keicho, N.; Yoshikura, H. Identification of new parvovirus DNA sequence in swine sera from Myanmar. Jpn. J. Infect. Dis. 2002, 54, 244–245. [Google Scholar] [CrossRef]
- Kim, K.H.; Bae, J.W. Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl. Environ. Microbiol. 2011, 77, 7663–7668. [Google Scholar] [CrossRef]
- Asplund, M.; Kjartansdóttir, K.; Mollerup, S.; Vinner, L.; Fridholm, H.; Herrera, J.; Friis-Nielsen, J.; Hansen, T.; Jensen, R.; Nielsen, I.; et al. Contaminating viral sequences in high-throughput sequencing viromics: A linkage study of 700 sequencing libraries. Clin. Microbiol. Infect. 2019, 25, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Keeler, E.; McFarland, A.; Scott, P.; Collman, R.G.; Bushman, F.D. The virome of the kitome: Small circular virus-like genomes in laboratory reagents. Microbiol. Resour. Announc. 2024, 13, e0126123. [Google Scholar] [CrossRef] [PubMed]
- Sangiovanni, M.; Granata, I.; Thind, A.S.; Guarracino, M.R. From trash to treasure: Detecting unexpected contamination in unmapped NGS data. BMC Bioinform. 2019, 20, 168. [Google Scholar] [CrossRef]
- Bartoszewicz, J.M.; Seidel, A.; Renard, B.Y. Interpretable detection of novel human viruses from genome sequencing data. NAR Genom. Bioinform. 2021, 3, lqab004. [Google Scholar] [CrossRef]
- Buggiotti, L.; Cheng, Z.; Wathes, D.C.; GplusE Consortium. Mining the Unmapped Reads in Bovine RNA-Seq Data Reveals the Prevalence of Bovine Herpes Virus-6 in European Dairy Cows and the Associated Changes in Their Phenotype and Leucocyte Transcriptome. Viruses 2020, 12, 1451. [Google Scholar] [CrossRef]
- Usman, T.; Hadlich, F.; Demasius, W.; Weikard, R.; Kühn, C. Unmapped reads from cattle RNAseq data: A source for missing and misassembled sequences in the reference assemblies and for detection of pathogens in the host. Genomics 2017, 109, 36–42. [Google Scholar] [CrossRef]
- Sachsenröder, J.; Twardziok, S.; Hammerl, J.A.; Janczyk, P.; Wrede, P.; Hertwig, S.; Johne, R. Simultaneous identification of DNA and RNA viruses present in pig faeces using process-controlled deep sequencing. PLoS ONE 2012, 7, e34631. [Google Scholar] [CrossRef]
- Ogawa, H.; Taira, O.; Hirai, T.; Takeuchi, H.; Nagao, A.; Ishikawa, Y.; Tuchiya, K.; Nunoya, T.; Ueda, S. Multiplex PCR and multiplex RT-PCR for inclusive detection of major swine DNA and RNA viruses in pigs with multiple infections. J. Virol. Methods 2009, 160, 210–214. [Google Scholar] [CrossRef]
