
Lymphoma in Border Collies: Genome-Wide Association and Pedigree Analysis


Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Animals
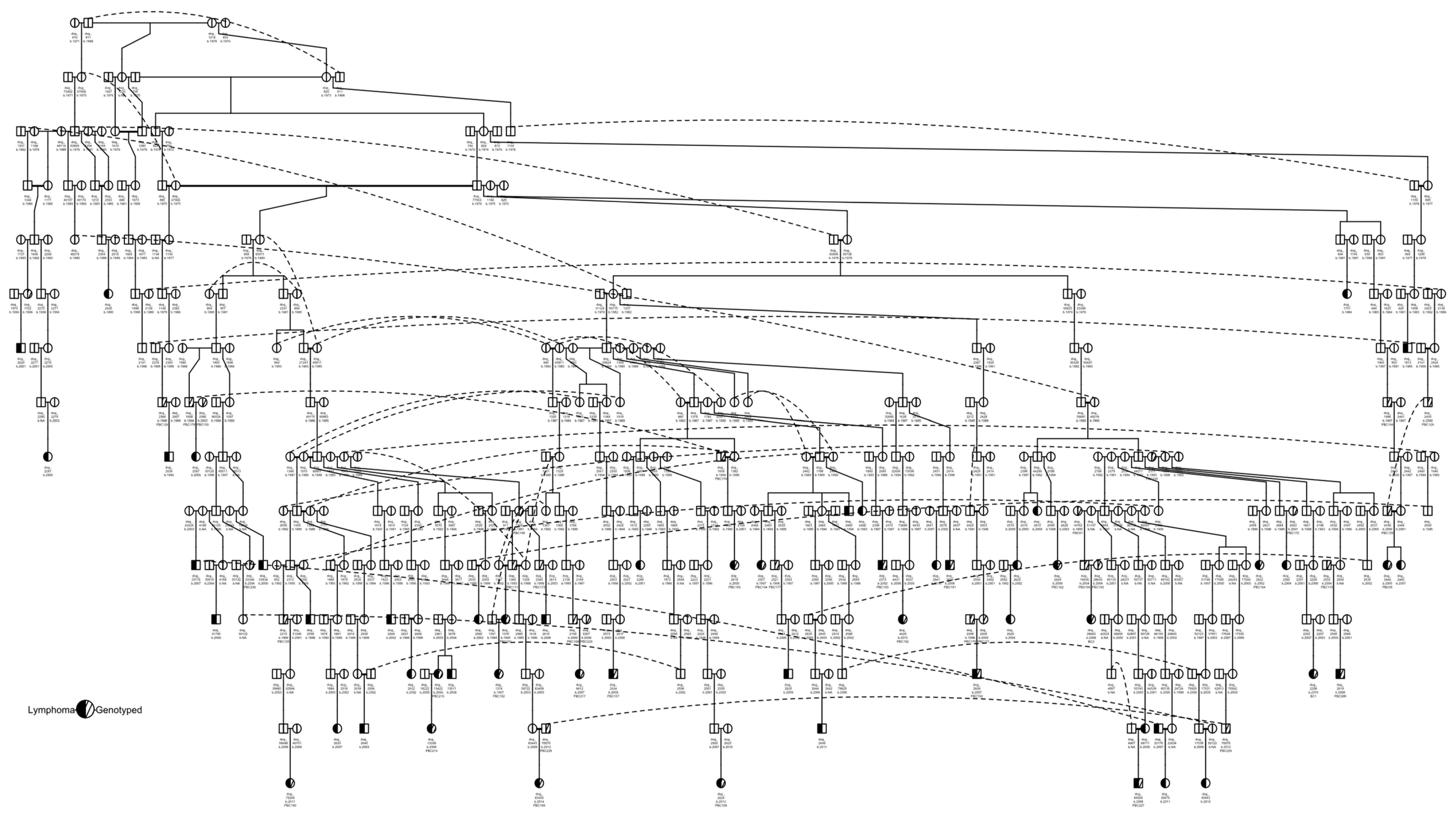
2.2. Pedigree Construction
2.3. Genotyping and Imputation
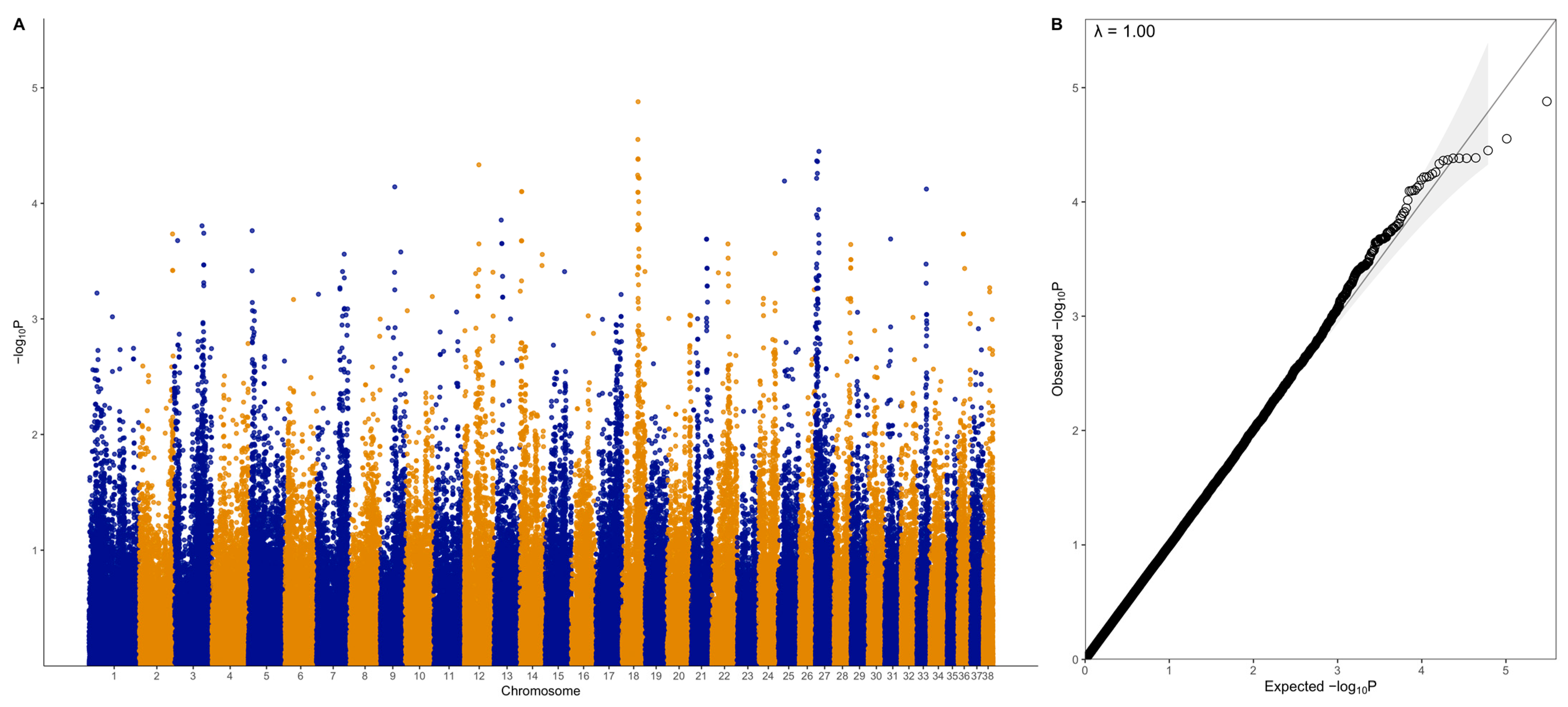
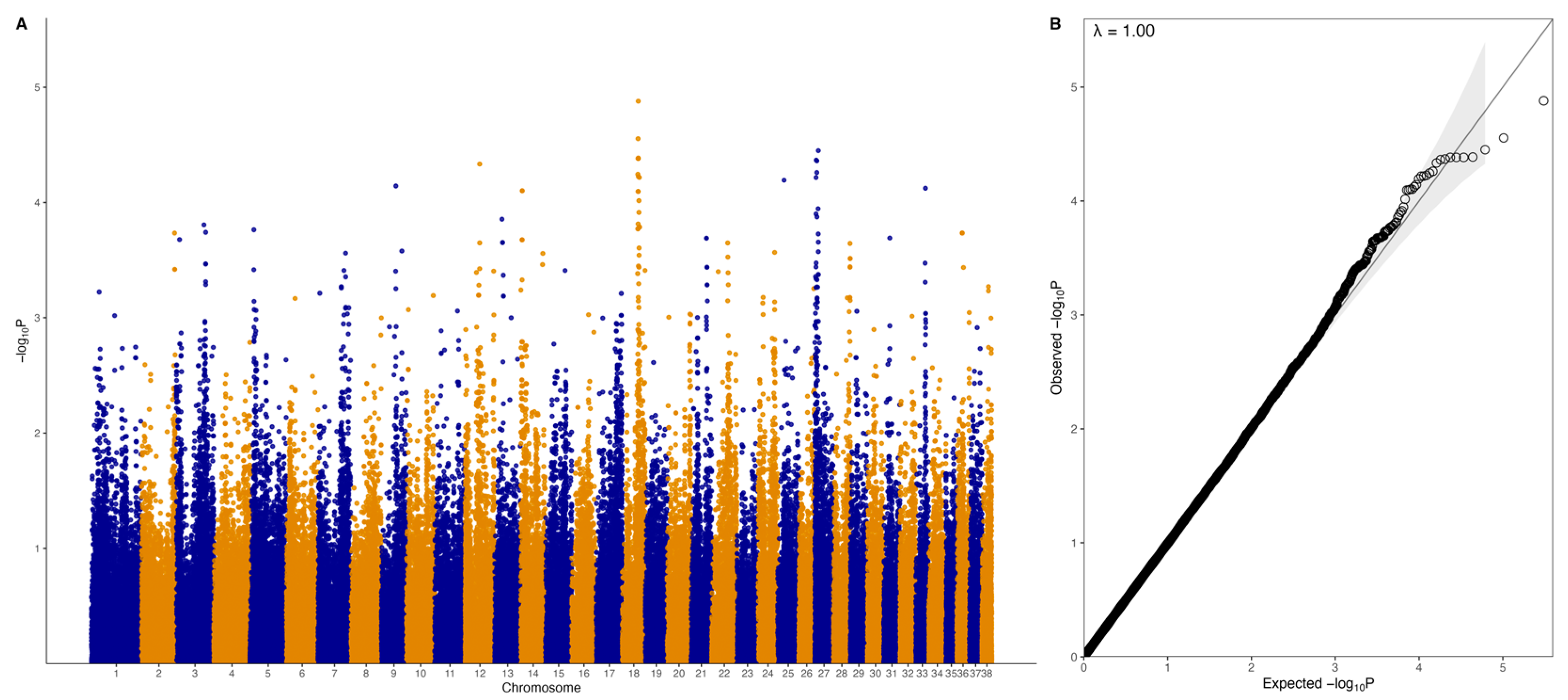
2.4. Genome-Wide Association Study (GWAS)
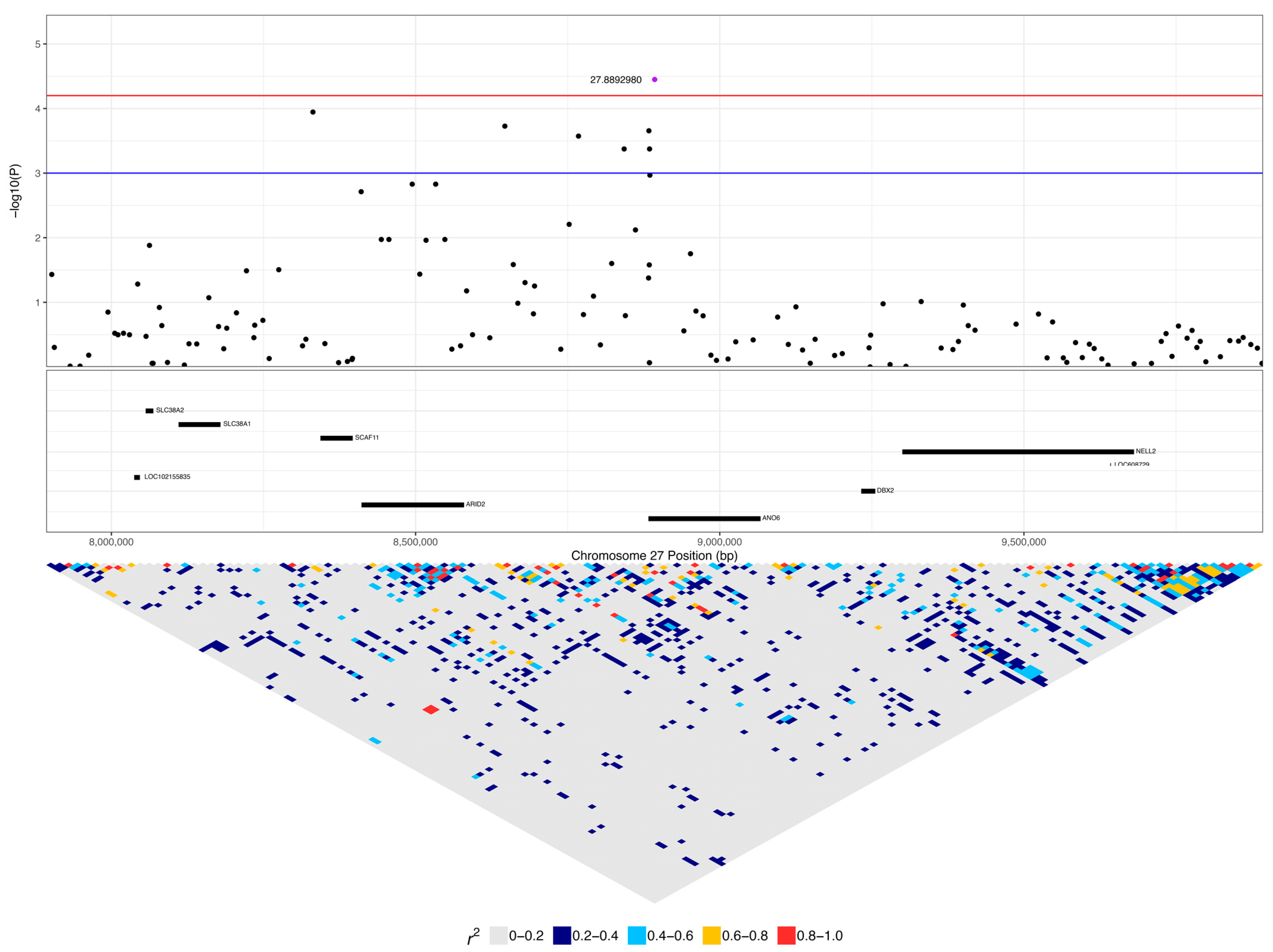
2.5. Haplotype Analysis
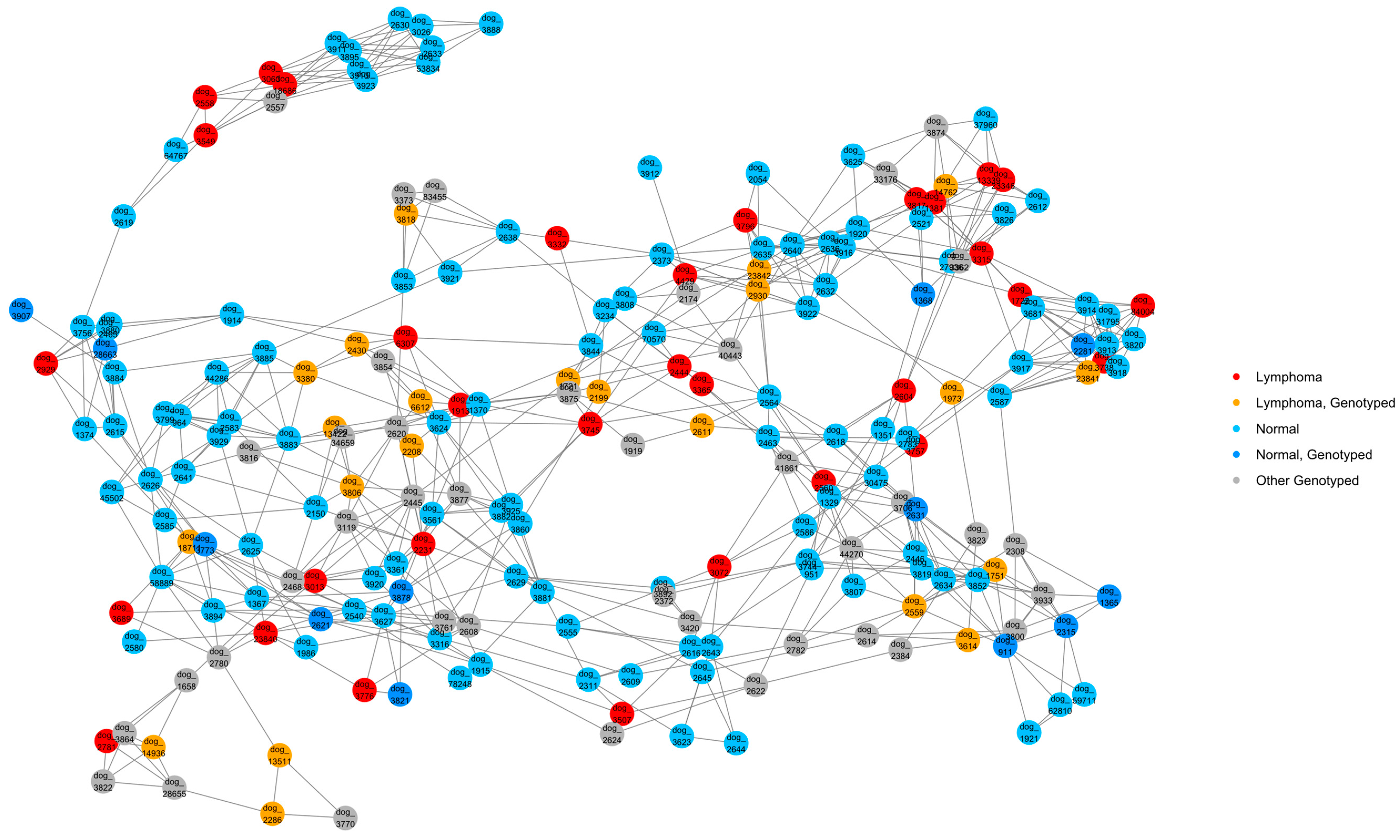
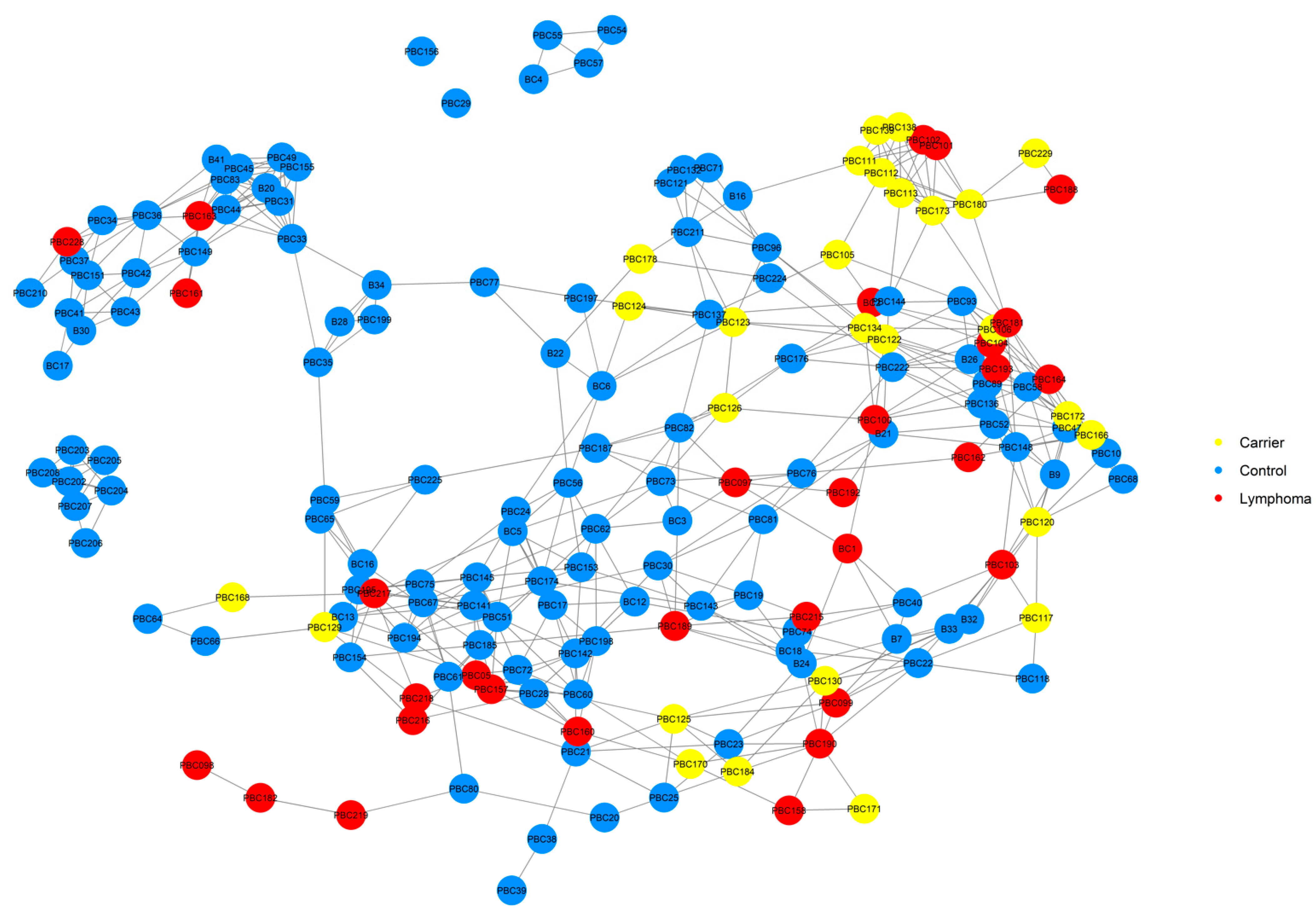
2.6. Relationship Networks (NetView)
2.7. Restricted Maximum Likelihood (REML) Analysis
3. Results
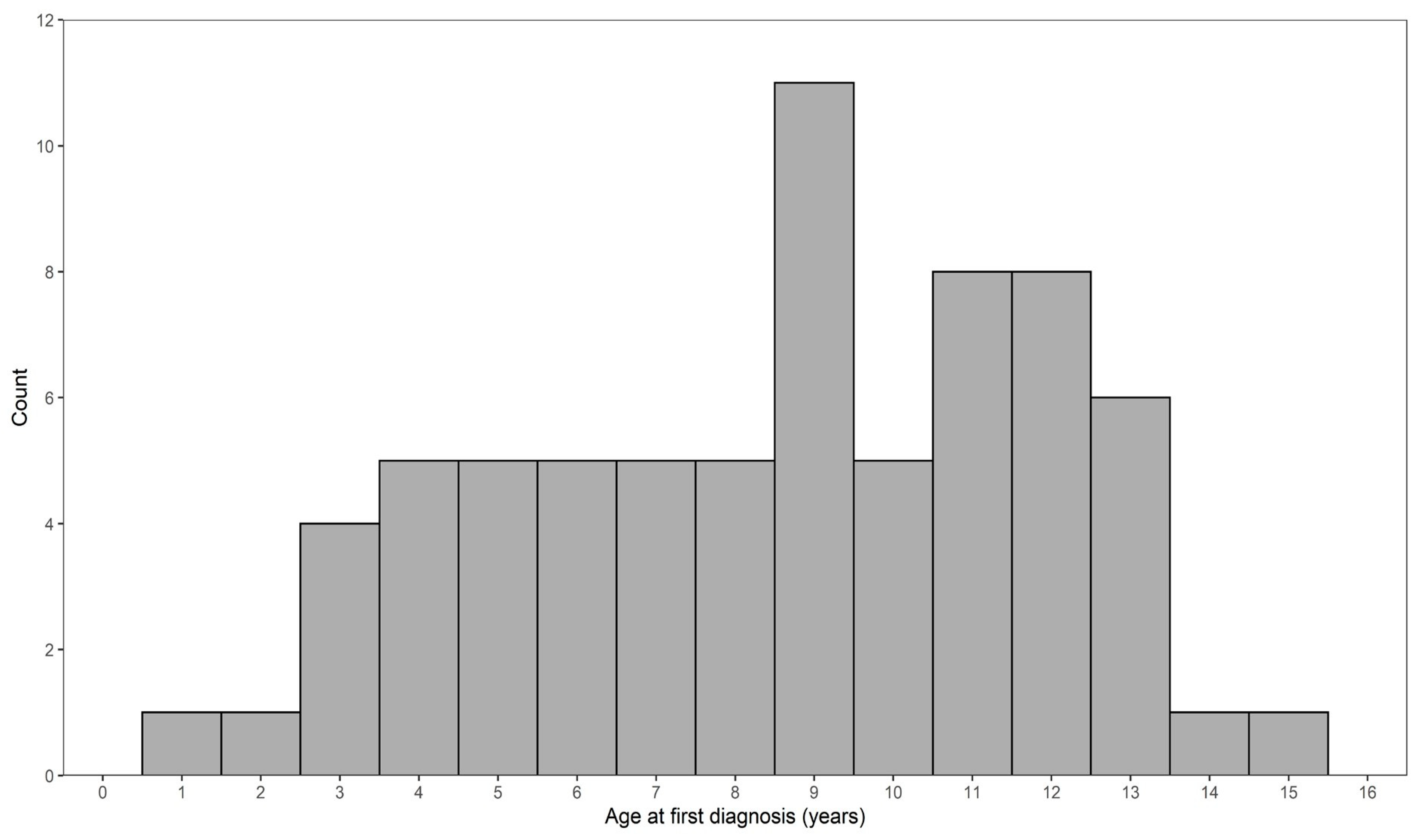
3.1. Pedigree Investigations
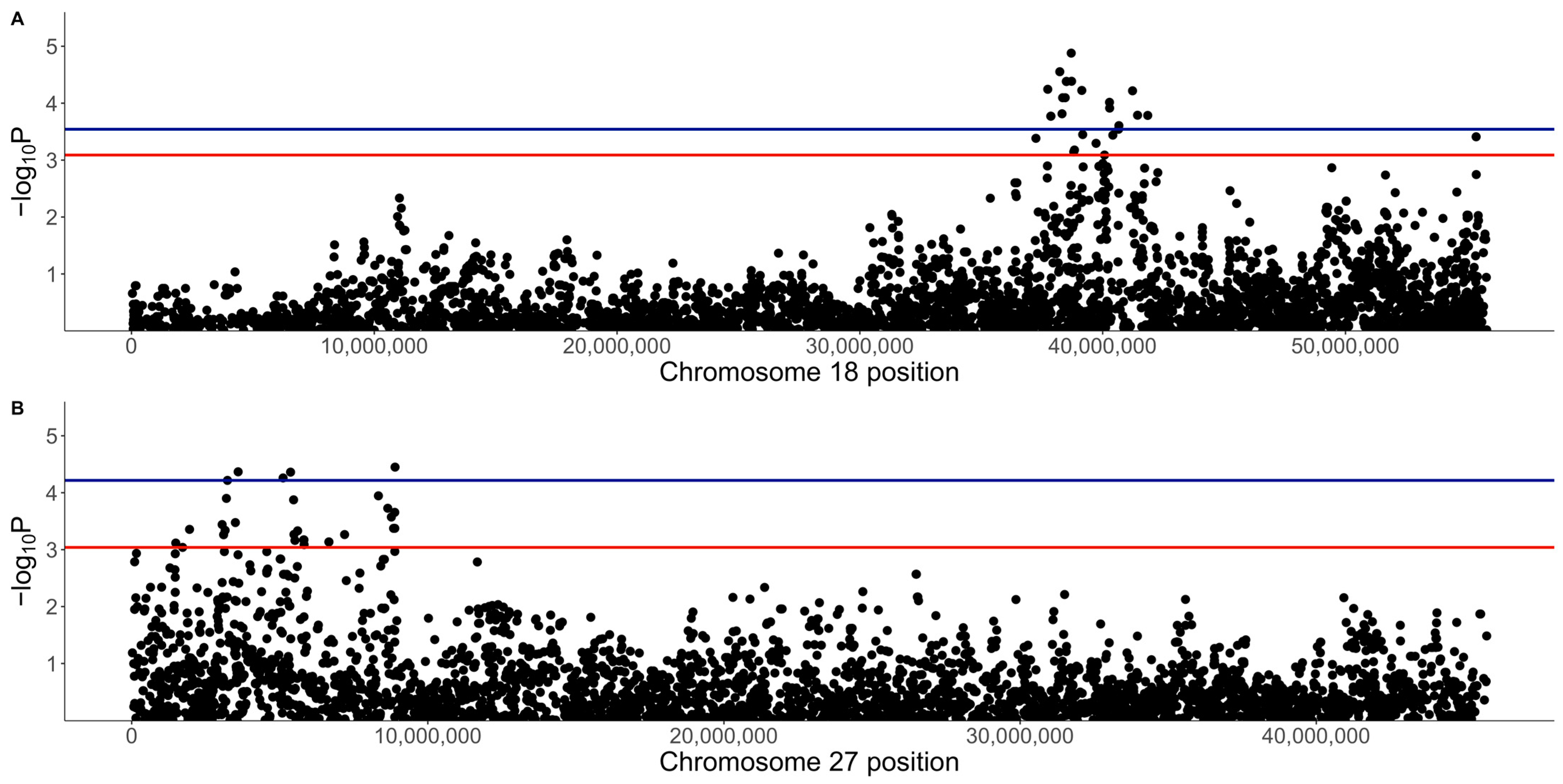
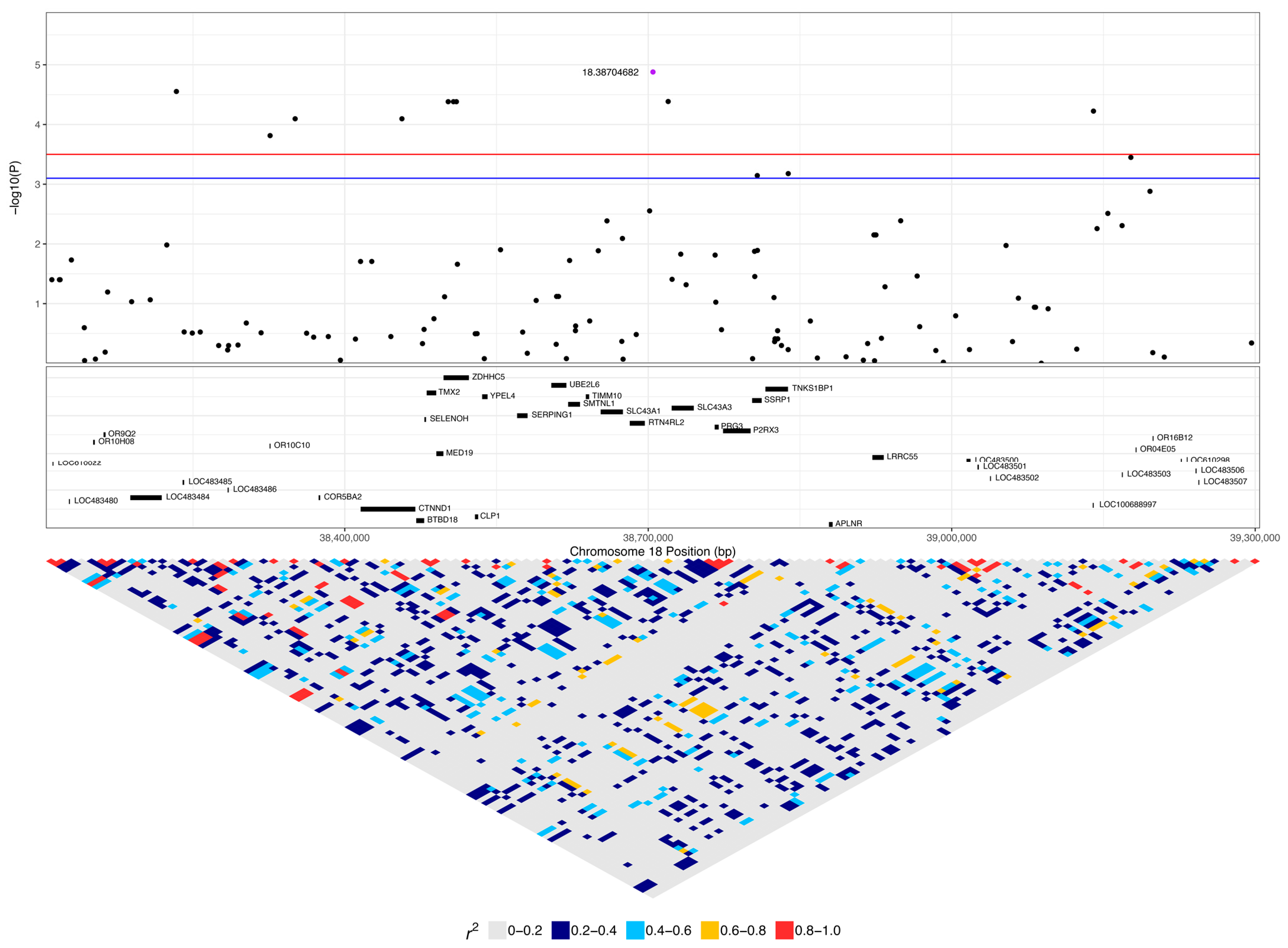
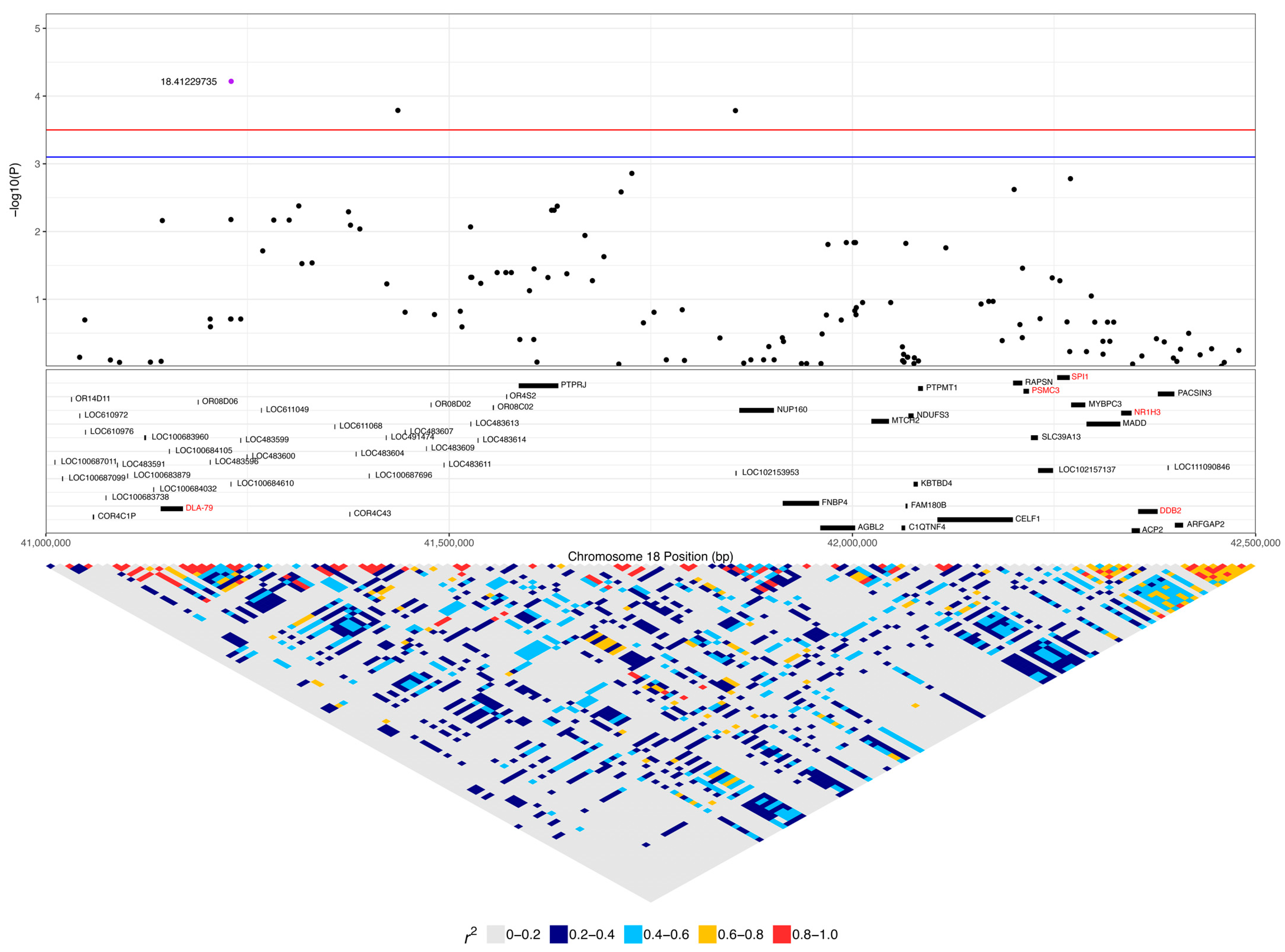
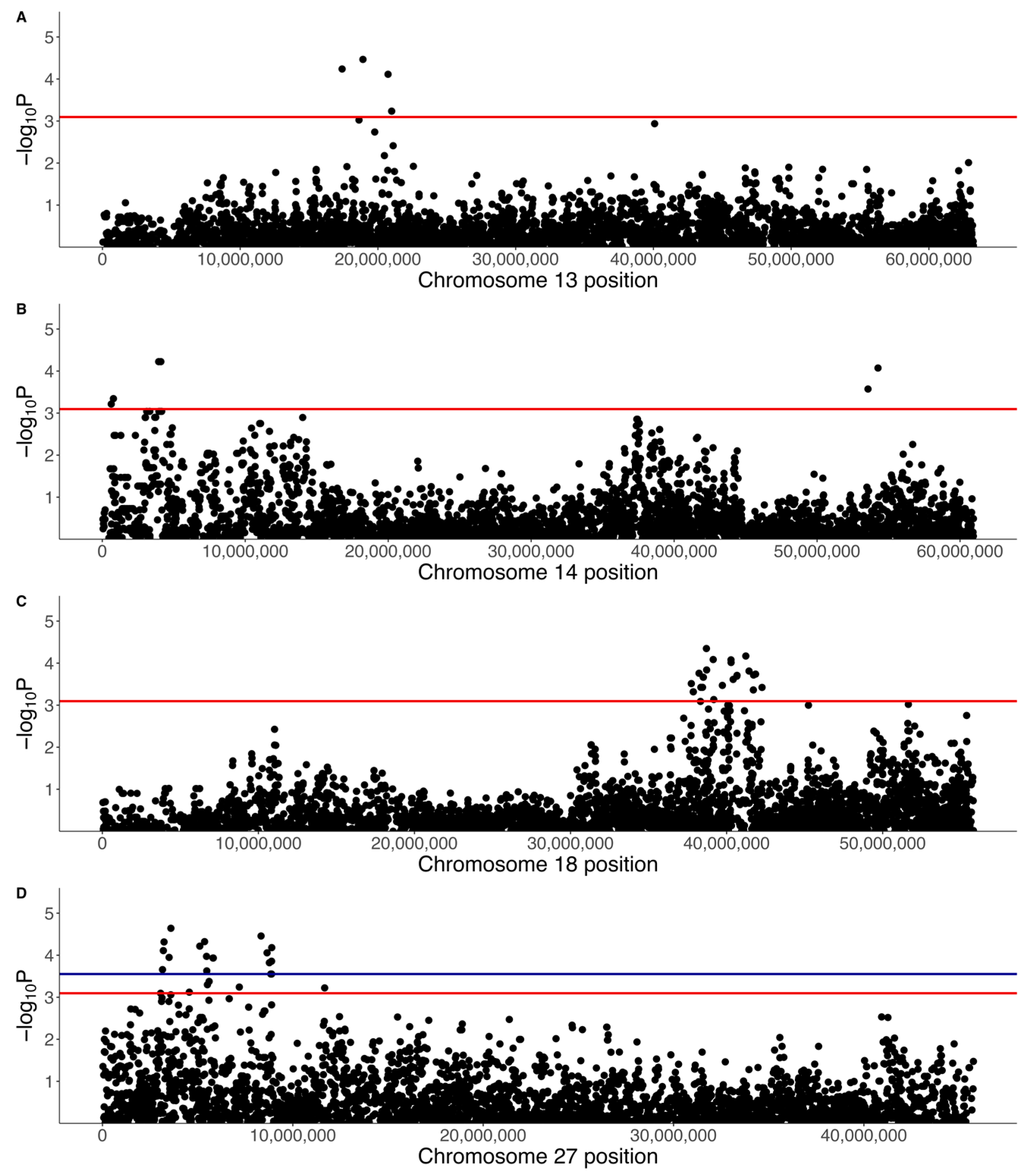
3.2. GWAS with Quantitative Phenotype
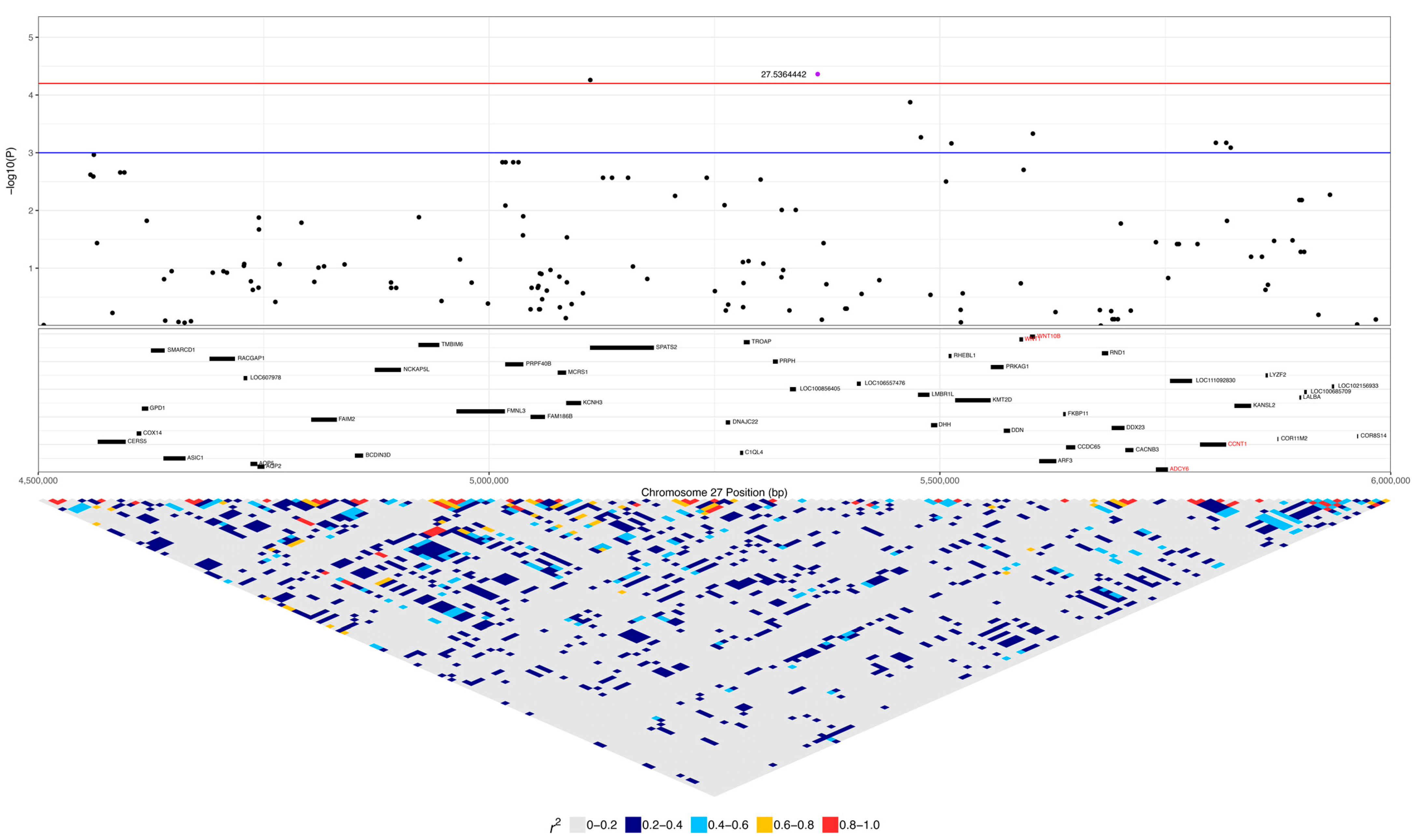
3.3. GWAS with Binary Phenotype
3.4. Phenotypic Variance Explained by Genetic Variance (Heritability)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dobson, J.M.; Samuel, S.; Milstein, H.; Rogers, K.; Wood, J.L. Canine neoplasia in the UK: Estimates of incidence rates from a population of insured dogs. J. Small Anim. Pract. 2002, 43, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.S.; Henley, W.E.; Harding, E.F.; Dobson, J.M.; Wood, J.L. Breed incidence of lymphoma in a UK population of insured dogs. Vet. Comp. Oncol. 2003, 1, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Comazzi, S.; Marelli, S.; Cozzi, M.; Rizzi, R.; Finotello, R.; Henriques, J.; Pastor, J.; Ponce, F.; Rohrer-Bley, C.; Rutgen, B.C.; et al. Breed-associated risks for developing canine lymphoma differ among countries: An European canine lymphoma network study. BMC Vet. Res. 2018, 14, 232. [Google Scholar] [CrossRef] [PubMed]
- Pinello, K.C.; Niza-Ribeiro, J.; Fonseca, L.; de Matos, A.J. Incidence, characteristics and geographical distributions of canine and human non-Hodgkin’s lymphoma in the Porto region (North West Portugal). Vet. J. 2019, 245, 70–76. [Google Scholar] [CrossRef]
- Modiano, J.F.; Breen, M.; Burnett, R.C.; Parker, H.G.; Inusah, S.; Thomas, R.; Avery, P.R.; Lindblad-Toh, K.; Ostrander, E.A.; Cutter, G.C.; et al. Distinct B-cell and T-cell lymphoproliferative disease prevalence among dog breeds indicates heritable risk. Cancer Res. 2005, 65, 5654–5661. [Google Scholar] [CrossRef]
- Pastor, M.; Chalvet-Monfray, K.; Marchal, T.; Keck, G.; Magnol, J.P.; Fournel-Fleury, C.; Ponce, F. Genetic and environmental risk indicators in canine non-Hodgkin’s lymphomas: Breed associations and geographic distribution of 608 cases diagnosed throughout France over 1 year. J. Vet. Intern. Med. 2009, 23, 301–310. [Google Scholar] [CrossRef]
- Ito, D.; Frantz, A.M.; Modiano, J.F. Canine lymphoma as a comparative model for human non-Hodgkin lymphoma: Recent progress and applications. Vet. Immunol. Immunopathol. 2014, 159, 192–201. [Google Scholar] [CrossRef]
- Richards, K.L.; Suter, S.E. Man’s best friend: What can pet dogs teach us about non-Hodgkin’s lymphoma? Immunol. Rev. 2015, 263, 173–191. [Google Scholar] [CrossRef]