- Brunborg, I.M.; Moldal, T.; Jonassen, C.M. Quantitation of porcine circovirus type 2 isolated from serum/plasma and tissue samples of healthy pigs and pigs with postweaning multisystemic wasting syndrome using a TaqMan-based real-time PCR. J. Virol. Methods 2004, 122, 171–178. [Google Scholar] [CrossRef]
- Chianini, F.; Majó, N.; Segalés, J.; Domınguez, J.; Domingo, M. Immunohistochemical characterisation of PCV2 associate lesions in lymphoid and non-lymphoid tissues of pigs with natural postweaning multisystemic wasting syndrome (PMWS). Vet. Immunol. Immunopathol. 2003, 94, 63–75. [Google Scholar] [CrossRef]
- Zhao, D.; Yang, B.; Yuan, X.; Shen, C.; Zhang, D.; Shi, X.; Zhang, T.; Cui, H.; Yang, J.; Chen, X.; et al. Advanced Research in Porcine Reproductive and Respiratory Syndrome Virus Co-infection With Other Pathogens in Swine. Front. Vet. Sci. 2021, 8, 699561. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, P.; Xie, C.; Ha, Z.; Shi, N.; Zhang, H.; Li, Z.; Han, J.; Xie, Y.; Qiu, X.; et al. Synergistic Pathogenicity by Coinfection and Sequential Infection with NADC30-like PRRSV and PCV2 in Post-Weaned Pigs. Viruses 2022, 14, 193. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Bermudez, D.S.; Diaz, A.; Polo, G.; Mogollon, J.D.; Jaime, J. Infection and coinfection of porcine-selected viruses (PPV1 to PPV8, PCV2 to PCV4, and PRRSV) in gilts and their associations with reproductive performance. Vet. Sci. 2024, 11, 185. [Google Scholar] [CrossRef]
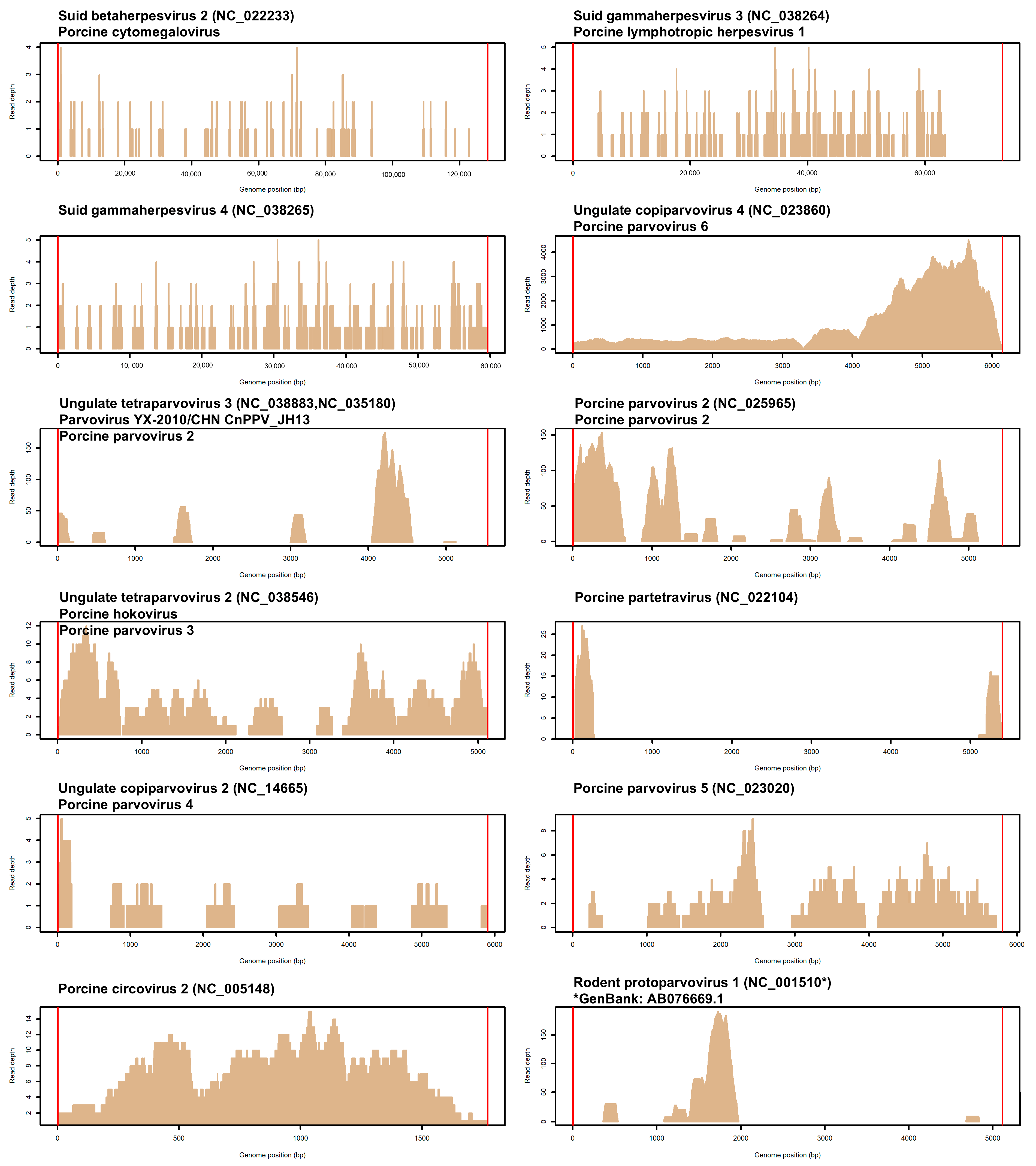

{kind=link}
| Pig | Site 1 | Sequenced Reads | Unmapped Reads (%) 2 | Reads of Viral Origin (%) 3 |
|---|---|---|---|---|
| S1 | #1 | 448,120,122 | 666,430 (0.15%) | 934 (0.140%) |
| S2 | #1 | 327,414,198 | 411,866 (0.13%) | 44 (0.011%) |
| S3 | #1 | 315,609,910 | 407,978 (0.13%) | 10 (0.002%) |
| S4 | #2 | 441,166,336 | 521,010 (0.12%) | 378 (0.073%) |
| S5 | #2 | 400,074,484 | 545,276 (0.14%) | 84 (0.015%) |
| S6 | #2 | 334,788,158 | 492,688 (0.15%) | 22 (0.004%) |
| S7 | #3 | 448,195,724 | 606,724 (0.14%) | 26,720 (4.40%) |
| S8 | #3 | 448,168,478 | 572,470 (0.13%) | 14,618 (2.55%) |
| S9 | #3 | 416,512,760 | 526,100 (0.13%) | 572 (0.108%) |
| Site #1 3 | Site #2 3 | Site #3 3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Virus Name | Alternative Name 1 | NCBI ID | Depth 2 | S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | S9 |
| Suid betaherpesvirus 2 | Porcine cytomegalovirus (PCMV) | NC_022233 | 0.119 | - | 40 a | - | 22 b | - | 20 b | 12 b | 8 b | - |
| Suid gammaherpesvirus 3 | Porcine lymphotropic herpesvirus 1 (PLHV1) | NC_038264 | 0.172 | - | - | 6 a | - | - | - | - | 6 a | 72 b |