- Thomas, R.; Smith, K.C.; Ostrander, E.A.; Galibert, F.; Breen, M. Chromosome aberrations in canine multicentric lymphomas detected with comparative genomic hybridisation and a panel of single locus probes. Br. J. Cancer 2003, 89, 1530–1537. [Google Scholar] [CrossRef]
- Ferraresso, S.; Bresolin, S.; Arico, A.; Comazzi, S.; Gelain, M.E.; Riondato, F.; Bargelloni, L.; Marconato, L.; Kronnie, G.; Aresu, L. Epigenetic silencing of TFPI-2 in canine diffuse large B-cell lymphoma. PLoS ONE 2014, 9, e92707. [Google Scholar] [CrossRef]
- Ferraresso, S.; Arico, A.; Sanavia, T.; Da Ros, S.; Milan, M.; Cascione, L.; Comazzi, S.; Martini, V.; Giantin, M.; Di Camillo, B.; et al. DNA methylation profiling reveals common signatures of tumorigenesis and defines epigenetic prognostic subtypes of canine Diffuse Large B-cell Lymphoma. Sci. Rep. 2017, 7, 11591. [Google Scholar] [CrossRef] [PubMed]
- Cascione, L.; Giudice, L.; Ferraresso, S.; Marconato, L.; Giannuzzi, D.; Napoli, S.; Bertoni, F.; Giugno, R.; Aresu, L. Long Non-Coding RNAs as Molecular Signatures for Canine B-Cell Lymphoma Characterization. Noncoding RNA 2019, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Craig, K.K.L.; Wood, G.A.; Keller, S.M.; Mutsaers, A.J.; Wood, R.D. MicroRNA profiling in canine multicentric lymphoma. PLoS ONE 2019, 14, e0226357. [Google Scholar] [CrossRef]
- Epiphanio, T.M.F.; Fernandes, N.; de Oliveira, T.F.; Lopes, P.A.; Ressio, R.A.; Goncalves, S.; Scattone, N.V.; Tedardi, M.V.; Kulikowski, L.D.; Damasceno, J.; et al. Global DNA methylation of peripheral blood leukocytes from dogs bearing multicentric non-Hodgkin lymphomas and healthy dogs: A comparative study. PLoS ONE 2019, 14, e0211898. [Google Scholar] [CrossRef] [PubMed]
- Elvers, I.; Turner-Maier, J.; Swofford, R.; Koltookian, M.; Johnson, J.; Stewart, C.; Zhang, C.Z.; Schumacher, S.E.; Beroukhim, R.; Rosenberg, M.; et al. Exome sequencing of lymphomas from three dog breeds reveals somatic mutation patterns reflecting genetic background. Genome Res. 2015, 25, 1634–1645. [Google Scholar] [CrossRef]
- Veldhoen, N.; Stewart, J.; Brown, R.; Milner, J. Mutations of the p53 gene in canine lymphoma and evidence for germ line p53 mutations in the dog. Oncogene 1998, 16, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Schofield, I.; Stevens, K.B.; Pittaway, C.; O’Neill, D.G.; Fecht, D.; Dobson, J.M.; Brodbelt, D.C. Geographic distribution and environmental risk factors of lymphoma in dogs under primary-care in the UK. J. Small Anim. Pract. 2019, 60, 746–754. [Google Scholar] [CrossRef]
- Pinello, K.C.; Santos, M.; Leite-Martins, L.; Niza-Ribeiro, J.; de Matos, A.J. Immunocytochemical study of canine lymphomas and its correlation with exposure to tobacco smoke. Vet. World 2017, 10, 1307–1313. [Google Scholar] [CrossRef]
- Takashima-Uebelhoer, B.B.; Barber, L.G.; Zagarins, S.E.; Procter-Gray, E.; Gollenberg, A.L.; Moore, A.S.; Bertone-Johnson, E.R. Household chemical exposures and the risk of canine malignant lymphoma, a model for human non-Hodgkin’s lymphoma. Environ. Res. 2012, 112, 171–176. [Google Scholar] [CrossRef]
- Gavazza, A.; Presciuttini, S.; Barale, R.; Lubas, G.; Gugliucci, B. Association between canine malignant lymphoma, living in industrial areas, and use of chemicals by dog owners. J. Vet. Intern. Med. 2001, 15, 190–195. [Google Scholar] [CrossRef]
- Marconato, L.; Leo, C.; Girelli, R.; Salvi, S.; Abramo, F.; Bettini, G.; Comazzi, S.; Nardi, P.; Albanese, F.; Zini, E. Association between waste management and cancer in companion animals. J. Vet. Intern. Med. 2009, 23, 564–569. [Google Scholar] [CrossRef]
- Tonomura, N.; Elvers, I.; Thomas, R.; Megquier, K.; Turner-Maier, J.; Howald, C.; Sarver, A.L.; Swofford, R.; Frantz, A.M.; Ito, D.; et al. Genome-wide Association Study Identifies Shared Risk Loci Common to Two Malignancies in Golden Retrievers. PLoS Genet. 2015, 11, e1004922. [Google Scholar] [CrossRef] [PubMed]
- Hayward, J.J.; Castelhano, M.G.; Oliveira, K.C.; Corey, E.; Balkman, C.; Baxter, T.L.; Casal, M.L.; Center, S.A.; Fang, M.; Garrison, S.J.; et al. Complex disease and phenotype mapping in the domestic dog. Nat. Commun. 2016, 7, 10460. [Google Scholar] [CrossRef]
- Labadie, J.D.; Elvers, I.; Feigelson, H.S.; Magzamen, S.; Yoshimoto, J.; Dossey, J.; Burnett, R.; Avery, A.C. Genome-wide association analysis of canine T zone lymphoma identifies link to hypothyroidism and a shared association with mast-cell tumors. BMC Genom. 2020, 21, 464. [Google Scholar] [CrossRef]
- Dutrow, E.V.; Serpell, J.A.; Ostrander, E.A. Domestic dog lineages reveal genetic drivers of behavioral diversification. Cell 2022, 185, 4737–4755.e18. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.T.; Williamson, P.; Khatkar, M.S. Analysis of dog breed diversity using a composite selection index. Sci. Rep. 2023, 13, 1674. [Google Scholar] [CrossRef]
- Jagannathan, V.; Drogemuller, C.; Leeb, T.; Dog Biomedical Variant Database, C. A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef]
- Mortlock, S.A.; Khatkar, M.S.; Williamson, P. Comparative Analysis of Genome Diversity in Bullmastiff Dogs. PLoS ONE 2016, 11, e0147941. [Google Scholar] [CrossRef] [PubMed]
- Mortlock, S.-A.; Booth, R.; Mazrier, H.; Khatkar, M.; Williamson, P. Visualization of Genome Diversity in German Shepherd Dogs. Bioinform. Biol. Insights 2016, 9, 37–42. [Google Scholar] [CrossRef]
- Soh, P.X.Y.; Hsu, W.T.; Khatkar, M.S.; Williamson, P. Evaluation of genetic diversity and management of disease in Border Collie dogs. Sci. Rep. 2021, 11, 6243. [Google Scholar] [CrossRef]
- Vaysse, A.; Ratnakumar, A.; Derrien, T.; Axelsson, E.; Rosengren Pielberg, G.; Sigurdsson, S.; Fall, T.; Seppala, E.H.; Hansen, M.S.; Lawley, C.T.; et al. Identification of genomic regions associated with phenotypic variation between dog breeds using selection mapping. PLoS Genet. 2011, 7, e1002316. [Google Scholar] [CrossRef]
- Shariflou, M.R.; James, J.W.; Nicholas, F.W.; Wade, C.M. A genealogical survey of Australian registered dog breeds. Vet. J. 2011, 189, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.G. Genomic analyses of modern dog breeds. Mamm. Genome 2012, 23, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.G.; Dreger, D.L.; Rimbault, M.; Davis, B.W.; Mullen, A.B.; Carpintero-Ramirez, G.; Ostrander, E.A. Genomic Analyses Reveal the Influence of Geographic Origin, Migration, and Hybridization on Modern Dog Breed Development. Cell Rep. 2017, 19, 697–708. [Google Scholar] [CrossRef]