| Suid gammaherpesvirus 4 | Porcine lymphotropic herpesvirus 2 (PLHV2) | NC_038265 | 0.593 | - | - | - | 236 | - | - | - | - | - |
| Ungulate copiparvovirus 4 | Porcine parvovirus 6 (PPV6) | NC_023860 | 1027 | - | - | - | - | - | - | 26,616 a | 14,596 a | 496 b |
| Ungulate tetraparvovirus 3 | Parvovirus YX-2010/CHN CnPPV_JH13; Porcine parvovirus 2 (PPV2) | NC_038883; NC_035180 | 5.400 | 186 a | - | - | 10 b | - | - | - | - | - |
| Porcine parvovirus 2 | - | NC_025965 | 11.88 | 394 a | - | - | 36 b | - | - | - | - | - |
| Ungulate tetraparvovirus 2 | Porcine hokovirus (p-PARV4) PPV3 | NC_038546 | 2.932 | - | - | - | 34 | 66 | - | - | - | - |
| Porcine partetravirus | - | NC_022104 | 0.777 | - | - | 14 | - | 14 | - | - | - | - |
| Ungulate copiparvovirus 2 | Porcine parvovirus 4 (PPV4) | NC_014665 | 0.406 | - | - | - | - | - | - | 16 | - | - |
| Porcine parvovirus 5 | - | NC_023020 | 1.756 | - | - | - | - | - | - | 68 | - | - |
| Porcine circovirus 2 | - | NC_005148 | 6.441 | 76 | - | - | - | - | - | - | - | - |
| Rodent protoparvovirus 1 | Minute virus of mice | NC_001510 | 8.503 | 268 a | - | - | 24 b | - | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bovo, S.; Ribani, A.; Schiavo, G.; Taurisano, V.; Bolner, M.; Bertolini, F.; Fontanesi, L. Mining Porcine Blood Whole-DNA Sequencing Datasets to Uncover Pig Viromes: An Exploratory Application to Identify Potential Infecting Agents of an Undefined Disease Outbreak. Vet. Sci. 2025, 12, 513. https://doi.org/10.3390/vetsci12060513
Bovo S, Ribani A, Schiavo G, Taurisano V, Bolner M, Bertolini F, Fontanesi L. Mining Porcine Blood Whole-DNA Sequencing Datasets to Uncover Pig Viromes: An Exploratory Application to Identify Potential Infecting Agents of an Undefined Disease Outbreak. Veterinary Sciences. 2025; 12(6):513. https://doi.org/10.3390/vetsci12060513
Chicago/Turabian StyleBovo, Samuele, Anisa Ribani, Giuseppina Schiavo, Valeria Taurisano, Matteo Bolner, Francesca Bertolini, and Luca Fontanesi. 2025. "Mining Porcine Blood Whole-DNA Sequencing Datasets to Uncover Pig Viromes: An Exploratory Application to Identify Potential Infecting Agents of an Undefined Disease Outbreak" Veterinary Sciences 12, no. 6: 513. https://doi.org/10.3390/vetsci12060513
APA StyleBovo, S., Ribani, A., Schiavo, G., Taurisano, V., Bolner, M., Bertolini, F., & Fontanesi, L. (2025). Mining Porcine Blood Whole-DNA Sequencing Datasets to Uncover Pig Viromes: An Exploratory Application to Identify Potential Infecting Agents of an Undefined Disease Outbreak. Veterinary Sciences, 12(6), 513. https://doi.org/10.3390/vetsci12060513