- Hédan, B.; Cadieu, É.; Rimbault, M.; Vaysse, A.; Dufaure de Citres, C.; Devauchelle, P.; Botherel, N.; Abadie, J.; Quignon, P.; Derrien, T.; et al. Identification of common predisposing loci to hematopoietic cancers in four dog breeds. PLoS Genet. 2021, 17, e1009395. [Google Scholar] [CrossRef] [PubMed]
- Bennett, P.F.; Taylor, R.; Williamson, P. Demographic risk factors for lymphoma in Australian dogs: 6201 cases. J. Vet. Intern. Med. 2018, 32, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.Y.; Soh, P.; Bennett, P.F.; Williamson, P. Lymphoma in Australian Border Collies: Survey results and pedigree analyses. Aust. Vet. J. 2019, 97, 14–22. [Google Scholar] [CrossRef]
- Sinnwell, J.P.; Therneau, T.M.; Schaid, D.J. The kinship2 R package for pedigree data. Hum. Hered. 2014, 78, 91–93. [Google Scholar] [CrossRef]
- Rainer, J.; Taliun, D.; D’Elia, Y.; Pattaro, C.; Domingues, F.S.; Weichenberger, C.X. FamAgg: An R package to evaluate familial aggregation of traits in large pedigrees. Bioinformatics 2016, 32, 1583–1585. [Google Scholar] [CrossRef]
- Coster, A.J.R. Pedigree: Pedigree Functions. R Package, Version 1.4. 2013, Volume 1. Available online: https://cran.r-project.org/web/packages/pedigree/index.html (accessed on 8 January 2020).
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Wijnrocx, K.; Francois, L.; Stinckens, A.; Janssens, S.; Buys, N. Half of 23 Belgian dog breeds has a compromised genetic diversity, as revealed by genealogical and molecular data analysis. J. Anim. Breed. Genet. 2016, 133, 375–383. [Google Scholar] [CrossRef]
- Bennett, P.; Williamson, P.; Taylor, R. Review of Canine Lymphoma Treated with Chemotherapy-Outcomes and Prognostic Factors. Vet. Sci. 2023, 10, 342. [Google Scholar] [CrossRef] [PubMed]
- Vail, D.M. Levels of evidence in canine oncology trials--a case in point. Vet. Comp. Oncol. 2013, 11, 167–168. [Google Scholar] [CrossRef] [PubMed]
- Pratap, S.; Scordino, T.S. Molecular and cellular genetics of non-Hodgkin lymphoma: Diagnostic and prognostic implications. Exp. Mol. Pathol. 2019, 106, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef]
- Turner, D. qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. J. Open. Source Softw. 2018, 3, 731. [Google Scholar] [CrossRef]
- He, F.; Ding, S.; Wang, H.; Qin, F. IntAssoPlot: An R Package for Integrated Visualization of Genome-Wide Association Study Results with Gene Structure and Linkage Disequilibrium Matrix. Front. Genet. 2020, 11, 260. [Google Scholar] [CrossRef]
- Sabik, O.L.; Calabrese, G.M.; Taleghani, E.; Ackert-Bicknell, C.L.; Farber, C.R. Identification of a Core Module for Bone Mineral Density through the Integration of a Co-expression Network and GWAS Data. Cell Rep. 2020, 32, 108145. [Google Scholar] [CrossRef]
- Tenenbaum, D. KEGGREST. R Package Version 1.24.1. Available online: https://bioconductor.riken.jp/packages/3.8/bioc/manuals/KEGGREST/man/KEGGREST.pdf. (accessed on 8 January 2020).
- Esau, D. Viral Causes of Lymphoma: The History of Epstein-Barr Virus and Human T-Lymphotropic Virus 1. Virol. Res. Treat. 2017, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Torres, H.A.; Kontoyiannis, D.P.; Aguilera, E.A.; Younes, A.; Luna, M.A.; Tarrand, J.J.; Nogueras, G.M.; Raad, I.I.; Chemaly, R.F. Cytomegalovirus infection in patients with lymphoma: An important cause of morbidity and mortality. Clin. Lymphoma Myeloma 2006, 6, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Mehravaran, H.; Makvandi, M.; Samarbaf Zade, A.; Neisi, N.; Kiani, H.; Radmehr, H.; Shahani, T.; Hoseini, S.Z.; Ranjbari, N.; Nahid Samiei, R. Association of Human Cytomegalovirus with Hodgkin’s Disease and Non-Hodgkin’s lymphomas. Asian Pac. J. Cancer Prev. 2017, 18, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Steinig, E.J.; Neuditschko, M.; Khatkar, M.S.; Raadsma, H.W.; Zenger, K.R. netview p: A network visualization tool to unravel complex population structure using genome-wide SNPs. Mol. Ecol. Resour. 2016, 16, 216–227. [Google Scholar] [CrossRef]
- Allaire, J.J.; Gandrud, C.; Russell, K.; Yetman, C. networkD3: D3 JavaScript Network Graphs from R. R Package Version 0.4. Available online: https://cran.r-project.org/package=networkD3 (accessed on 8 January 2020).
- Lee, S.H.; Wray, N.R.; Goddard, M.E.; Visscher, P.M. Estimating missing heritability for disease from genome-wide association studies. Am. J. Hum. Genet. 2011, 88, 294–305. [Google Scholar] [CrossRef]
- Van Rooyen, L.J.; Hooijberg, E.; Reyers, F. Breed prevalence of canine lymphoma in South Africa. J. S. Afr. Vet. Assoc. 2018, 89, e1–e11. [Google Scholar] [CrossRef]
- Onions, D.E. A prospective survey of familial canine lymphosarcoma. J. Natl. Cancer Inst. 1984, 72, 909–912. [Google Scholar]
- Lobetti, R.G. Lymphoma in 3 related Rottweilers from a single household. J. S. Afr. Vet. Assoc. 2009, 80, 103–105. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cole, K.A.; Chuaqui, R.F.; Katz, K.; Pack, S.; Zhuang, Z.; Cole, C.E.; Lyne, J.C.; Linehan, W.M.; Liotta, L.A.; Emmert-Buck, M.R. cDNA sequencing and analysis of POV1 (PB39): A novel gene up-regulated in prostate cancer. Genomics 1998, 51, 282–287. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, H.; Yang, W.; Perez-Andreu, V.; Devidas, M.; Fan, Y.; Cheng, C.; Pei, D.; Scheet, P.; Burchard, E.G.; Eng, C.; et al. Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J. Natl. Cancer Inst. 2013, 105, 733–742. [Google Scholar] [CrossRef]
- Shimozono, N.; Jinnin, M.; Masuzawa, M.; Masuzawa, M.; Wang, Z.; Hirano, A.; Tomizawa, Y.; Etoh-Kira, T.; Kajihara, I.; Harada, M.; et al. NUP160-SLC43A3 is a novel recurrent fusion oncogene in angiosarcoma. Cancer Res. 2015, 75, 4458–4465. [Google Scholar] [CrossRef]
- Liao, J.; Tao, X.; Ding, Q.; Liu, J.; Yang, X.; Yuan, F.E.; Yang, J.A.; Liu, B.; Xiang, G.A.; Chen, Q. SSRP1 silencing inhibits the proliferation and malignancy of human glioma cells via the MAPK signaling pathway. Oncol. Rep. 2017, 38, 2667–2676. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; He, K.; Guo, Q.; Chen, J.; Zhang, M.; Huang, K.; Yang, D.; Wu, L.; Deng, Y.; Luo, X.; et al. SSRP1 promotes colorectal cancer progression and is negatively regulated by miR-28-5p. J. Cell. Mol. Med. 2019, 23, 3118–3129. [Google Scholar] [CrossRef]
- Jeronimo, C.; Robert, F. The histone chaperone FACT: A guardian of chromatin structure integrity. Transcription 2022, 13, 16–38. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; He, K.; Luo, T.; Deng, Y.; Wang, H.; Liu, H.; Zhang, J.; Chen, K.; Xiao, J.; Duan, X.; et al. SSRP1 Contributes to the Malignancy of Hepatocellular Carcinoma and Is Negatively Regulated by miR-497. Mol. Ther. 2016, 24, 903–914. [Google Scholar] [CrossRef]
- Wagner, J.L. Molecular Organization of the Canine Major Histocompatibility Complex. J. Hered. 2003, 94, 23–26. [Google Scholar] [CrossRef][Green Version]
- Wang, S.S.; Abdou, A.M.; Morton, L.M.; Thomas, R.; Cerhan, J.R.; Gao, X.; Cozen, W.; Rothman, N.; Davis, S.; Severson, R.K.; et al. Human leukocyte antigen class I and II alleles in non-Hodgkin lymphoma etiology. Blood 2010, 115, 4820–4823. [Google Scholar] [CrossRef]
- Zhong, C.; Gragert, L.; Maiers, M.; Hill, B.T.; Garcia-Gomez, J.; Gendzekhadze, K.; Senitzer, D.; Song, J.; Weisenburger, D.; Goldstein, L.; et al. The association between HLA and non-Hodgkin lymphoma subtypes, among a transplant-indicated population. Leuk. Lymphoma 2019, 60, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Amiot, L.; Onno, M.; Lamy, T.; Dauriac, C.; Le Prise, P.Y.; Fauchet, R.; Drenou, B. Loss of HLA molecules in B lymphomas is associated with an aggressive clinical course. Br. J. Haematol. 1998, 100, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Hao, Q.; Liang, Y.; Kong, E. Protein palmitoylation in cancer: Molecular functions and therapeutic potential. Mol. Oncol. 2023, 17, 3–26. [Google Scholar] [CrossRef]
- Yamashita, A.; Taniwaki, T.; Kaikoi, Y.; Yamazaki, T. Protective role of the endoplasmic reticulum protein mitsugumin23 against ultraviolet C-induced cell death. FEBS Lett. 2013, 587, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Oh, I.H.; McCrea, P.D. Phosphorylation and isoform use in p120-catenin during development and tumorigenesis. Biochim. Biophys. Acta 2016, 1863, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Noordhuis, M.G.; Fehrmann, R.S.; Wisman, G.B.; Nijhuis, E.R.; van Zanden, J.J.; Moerland, P.D.; Ver Loren van Themaat, E.; Volders, H.H.; Kok, M.; ten Hoor, K.A.; et al. Involvement of the TGF-beta and beta-catenin pathways in pelvic lymph node metastasis in early-stage cervical cancer. Clin. Cancer Res. 2011, 17, 1317–1330. [Google Scholar] [CrossRef]
- Wang, W.; Fei, Y.; Liu, S.L. CTNND(1) 755 T>G Promoter Polymorphism and Risk of Pancreatic Carcinoma in Chinese. J. Clin. Lab. Anal. 2017, 31, e22055. [Google Scholar] [CrossRef]
- Ousingsawat, J.; Schreiber, R.; Kunzelmann, K. TMEM16F/Anoctamin 6 in Ferroptotic Cell Death. Cancers 2019, 11, 625. [Google Scholar] [CrossRef]
- Ousingsawat, J.; Wanitchakool, P.; Schreiber, R.; Kunzelmann, K. Contribution of TMEM16F to pyroptotic cell death. Cell Death Dis. 2018, 9, 300. [Google Scholar] [CrossRef]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Rim, E.Y.; Clevers, H.; Nusse, R. The Wnt Pathway: From Signaling Mechanisms to Synthetic Modulators. Annu. Rev. Biochem. 2022, 91, 571–598. [Google Scholar] [CrossRef]
- Janovská, P.; Bryja, V. Wnt signalling pathways in chronic lymphocytic leukaemia and B-cell lymphomas. Br. J. Pharmacol. 2017, 174, 4701–4715. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Grainger, S.; Traver, D.; Willert, K. Wnt Signaling in Hematological Malignancies. Prog. Mol. Biol. Transl. Sci. 2018, 153, 321–341. [Google Scholar] [CrossRef]
- Linke, F.; Zaunig, S.; Nietert, M.M.; von Bonin, F.; Lutz, S.; Dullin, C.; Janovská, P.; Beissbarth, T.; Alves, F.; Klapper, W.; et al. WNT5A: A motility-promoting factor in Hodgkin lymphoma. Oncogene 2017, 36, 13–23. [Google Scholar] [CrossRef]
- El Ayachi, I.; Fatima, I.; Wend, P.; Alva-Ornelas, J.A.; Runke, S.; Kuenzinger, W.L.; Silva, J.; Silva, W.; Gray, J.K.; Lehr, S.; et al. The WNT10B Network Is Associated with Survival and Metastases in Chemoresistant Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 982–993. [Google Scholar] [CrossRef]
- Lazzaroni, F.; Del Giacco, L.; Biasci, D.; Turrini, M.; Prosperi, L.; Brusamolino, R.; Cairoli, R.; Beghini, A. Intronless WNT10B-short variant underlies new recurrent allele-specific rearrangement in acute myeloid leukaemia. Sci. Rep. 2016, 6, 37201. [Google Scholar] [CrossRef]
- Choi, J.H.; Zhong, X.; McAlpine, W.; Liao, T.C.; Zhang, D.; Fang, B.; Russell, J.; Ludwig, S.; Nair-Gill, E.; Zhang, Z.; et al. LMBR1L regulates lymphopoiesis through Wnt/β-catenin signaling. Science 2019, 364, eaau0812. [Google Scholar] [CrossRef]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef]
- Ye, H.; Lu, L.; Ge, B.; Gao, S.; Ma, Y.; Liang, B.; Yu, K.; Yang, K. MLL2 protein is a prognostic marker for gastrointestinal diffuse large B-cell lymphoma. Int. J. Clin. Exp. Pathol. 2015, 8, 13043–13050. [Google Scholar]
- Ortega-Molina, A.; Boss, I.W.; Canela, A.; Pan, H.; Jiang, Y.; Zhao, C.; Jiang, M.; Hu, D.; Agirre, X.; Niesvizky, I.; et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat. Med. 2015, 21, 1199–1208. [Google Scholar] [CrossRef]
- da Silva Almeida, A.C.; Abate, F.; Khiabanian, H.; Martinez-Escala, E.; Guitart, J.; Tensen, C.P.; Vermeer, M.H.; Rabadan, R.; Ferrando, A.; Palomero, T. The mutational landscape of cutaneous T cell lymphoma and Sézary syndrome. Nat. Genet. 2015, 47, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Klein-Hitpass, L.; Grabellus, F.; Arnold, G.; Klapper, W.; Pförtner, R.; Dührsen, U.; Eckstein, A.; Dürig, J.; Küppers, R. Recurrent mutations in NF-κB pathway components, KMT2D, and NOTCH1/2 in ocular adnexal MALT-type marginal zone lymphomas. Oncotarget 2016, 7, 62627–62639. [Google Scholar] [CrossRef] [PubMed]
- Froimchuk, E.; Jang, Y.; Ge, K. Histone H3 lysine 4 methyltransferase KMT2D. Gene 2017, 627, 337–342. [Google Scholar] [CrossRef]
- Ji, M.M.; Huang, Y.H.; Huang, J.Y.; Wang, Z.F.; Fu, D.; Liu, H.; Liu, F.; Leboeuf, C.; Wang, L.; Ye, J.; et al. Histone modifier gene mutations in peripheral T-cell lymphoma not otherwise specified. Haematologica 2018, 103, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Bellan, C.; De Falco, G.; Lazzi, S.; Micheli, P.; Vicidomini, S.; Schürfeld, K.; Amato, T.; Palumbo, A.; Bagella, L.; Sabattini, E.; et al. CDK9/CYCLIN T1 expression during normal lymphoid differentiation and malignant transformation. J. Pathol. 2004, 203, 946–952. [Google Scholar] [CrossRef]
- De Falco, G.; Leucci, E.; Onnis, A.; Bellan, C.; Tigli, C.; Wirths, S.; Cerino, G.; Cocco, M.; Crupi, D.; De Luca, A.; et al. Cdk9/Cyclin T1 complex: A key player during the activation/differentiation process of normal lymphoid B cells. J. Cell. Physiol. 2008, 215, 276–282. [Google Scholar] [CrossRef]
- Šmerc, A.; Sodja, E.; Legiša, M. Posttranslational modification of 6-phosphofructo-1-kinase as an important feature of cancer metabolism. PLoS ONE 2011, 6, e19645. [Google Scholar] [CrossRef]
- Yang, Y.; Ishak Gabra, M.B.; Hanse, E.A.; Lowman, X.H.; Tran, T.Q.; Li, H.; Milman, N.; Liu, J.; Reid, M.A.; Locasale, J.W.; et al. MiR-135 suppresses glycolysis and promotes pancreatic cancer cell adaptation to metabolic stress by targeting phosphofructokinase-1. Nat. Commun. 2019, 10, 809. [Google Scholar] [CrossRef] [PubMed]
- Nadanaka, S.; Kitagawa, H. Heparan sulphate biosynthesis and disease. J. Biochem. 2008, 144, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.W.; Huang, X.; Liu, S.; Lu, Y. EXT1, Regulated by MiR-665, Promotes Cell Apoptosis via ERK1/2 Signaling Pathway in Acute Lymphoblastic Leukemia. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 6491–6503. [Google Scholar] [CrossRef]
- Caron, A.; Briscoe, D.M.; Richard, D.; Laplante, M. DEPTOR at the Nexus of Cancer, Metabolism, and Immunity. Physiol. Rev. 2018, 98, 1765–1803. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J.; Zeng, Z.; Que, W.; Zhou, L. Knockdown of DEPTOR induces apoptosis, increases chemosensitivity to doxorubicin and suppresses autophagy in RPMI-8226 human multiple myeloma cells in vitro. Int. J. Mol. Med. 2013, 31, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Zandvliet, M. Canine lymphoma: A review. Vet. Q. 2016, 36, 76–104. [Google Scholar] [CrossRef]
- Jabłońska, E.; Białopiotrowicz, E.; Szydłowski, M.; Prochorec-Sobieszek, M.; Juszczyński, P.; Szumera-Ciećkiewicz, A. DEPTOR is a microRNA-155 target regulating migration and cytokine production in diffuse large B-cell lymphoma cells. Exp. Hematol. 2020, 88, 56–67.e52. [Google Scholar] [CrossRef]
- Dagan, L.N.; Jiang, X.; Bhatt, S.; Cubedo, E.; Rajewsky, K.; Lossos, I.S. miR-155 regulates HGAL expression and increases lymphoma cell motility. Blood 2012, 119, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Due, H.; Svendsen, P.; Bødker, J.S.; Schmitz, A.; Bøgsted, M.; Johnsen, H.E.; El-Galaly, T.C.; Roug, A.S.; Dybkær, K. miR-155 as a Biomarker in B-Cell Malignancies. BioMed Res. Int. 2016, 2016, 9513037. [Google Scholar] [CrossRef]
- Ahmadvand, M.; Eskandari, M.; Pashaiefar, H.; Yaghmaie, M.; Manoochehrabadi, S.; Khakpour, G.; Sheikhsaran, F.; Montazer Zohour, M. Over expression of circulating miR-155 predicts prognosis in diffuse large B-cell lymphoma. Leuk. Res. 2018, 70, 45–48. [Google Scholar] [CrossRef]
- Huskova, H.; Korecka, K.; Karban, J.; Vargova, J.; Vargova, K.; Dusilkova, N.; Trneny, M.; Stopka, T. Oncogenic microRNA-155 and its target PU.1: An integrative gene expression study in six of the most prevalent lymphomas. Int. J. Hematol. 2015, 102, 441–450. [Google Scholar] [CrossRef]
- Tanaka, T.; Iino, M. Sec8 regulates cytokeratin8 phosphorylation and cell migration by controlling the ERK and p38 MAPK signalling pathways. Cell Signal. 2015, 27, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kikuchi, N.; Goto, K.; Iino, M. Sec6/8 regulates Bcl-2 and Mcl-1, but not Bcl-xl, in malignant peripheral nerve sheath tumor cells. Apoptosis Int. J. Program. Cell Death 2016, 21, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Briggs, J.M. Functional Implications of the spectrum of BCL2 mutations in Lymphoma. Mutat. Res. Rev. Mutat. Res. 2016, 769, 1–18. [Google Scholar] [CrossRef]
- Schuetz, J.M.; Johnson, N.A.; Morin, R.D.; Scott, D.W.; Tan, K.; Ben-Nierah, S.; Boyle, M.; Slack, G.W.; Marra, M.A.; Connors, J.M.; et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia 2012, 26, 1383–1390. [Google Scholar] [CrossRef]
- Liu, J.Z.; Erlich, Y.; Pickrell, J.K. Case-control association mapping by proxy using family history of disease. Nat. Genet. 2017, 49, 325–331. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location (Chr:BP) | p-Value | q-Value | A1 | A2 | A1 Freq | b | SE | Gene | Frequency of A1A1 (No. of Dogs) | Frequency of A1A2 (No. of Dogs) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lym. | Carr. | Cont. | Lym. | Carr. | Cont. | |||||||||
| 18:38704682 | 1.32 × 10−5 | 0.022 | A | G | 0.32 | 0.36 | 0.08 | - | 0.26 (8) | 0.19 (5) | 0.08 (9) | 0.55 (17) | 0.33 (9) | 0.37 (44) |
| 18:38233567 | 2.80 × 10−5 | 0.022 | A | G | 0.12 | 0.55 | 0.13 | - | 0.06 (2) | 0 (0) | 0 (0) | 0.42 (13) | 0.41 (11) | 0.13 (15) |
| 18:38719709 | 4.12 × 10−5 | 0.022 | G | A | 0.32 | 0.34 | 0.08 | - | 0.23 (7) | 0.22 (6) | 0.08 (9) | 0.55 (17) | 0.37 (10) | 0.34 (41) |
| 18:38502268 | 4.15 × 10−5 | 0.022 | A | G | 0.14 | 0.50 | 0.12 | ZDHHC5 | 0.1 (3) | 0 (0) | 0 (0) | 0.39 (12) | 0.41 (11) | 0.16 (19) |
| 18:38507461 | 4.15 × 10−5 | 0.022 | A | G | 0.14 | 0.50 | 0.12 | ZDHHC5 | 0.1 (3) | 0 (0) | 0 (0) | 0.39 (12) | 0.41 (11) | 0.16 (19) |
| 18:38510335 | 4.15 × 10−5 | 0.022 | A | G | 0.14 | 0.50 | 0.12 | ZDHHC5 | 0.1 (3) | 0 (0) | 0 (0) | 0.39 (12) | 0.41 (11) | 0.16 (19) |
| 18:37737740 | 5.70 × 10−5 | 0.022 | C | A | 0.14 | 0.49 | 0.12 | - | 0.06 (2) | 0 (0) | 0 (0) | 0.45 (14) | 0.41 (11) | 0.16 (19) |
| 18:39140112 | 5.97 × 10−5 | 0.022 | G | A | 0.25 | 0.38 | 0.09 | LOC100688997 | 0.16 (5) | 0.07 (2) | 0.03 (4) | 0.58 (18) | 0.33 (9) | 0.34 (41) |
| 18:41229735 | 6.06 × 10−5 | 0.022 | A | G | 0.44 | 0.33 | 0.08 | LOC100684610 (OR4C5) | 0.45 (14) | 0.22 (6) | 0.13 (15) | 0.42 (13) | 0.56 (15) | 0.49 (58) |
| 18:38350947 | 8.02 × 10−5 | 0.023 | A | G | 0.12 | 0.51 | 0.13 | - | 0.06 (2) | 0 (0) | 0 (0) | 0.42 (13) | 0.41 (11) | 0.13 (16) |
| 18:38456518 | 8.02 × 10−5 | 0.023 | A | G | 0.12 | 0.51 | 0.13 | CTNND1 | 0.06 (2) | 0 (0) | 0 (0) | 0.42 (13) | 0.41 (11) | 0.13 (16) |
| 18:40281122 | 9.65 × 10−5 | 0.026 | A | G | 0.42 | 0.31 | 0.08 | - | 0.42 (13) | 0.19 (5) | 0.13 (16) | 0.42 (13) | 0.56 (15) | 0.45 (53) |
| 18:40286669 | 1.22 × 10−4 | 0.030 | G | A | 0.43 | 0.31 | 0.08 | - | 0.42 (13) | 0.19 (5) | 0.13 (16) | 0.42 (13) | 0.56 (15) | 0.47 (56) |
| 18:38326192 | 1.54 × 10−4 | 0.030 | A | G | 0.14 | 0.46 | 0.12 | OR10C10 | 0.06 (2) | 0 (0) | 0 (0) | 0.42 (13) | 0.41 (11) | 0.17 (20) |
| 18:41436427 | 1.63 × 10−4 | 0.030 | C | A | 0.50 | −0.31 | 0.08 | - | 0.1 (3) | 0.07 (2) | 0.35 (42) | 0.42 (13) | 0.67 (18) | 0.45 (53) |
| 18:41854962 | 1.64 × 10−4 | 0.030 | C | G | 0.15 | 0.42 | 0.11 | - | 0.1 (3) | 0.04 (1) | 0.01 (1) | 0.32 (10) | 0.44 (12) | 0.17 (20) |
| 18:37862012 | 1.69 × 10−4 | 0.030 | A | T | 0.18 | 0.40 | 0.11 | - | 0.19 (6) | 0.04 (1) | 0 (0) | 0.32 (10) | 0.41 (11) | 0.24 (28) |
| 18:37867871 | 1.69 × 10−4 | 0.030 | A | G | 0.18 | 0.40 | 0.11 | OR5B21 | 0.19 (6) | 0.04 (1) | 0 (0) | 0.32 (10) | 0.41 (11) | 0.24 (28) |
| 18:40667579 | 2.48 × 10−4 | 0.042 | C | A | 0.47 | 0.30 | 0.08 | - | 0.45 (14) | 0.22 (6) | 0.15 (18) | 0.45 (14) | 0.56 (15) | 0.5 (60) |
| 18:40653765 | 2.87 × 10−4 | 0.046 | A | C | 0.47 | 0.30 | 0.08 | - | 0.45 (14) | 0.22 (6) | 0.15 (18) | 0.45 (14) | 0.56 (15) | 0.53 (63) |
| 18:39177075 | 3.56 × 10−4 | 0.053 | A | G | 0.32 | 0.32 | 0.09 | - | 0.23 (7) | 0.15 (4) | 0.08 (9) | 0.55 (17) | 0.44 (12) | 0.38 (45) |
| 18:40418483 | 3.64 × 10−4 | 0.053 | A | G | 0.47 | 0.29 | 0.08 | - | 0.42 (13) | 0.22 (6) | 0.16 (19) | 0.48 (15) | 0.52 (14) | 0.52 (62) |
| 18:55380537 | 3.89 × 10−4 | 0.054 | A | T | 0.44 | −0.30 | 0.08 | TMEM109 | 0 (0) | 0 (0) | 0.29 (35) | 0.48 (15) | 0.59 (16) | 0.47 (56) |
| 18:37249960 | 4.14 × 10−4 | 0.055 | A | G | 0.14 | 0.40 | 0.11 | LOC483451 | 0.1 (3) | 0 (0) | 0.01 (1) | 0.39 (12) | 0.37 (10) | 0.16 (19) |
| 18:39727438 | 5.07 × 10−4 | 0.065 | G | A | 0.31 | 0.29 | 0.08 | - | 0.29 (9) | 0.11 (3) | 0.08 (9) | 0.45 (14) | 0.48 (13) | 0.35 (42) |
| 18:38838399 | 6.64 × 10−4 | 0.082 | G | A | 0.48 | 0.26 | 0.08 | - | 0.45 (14) | 0.33 (9) | 0.21 (25) | 0.45 (14) | 0.37 (10) | 0.43 (51) |
| 18:38807806 | 7.17 × 10−4 | 0.085 | A | G | 0.15 | 0.41 | 0.12 | SSRP1 | 0.06 (2) | 0 (0) | 0 (0) | 0.45 (14) | 0.37 (10) | 0.2 (24) |
| 18:40076496 | 8.15 × 10−4 | 0.093 | G | A | 0.21 | 0.34 | 0.10 | - | 0.13 (4) | 0.04 (1) | 0.02 (2) | 0.48 (15) | 0.41 (11) | 0.29 (34) |
| Location (Chr:BP) | p-Value | q-Value | A1 | A2 | A1 Freq | b | SE | Gene | Frequency of A1A1 (No. of Dogs) | Frequency of A1A2 (No. of Dogs) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lym. | Carr. | Cont. | Lym. | Carr. | Cont. | |||||||||
| 27:8892980 | 3.55 × 10−5 | 0.038 | C | G | 0.22 | 0.36 | 0.09 | ANO6 | 0.16 (5) | 0.04 (1) | 0.04 (5) | 0.55 (17) | 0.37 (10) | 0.25 (30) |
| 27:3594560 | 4.29 × 10−5 | 0.038 | G | A | 0.15 | 0.42 | 0.10 | BIN2 | 0.13 (4) | 0.07 (2) | 0 (0) | 0.45 (14) | 0.26 (7) | 0.18 (21) |
| 27:5364442 | 4.35 × 10−5 | 0.038 | A | G | 0.18 | 0.39 | 0.10 | LOC111092735 | 0.16 (5) | 0.07 (2) | 0.01 (1) | 0.42 (13) | 0.33 (9) | 0.22 (26) |
| 27:5112245 | 5.50 × 10−5 | 0.038 | A | G | 0.24 | 0.34 | 0.09 | SPATS2 | 0.29 (9) | 0.11 (3) | 0.02 (2) | 0.39 (12) | 0.52 (14) | 0.27 (32) |
| 27:3234647 | 6.07 × 10−5 | 0.038 | G | A | 0.18 | 0.39 | 0.10 | SCN8A | 0.13 (4) | 0.07 (2) | 0.03 (3) | 0.48 (15) | 0.26 (7) | 0.19 (23) |
| 27:8331252 | 1.13 × 10−4 | 0.053 | G | A | 0.12 | 0.44 | 0.11 | - | 0.06 (2) | 0.04 (1) | 0 (0) | 0.45 (14) | 0.26 (7) | 0.13 (15) |
| 27:3197949 | 1.26 × 10−4 | 0.053 | G | A | 0.18 | 0.37 | 0.10 | SCN8A | 0.13 (4) | 0.07 (2) | 0.03 (3) | 0.48 (15) | 0.26 (7) | 0.21 (25) |
| 27:5467028 | 1.33 × 10−4 | 0.053 | G | A | 0.36 | 0.30 | 0.08 | - | 0.32 (10) | 0.11 (3) | 0.09 (11) | 0.52 (16) | 0.48 (13) | 0.41 (49) |
| 27:8646723 | 1.87 × 10−4 | 0.066 | G | A | 0.13 | 0.41 | 0.11 | - | 0.06 (2) | 0.04 (1) | 0 (0) | 0.45 (14) | 0.26 (7) | 0.17 (20) |
| 27:8883501 | 2.21 × 10−4 | 0.070 | A | G | 0.15 | 0.37 | 0.10 | ANO6 | 0.13 (4) | 0.04 (1) | 0.01 (1) | 0.42 (13) | 0.26 (7) | 0.17 (20) |
| 27:8767784 | 2.67 × 10−4 | 0.077 | T | A | 0.14 | 0.40 | 0.11 | LOC111092890 | 0.06 (2) | 0.04 (1) | 0 (0) | 0.45 (14) | 0.26 (7) | 0.18 (21) |
| 27:3501246 | 3.35 × 10−4 | 0.082 | C | A | 0.37 | −0.30 | 0.08 | - | 0.03 (1) | 0.11 (3) | 0.2 (24) | 0.29 (9) | 0.33 (9) | 0.47 (56) |
| 27:3054141 | 3.62 × 10−4 | 0.082 | A | G | 0.23 | 0.32 | 0.09 | - | 0.13 (4) | 0.07 (2) | 0.04 (5) | 0.55 (17) | 0.48 (13) | 0.25 (30) |
| 27:8842830 | 4.22 × 10−4 | 0.082 | A | C | 0.13 | 0.38 | 0.11 | LOC111092890 | 0.06 (2) | 0.04 (1) | 0.01 (1) | 0.45 (14) | 0.26 (7) | 0.14 (17) |
| 27:8884575 | 4.22 × 10−4 | 0.082 | A | G | 0.13 | 0.38 | 0.11 | ANO6 | 0.06 (2) | 0.04 (1) | 0.01 (1) | 0.45 (14) | 0.26 (7) | 0.14 (17) |
| 27:1953293 | 4.40 × 10−4 | 0.082 | G | A | 0.23 | 0.33 | 0.09 | LOC111092799 | 0.13 (4) | 0.07 (2) | 0.03 (3) | 0.52 (16) | 0.48 (13) | 0.3 (36) |
| 27:3154712 | 4.58 × 10−4 | 0.082 | A | G | 0.34 | 0.29 | 0.08 | FIGNL2 | 0.26 (8) | 0.15 (4) | 0.08 (10) | 0.55 (17) | 0.41 (11) | 0.4 (48) |
| 27:5603116 | 4.67 × 10−4 | 0.082 | A | G | 0.39 | 0.28 | 0.08 | WNT10B | 0.42 (13) | 0.26 (7) | 0.05 (6) | 0.35 (11) | 0.48 (13) | 0.52 (62) |
| 27:5478927 | 5.41 × 10−4 | 0.083 | A | G | 0.31 | 0.29 | 0.08 | LMBR1L | 0.23 (7) | 0.22 (6) | 0.03 (4) | 0.52 (16) | 0.33 (9) | 0.43 (51) |
| 27:7188905 | 5.43 × 10−4 | 0.083 | A | G | 0.28 | 0.30 | 0.09 | - | 0.23 (7) | 0.11 (3) | 0.04 (5) | 0.48 (15) | 0.44 (12) | 0.34 (41) |
| 27:3104073 | 5.47 × 10−4 | 0.083 | A | G | 0.23 | 0.31 | 0.09 | ANKRD33 | 0.13 (4) | 0.07 (2) | 0.03 (4) | 0.52 (16) | 0.48 (13) | 0.26 (31) |
| 27:5806033 | 6.73 × 10−4 | 0.089 | A | G | 0.22 | 0.32 | 0.10 | CCNT1 | 0.13 (4) | 0.22 (6) | 0.01 (1) | 0.55 (17) | 0.33 (9) | 0.25 (30) |
| 27:5817551 | 6.73 × 10−4 | 0.089 | G | A | 0.22 | 0.32 | 0.10 | - | 0.13 (4) | 0.22 (6) | 0.01 (1) | 0.55 (17) | 0.33 (9) | 0.25 (30) |
| 27:5512765 | 6.89 × 10−4 | 0.089 | A | G | 0.36 | 0.28 | 0.08 | RHEBL1 | 0.29 (9) | 0.11 (3) | 0.08 (9) | 0.52 (16) | 0.48 (13) | 0.47 (56) |
| 27:6657558 | 7.32 × 10−4 | 0.089 | G | A | 0.23 | 0.31 | 0.09 | - | 0.19 (6) | 0.07 (2) | 0.03 (3) | 0.45 (14) | 0.37 (10) | 0.31 (37) |
| 27:6660738 | 7.32 × 10−4 | 0.089 | A | G | 0.23 | 0.31 | 0.09 | - | 0.19 (6) | 0.07 (2) | 0.03 (3) | 0.45 (14) | 0.37 (10) | 0.31 (37) |
| 27:1488082 | 7.65 × 10−4 | 0.090 | G | A | 0.35 | 0.27 | 0.08 | LOC486504 | 0.29 (9) | 0.11 (3) | 0.12 (14) | 0.45 (14) | 0.44 (12) | 0.39 (46) |
| 27:5822515 | 8.18 × 10−4 | 0.093 | G | A | 0.22 | 0.31 | 0.09 | - | 0.13 (4) | 0.22 (6) | 0.01 (1) | 0.55 (17) | 0.37 (10) | 0.25 (30) |
| 27:1719544 | 9.13 × 10−4 | 0.100 | A | G | 0.34 | 0.27 | 0.08 | TARBP2 | 0.26 (8) | 0.07 (2) | 0.17 (20) | 0.45 (14) | 0.41 (11) | 0.31 (37) |
| No. of SNPs | Start SNP | End SNP | Haplotype | p-Value | Genes in Region | Frequency of Homozygosity (No. of Dogs) | ||
|---|---|---|---|---|---|---|---|---|
| Lym. | Carr. | Cont. | ||||||
| 12 | 18:38038090 | 18:38129762 | AACAAAACAACG | 7.75 × 10−8 | LOC106559970, OR08G08, LOC483477, COR5BC3, LOC483479, LOC610022, LOC483480 | 0.06 (2) | 0 (0) | 0 (0) |
| 2 | 18:39123584 | 18:39140112 | AG | 1.65 × 10−7 | LOC100688997 | 0.1 (3) | 0.07 (2) | 0.02 (2) |
| 2 | 18:37724157 | 18:37737740 | CC | 5.77 × 10−7 | LOC100686230 | 0.06 (2) | 0 (0) | 0 (0) |
| 5 | 18:38383741 | 18:38426687 | GGGAG | 6.09 × 10−7 | CTNND1 | 0.06 (2) | 0 (0) | 0 (0) |
| 3 | 18:38456518 | 18:38478390 | AAC | 6.09 × 10−7 | CTNND1, BTBD18 | 0.06 (2) | 0 (0) | 0 (0) |
| 5 | 18:38498673 | 18:38511357 | AAAAT | 2.31 × 10−6 | ZDHHC5 | 0.1 (3) | 0 (0) | 0 (0) |
| 2 | 18:55380537 | 18:55385901 | TG | 3.36 × 10−6 | TMEM109 | 0.52 (16) | 0.41 (11) | 0.24 (28) |
| 2 | 18:37862012 | 18:37867871 | AA | 6.12 × 10−6 | OR5B21 | 0.19 (6) | 0.04 (1) | 0 (0) |
| 2 | 18:37862012 | 18:37867871 | TG | 6.12 × 10−6 | OR5B21 | 0.48 (15) | 0.56 (15) | 0.76 (91) |
| 2 | 18:38317284 | 18:38326192 | AA | 1.07 × 10−5 | OR10C10 | 0.06 (2) | 0 (0) | 0 (0) |
| 14 | 18:13130318 | 18:13325298 | AGAAAAAAGAAGAG | 1.27 × 10−5 | COG5 | 0 (0) | 0 (0) | 0 (0) |
| 3 | 18:41713196 | 18:41740841 | GGG | 4.05 × 10−5 | LOC106559997 | 0.06 (2) | 0.07 (2) | 0.33 (39) |
| 3 | 18:38805092 | 18:38807806 | AGA | 4.45 × 10−5 | SSRP1 | 0.06 (2) | 0 (0) | 0 (0) |
| 2 | 18:41422687 | 18:41436427 | AC | 6.03 × 10−5 | - | 0.1 (3) | 0.07 (2) | 0.35 (42) |
| 3 | 18:39168396 | 18:39195972 | AAA | 6.86 × 10−5 | LOC483503, OR04E05 | 0.1 (3) | 0.04 (1) | 0.01 (1) |
| 5 | 18:39846804 | 18:39860197 | AGGAA | 8.23 × 10−5 | LOC483541, LOC100685982 | 0.16 (5) | 0.04 (1) | 0.01 (1) |
| 2 | 18:40663070 | 18:40667579 | GC | 8.38 × 10−5 | - | 0.45 (14) | 0.22 (6) | 0.15 (18) |
| 3 | 18:38719709 | 18:38732149 | GAA | 8.47 × 10−5 | SLC43A3 | 0.23 (7) | 0.22 (6) | 0.08 (9) |
| No. of SNPs | Start SNP | End SNP | Haplotype | p-Value | Genes in Region | Frequency of Homozygosity (No. of Dogs) | ||
|---|---|---|---|---|---|---|---|---|
| Lym. | Carr. | Cont. | ||||||
| 9 | 27:5104085 | 27:5241391 | AAAAGCGAA | 9.37 × 10−9 | SPATS2 | 0.29 (9) | 0.07 (2) | 0.02 (2) |
| 6 | 27:4662318 | 27:4728424 | GAAGGA | 1.27 × 10−8 | ASIC1, RACGAP1, LOC111092823 | 0.13 (4) | 0.07 (2) | 0 (0) |
| 5 | 27:5304179 | 27:5340118 | GAAAC | 2.04 × 10−8 | PRPH, LOC100856405 | 0.16 (5) | 0.07 (2) | 0.01 (1) |
| 4 | 27:5919807 | 27:5983459 | AGAA | 8.05 × 10−8 | cOR8S6P, LOC102156933, LOC100855426, COR8S14, LOC111092874 | 0.1 (3) | 0.07 (2) | 0 (0) |
| 4 | 27:8010819 | 27:8043205 | AGAA | 1.11 × 10−7 | LOC102154451, LOC102155835 | 0.06 (2) | 0.04 (1) | 0 (0) |
| 5 | 27:3590788 | 27:3615721 | AGAGA | 1.33 × 10−7 | BIN2, SMAGP | 0.13 (4) | 0.07 (2) | 0 (0) |
| 5 | 27:5762992 | 27:5817551 | GAGAG | 1.74 × 10−7 | LOC111092830, CCNT1 | 0.13 (4) | 0.22 (6) | 0.01 (1) |
| 9 | 27:8396219 | 27:8548130 | AGCGCGGGG | 2.27 × 10−7 | SCAF11, ARID2 | 0.06 (2) | 0.04 (1) | 0 (0) |
| 2 | 27:8314156 | 27:8331252 | AG | 2.57 × 10−7 | LOC111092841 | 0.06 (2) | 0.04 (1) | 0 (0) |
| 13 | 27:3197949 | 27:3347721 | GTCGAGGAG GGGC | 3.97 × 10−7 | SCN8A | 0.13 (4) | 0.07 (2) | 0 (0) |
| 4 | 27:5919807 | 27:5983459 | AAAA | 2.70 × 10−6 | cOR8S6P, LOC102156933, LOC100855426, COR8S14, LOC111092874 | 0.16 (5) | 0.11 (3) | 0.45 (53) |
| 3 | 27:5261130 | 27:5265185 | GGA | 4.07 × 10−6 | DNAJC22 | 0.29 (9) | 0.19 (5) | 0.08 (9) |
| 4 | 27:8583844 | 27:8646723 | CAAG | 5.07 × 10−6 | LOC102155300 | 0.06 (2) | 0.04 (1) | 0 (0) |
| 3 | 27:8842830 | 27:8861426 | AAA | 8.77 × 10−6 | LOC111092890 | 0.06 (2) | 0.04 (1) | 0.01 (1) |
| 6 | 27:8883501 | 27:8885135 | AGAAAA | 8.77 × 10−6 | ANO6 | 0.06 (2) | 0.04 (1) | 0.01 (1) |
| 2 | 27:8767784 | 27:8775933 | TG | 1.81 × 10−5 | LOC111092890 | 0.06 (2) | 0.04 (1) | 0 (0) |
| 3 | 27:5281537 | 27:5282177 | CAG | 2.24 × 10−5 | C1QL4 | 0.23 (7) | 0.22 (6) | 0.05 (6) |
| 6 | 27:21881521 | 27:21942273 | AAAGGG | 2.33 × 10−5 | LOC111092834 | 0.03 (1) | 0.04 (1) | 0.02 (2) |
| 3 | 27:5861077 | 27:5870632 | GGA | 2.99 × 10−5 | LYZF2 | 0.35 (11) | 0.3 (8) | 0.12 (14) |
| 4 | 27:1460779 | 27:1471371 | AAGG | 3.26 × 10−5 | LOC486504 | 0.32 (10) | 0.22 (6) | 0.06 (7) |
| 3 | 27:1250330 | 27:1251390 | AGA | 3.96 × 10−5 | LOC607625 | 0.1 (3) | 0.07 (2) | 0.01 (1) |
| 5 | 27:4735827 | 27:4744789 | AAGCA | 3.96 × 10−5 | AQP5, AQP2 | 0.35 (11) | 0.22 (6) | 0.09 (11) |
| 3 | 27:5261130 | 27:5265185 | AGA | 4.21 × 10−5 | DNAJC22 | 0.03 (1) | 0 (0) | 0.13 (15) |
| 3 | 27:3102259 | 27:3104073 | GAA | 4.74 × 10−5 | ANKRD33 | 0.13 (4) | 0.07 (2) | 0.03 (4) |
| 5 | 27:11605144 | 27:11653990 | CGAGA | 5.17 × 10−5 | LOC106557903 | 0.1 (3) | 0.3 (8) | 0.05 (6) |
| 2 | 27:2965369 | 27:2966261 | GC | 5.84 × 10−5 | - | 0.87 (27) | 0.74 (20) | 0.53 (63) |
| 5 | 27:21532742 | 27:21616626 | GCAGC | 7.16 × 10−5 | RASSF8, LOC106557908 | 0.03 (1) | 0.04 (1) | 0.02 (2) |
| 19 | 27:4373882 | 27:4565172 | AAAGAGGAGGAGAGAAAAG | 8.25 × 10−5 | LARP4, FAM186A, LIMA1 | 0.13 (4) | 0.07 (2) | 0.03 (4) |
| 4 | 27:6639027 | 27:6661792 | CGAA | 9.24 × 10−5 | PFKM | 0.19 (6) | 0.07 (2) | 0.03 (3) |
| Location (Chr:BP) | p-Value | q-Value | A1 | A2 | A1 Freq | b | SE | Gene | Frequency of A1A1 (No. of Dogs) | Frequency of A1A2 (No. of Dogs) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lym. | Cont. | Lym. | Cont. | |||||||||
| 13:17399337 | 5.78 × 10−5 | 0.087 | A | C | 0.02 | 0.63 | 0.16 | EXT1 | 0 (0) | 0 (0) | 0.19 (6) | 0.01 (1) |
| 13:18910196 | 3.42 × 10−5 | 0.077 | A | G | 0.02 | 0.69 | 0.17 | DEPTOR | 0 (0) | 0 (0) | 0.16 (5) | 0.01 (1) |
| 13:18913685 | 3.42 × 10−5 | 0.077 | A | G | 0.02 | 0.69 | 0.17 | DEPTOR | 0 (0) | 0 (0) | 0.16 (5) | 0.01 (1) |
| 13:20734477 | 7.72 × 10−5 | 0.087 | A | G | 0.03 | 0.53 | 0.13 | LOC111098540 | 0 (0) | 0 (0) | 0.23 (7) | 0.03 (3) |
| 14:3945297 | 5.97 × 10−5 | 0.084 | A | G | 0.07 | 0.36 | 0.09 | EXOC4 | 0.1 (3) | 0 (0) | 0.19 (6) | 0.08 (10) |
| 14:4095927 | 5.97 × 10−5 | 0.084 | A | G | 0.07 | 0.36 | 0.09 | - | 0.1 (3) | 0 (0) | 0.19 (6) | 0.08 (10) |
| 14:54260890 | 8.46 × 10−5 | 0.084 | A | G | 0.02 | 0.72 | 0.18 | - | 0 (0) | 0 (0) | 0.13 (4) | 0.01 (1) |
| 18:37737740 | 3.07 × 10−4 | 0.066 | C | A | 0.12 | 0.25 | 0.07 | - | 0.06 (2) | 0 (0) | 0.45 (14) | 0.16 (19) |
| 18:37862012 | 4.81 × 10−4 | 0.074 | A | T | 0.17 | 0.21 | 0.06 | - | 0.19 (6) | 0 (0) | 0.32 (10) | 0.24 (28) |
| 18:37867871 | 4.81 × 10−4 | 0.074 | A | G | 0.17 | 0.21 | 0.06 | OR5B21 | 0.19 (6) | 0 (0) | 0.32 (10) | 0.24 (28) |
| 18:38233567 | 1.74 × 10−4 | 0.053 | A | G | 0.11 | 0.29 | 0.08 | - | 0.06 (2) | 0 (0) | 0.42 (13) | 0.13 (15) |
| 18:38350947 | 3.76 × 10−4 | 0.066 | A | G | 0.11 | 0.27 | 0.08 | - | 0.06 (2) | 0 (0) | 0.42 (13) | 0.13 (16) |
| 18:38456518 | 3.76 × 10−4 | 0.066 | A | G | 0.11 | 0.27 | 0.08 | CTNND1 | 0.06 (2) | 0 (0) | 0.42 (13) | 0.13 (16) |
| 18:38502268 | 2.16 × 10−4 | 0.053 | A | G | 0.12 | 0.26 | 0.07 | ZDHHC5 | 0.1 (3) | 0 (0) | 0.39 (12) | 0.16 (19) |
| 18:38507461 | 2.16 × 10−4 | 0.053 | A | G | 0.12 | 0.26 | 0.07 | ZDHHC5 | 0.1 (3) | 0 (0) | 0.39 (12) | 0.16 (19) |
| 18:38510335 | 2.16 × 10−4 | 0.053 | A | G | 0.12 | 0.26 | 0.07 | ZDHHC5 | 0.1 (3) | 0 (0) | 0.39 (12) | 0.16 (19) |
| 18:38704682 | 4.49 × 10−5 | 0.053 | A | G | 0.32 | 0.20 | 0.05 | - | 0.26 (8) | 0.08 (9) | 0.55 (17) | 0.37 (44) |
| 18:38719709 | 1.45 × 10−4 | 0.053 | G | A | 0.30 | 0.18 | 0.05 | - | 0.23 (7) | 0.08 (9) | 0.55 (17) | 0.34 (41) |
| 18:39140112 | 8.21 × 10−5 | 0.053 | G | A | 0.26 | 0.22 | 0.06 | LOC100688997 | 0.16 (5) | 0.03 (4) | 0.58 (18) | 0.34 (41) |
| 18:39727438 | 3.38 × 10−4 | 0.066 | G | A | 0.31 | 0.17 | 0.05 | - | 0.29 (9) | 0.08 (9) | 0.45 (14) | 0.35 (42) |
| 18:40281122 | 8.35 × 10−5 | 0.053 | A | G | 0.41 | 0.18 | 0.05 | - | 0.42 (13) | 0.13 (16) | 0.42 (13) | 0.45 (53) |
| 18:40286669 | 9.66 × 10−5 | 0.053 | G | A | 0.42 | 0.18 | 0.05 | - | 0.42 (13) | 0.13 (16) | 0.42 (13) | 0.47 (56) |
| 18:40418483 | 2.44 × 10−4 | 0.056 | A | G | 0.47 | 0.17 | 0.05 | - | 0.42 (13) | 0.16 (19) | 0.48 (15) | 0.52 (62) |
| 18:40653765 | 2.05 × 10−4 | 0.053 | A | C | 0.47 | 0.18 | 0.05 | - | 0.45 (14) | 0.15 (18) | 0.45 (14) | 0.53 (63) |
| 18:40667579 | 1.95 × 10−4 | 0.053 | C | A | 0.46 | 0.17 | 0.05 | - | 0.45 (14) | 0.15 (18) | 0.45 (14) | 0.5 (60) |
| 18:41229735 | 6.77 × 10−5 | 0.053 | A | G | 0.43 | 0.19 | 0.05 | LOC100684610 | 0.45 (14) | 0.13 (15) | 0.42 (13) | 0.49 (58) |
| 18:41436427 | 1.54 × 10−4 | 0.053 | C | A | 0.52 | −0.17 | 0.05 | - | 0.1 (3) | 0.35 (42) | 0.42 (13) | 0.45 (53) |
| 18:41713196 | 4.34 × 10−4 | 0.073 | A | G | 0.15 | 0.24 | 0.07 | - | 0.06 (2) | 0.03 (3) | 0.45 (14) | 0.18 (21) |
| 18:41726488 | 1.92 × 10−4 | 0.053 | A | G | 0.15 | 0.26 | 0.07 | - | 0.06 (2) | 0.02 (2) | 0.45 (14) | 0.18 (22) |
| 18:41854962 | 1.82 × 10−4 | 0.053 | C | G | 0.13 | 0.24 | 0.06 | - | 0.1 (3) | 0.01 (1) | 0.32 (10) | 0.17 (20) |
| 18:42270324 | 3.78 × 10−4 | 0.066 | A | G | 0.07 | 0.33 | 0.09 | - | 0 (0) | 0.01 (1) | 0.35 (11) | 0.08 (9) |
| 27:3054141 | 8.03 × 10−4 | 0.096 | A | G | 0.22 | 0.18 | 0.05 | - | 0.13 (4) | 0.04 (5) | 0.55 (17) | 0.25 (30) |
| 27:3154712 | 2.20 × 10−4 | 0.041 | A | G | 0.34 | 0.18 | 0.05 | FIGNL2 | 0.26 (8) | 0.08 (10) | 0.55 (17) | 0.4 (48) |
| 27:3197949 | 7.78 × 10−5 | 0.027 | G | A | 0.18 | 0.23 | 0.06 | SCN8A | 0.13 (4) | 0.03 (3) | 0.48 (15) | 0.21 (25) |
| 27:3234647 | 4.84 × 10−5 | 0.027 | G | A | 0.17 | 0.24 | 0.06 | SCN8A | 0.13 (4) | 0.03 (3) | 0.48 (15) | 0.19 (23) |
| 27:3501246 | 1.13 × 10−4 | 0.027 | C | A | 0.38 | −0.18 | 0.05 | - | 0.03 (1) | 0.2 (24) | 0.29 (9) | 0.47 (56) |
| 27:3594560 | 2.29 × 10−5 | 0.027 | G | A | 0.14 | 0.27 | 0.06 | BIN2 | 0.13 (4) | 0 (0) | 0.45 (14) | 0.18 (21) |
| 27:4561667 | 7.57 × 10−4 | 0.095 | A | C | 0.23 | 0.18 | 0.05 | LIMA1 | 0.13 (4) | 0.05 (6) | 0.58 (18) | 0.25 (30) |
| 27:5112245 | 6.08 × 10−5 | 0.027 | A | G | 0.22 | 0.20 | 0.05 | SPATS2 | 0.29 (9) | 0.02 (2) | 0.39 (12) | 0.27 (32) |
| 27:5364442 | 4.76 × 10−5 | 0.027 | A | G | 0.17 | 0.24 | 0.06 | LOC111092735 | 0.16 (5) | 0.01 (1) | 0.42 (13) | 0.22 (26) |
| 27:5467028 | 1.07 × 10−4 | 0.027 | G | A | 0.36 | 0.18 | 0.05 | - | 0.32 (10) | 0.09 (11) | 0.52 (16) | 0.41 (49) |
| 27:5478927 | 2.36 × 10−4 | 0.042 | A | G | 0.30 | 0.19 | 0.05 | LMBR1L | 0.23 (7) | 0.03 (4) | 0.52 (16) | 0.43 (51) |
| 27:5512765 | 5.03 × 10−4 | 0.072 | A | G | 0.36 | 0.17 | 0.05 | RHEBL1 | 0.29 (9) | 0.08 (9) | 0.52 (16) | 0.47 (56) |
| 27:5603116 | 4.20 × 10−4 | 0.063 | A | G | 0.37 | 0.17 | 0.05 | WNT10B | 0.42 (13) | 0.05 (6) | 0.35 (11) | 0.52 (62) |
| 27:5806033 | 1.17 × 10−4 | 0.027 | A | G | 0.19 | 0.24 | 0.06 | CCNT1 | 0.13 (4) | 0.01 (1) | 0.55 (17) | 0.25 (30) |
| 27:5817551 | 1.17 × 10−4 | 0.027 | G | A | 0.19 | 0.24 | 0.06 | - | 0.13 (4) | 0.01 (1) | 0.55 (17) | 0.25 (30) |
| 27:5822515 | 1.17 × 10−4 | 0.027 | G | A | 0.19 | 0.24 | 0.06 | - | 0.13 (4) | 0.01 (1) | 0.55 (17) | 0.25 (30) |
| 27:7188905 | 5.73 × 10−4 | 0.078 | A | G | 0.27 | 0.18 | 0.05 | - | 0.23 (7) | 0.04 (5) | 0.48 (15) | 0.34 (41) |
| 27:8331252 | 3.49 × 10−5 | 0.027 | G | A | 0.11 | 0.30 | 0.07 | - | 0.06 (2) | 0 (0) | 0.45 (14) | 0.13 (15) |
| 27:8646723 | 8.78 × 10−5 | 0.027 | G | A | 0.13 | 0.28 | 0.07 | - | 0.06 (2) | 0 (0) | 0.45 (14) | 0.17 (20) |
| 27:8767784 | 1.51 × 10−4 | 0.030 | T | A | 0.13 | 0.26 | 0.07 | LOC111092890 | 0.06 (2) | 0 (0) | 0.45 (14) | 0.18 (21) |
| 27:8842830 | 2.80 × 10−4 | 0.044 | A | C | 0.12 | 0.24 | 0.07 | LOC111092890 | 0.06 (2) | 0.01 (1) | 0.45 (14) | 0.14 (17) |
| 27:8883501 | 1.39 × 10−4 | 0.030 | A | G | 0.14 | 0.24 | 0.06 | ANO6 | 0.13 (4) | 0.01 (1) | 0.42 (13) | 0.17 (20) |
| 27:8884575 | 2.80 × 10−4 | 0.044 | A | G | 0.12 | 0.24 | 0.07 | ANO6 | 0.06 (2) | 0.01 (1) | 0.45 (14) | 0.14 (17) |
| 27:8892980 | 6.60 × 10−5 | 0.027 | C | G | 0.22 | 0.21 | 0.05 | ANO6 | 0.16 (5) | 0.04 (5) | 0.55 (17) | 0.25 (30) |
| 27:11672173 | 6.00 × 10−4 | 0.078 | A | C | 0.16 | 0.23 | 0.07 | LOC106557903 | 0.03 (1) | 0 (0) | 0.55 (17) | 0.24 (28) |
| No. of SNPs | Start SNP | End SNP | Haplotype | Odds Ratio | p-Value | Genes in Region | Frequency of Homozygosity (No. of Dogs) | |
|---|---|---|---|---|---|---|---|---|
| Lym. | Cont. | |||||||
| 14 | 18:38233567 | 18:38383741 | AAGAGGGAAAAAGG | 5.61 | 7.66 × 10−5 | LOC483485, LOC610080, LOC106559985, OR10C10, LOC610127, COR5BA2 | 0.06 (2) | 0 (0) |
| 3 | 18:38502268 | 18:38510335 | AAA | 4.91 | 7.86 × 10−5 | ZDHHC5 | 0.1 (3) | 0 (0) |
| 3 | 18:38502268 | 18:38510335 | GGG | 0.204 | 7.86 × 10−5 | ZDHHC5 | 0.52 (16) | 0.84 (100) |
| 2 | 18:37862012 | 18:37867871 | AA | 3.98 | 9.97 × 10−5 | OR5B21 | 0.19 (6) | 0 (0) |
| 2 | 18:37862012 | 18:37867871 | TG | 0.251 | 9.97 × 10−5 | OR5B21 | 0.48 (15) | 0.76 (91) |
| No. of SNPs | Start SNP | End SNP | Haplotype | Odds Ratio | p-Value | Genes in Region | Frequency of Homozygosity (No. of Dogs) | |
|---|---|---|---|---|---|---|---|---|
| Lym. | Cont. | |||||||
| 8 | 27:5112245 | 27:5241391 | AAAGCGAA | 4.94 | 2.82 × 10−6 | SPATS2 | 0.29 (9) | 0.02 (2) |
| 2 | 27:3590788 | 27:3594560 | AG | 6.26 | 5.33 × 10−6 | BIN2 | 0.13 (4) | 0 (0) |
| 12 | 27:3211312 | 27:3347721 | TCGAGGAGGGGC | 5.95 | 7.53 × 10−6 | SCN8A | 0.13 (4) | 0 (0) |
| 2 | 27:5806033 | 27:5817551 | AG | 5.31 | 1.51 × 10−5 | CCNT1 | 0.13 (4) | 0.01 (1) |
| 2 | 27:5806033 | 27:5817551 | GA | 0.188 | 1.51 × 10−5 | CCNT1 | 0.32 (10) | 0.74 (88) |
| 4 | 27:8752431 | 27:8792430 | GTGG | 5.24 | 6.12 × 10−5 | LOC111092890 | 0.06 (2) | 0 (0) |
| 2 | 27:5592820 | 27:5603116 | CA | 3.65 | 6.83 × 10−5 | WNT10B | 0.42 (13) | 0.05 (6) |
| Chromosome | Prevalence | V(G)/Vp ± SE (%) | V(G)/Vp_L ± SE (%) | p-Value |
|---|---|---|---|---|
| All autosomes | 0.025 | 100 ± 20.52 | 106.09 ± 21.77 | 1.43 × 10−6 |
| 0.05 | 129.37 ± 26.55 | |||
| 0.1 | 160.4 ± 32.92 | |||
| Chr 18 | 0.025 | 31.27 ± 12.77 | 33.17 ± 13.54 | 1.21 × 10−3 |
| 0.05 | 40.45 ± 16.52 | |||
| 0.1 | 50.16 ± 20.48 | |||
| Chr 18 37 to 56 Mb region | 0.025 | 40.91 ± 12.1 | 43.4 ± 12.83 | 7.10 × 10−6 |
| 0.05 | 52.93 ± 15.65 | |||
| 0.1 | 65.63 ± 19.4 | |||
| Chr 27 | 0.025 | 40.71 ± 11.26 | 43.19 ± 11.94 | 1.34 × 10−6 |
| 0.05 | 52.67 ± 14.56 | |||
| 0.1 | 65.3 ± 18.06 | |||
| Chr 27 1 to 9 Mb region | 0.025 | 26.12 ± 9.6 | 27.71 ± 10.18 | 2.50 × 10−7 |
| 0.05 | 33.79 ± 12.42 | |||
| 0.1 | 41.9 ± 15.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soh, P.X.Y.; Khatkar, M.S.; Williamson, P. Lymphoma in Border Collies: Genome-Wide Association and Pedigree Analysis. Vet. Sci. 2023, 10, 581. https://doi.org/10.3390/vetsci10090581
Soh PXY, Khatkar MS, Williamson P. Lymphoma in Border Collies: Genome-Wide Association and Pedigree Analysis. Veterinary Sciences. 2023; 10(9):581. https://doi.org/10.3390/vetsci10090581
Chicago/Turabian StyleSoh, Pamela Xing Yi, Mehar Singh Khatkar, and Peter Williamson. 2023. "Lymphoma in Border Collies: Genome-Wide Association and Pedigree Analysis" Veterinary Sciences 10, no. 9: 581. https://doi.org/10.3390/vetsci10090581
APA StyleSoh, P. X. Y., Khatkar, M. S., & Williamson, P. (2023). Lymphoma in Border Collies: Genome-Wide Association and Pedigree Analysis. Veterinary Sciences, 10(9), 581. https://doi.org/10.3390/vetsci10090581