
Correlating Pure Component Properties with MOSCED Solubility Parameters: Enthalpy of Vaporization and Vapor Pressure



Abstract
1. Introduction
2. Method
2.1. Theory
2.2. Data Compilation and Regression
3. Results and Discussion
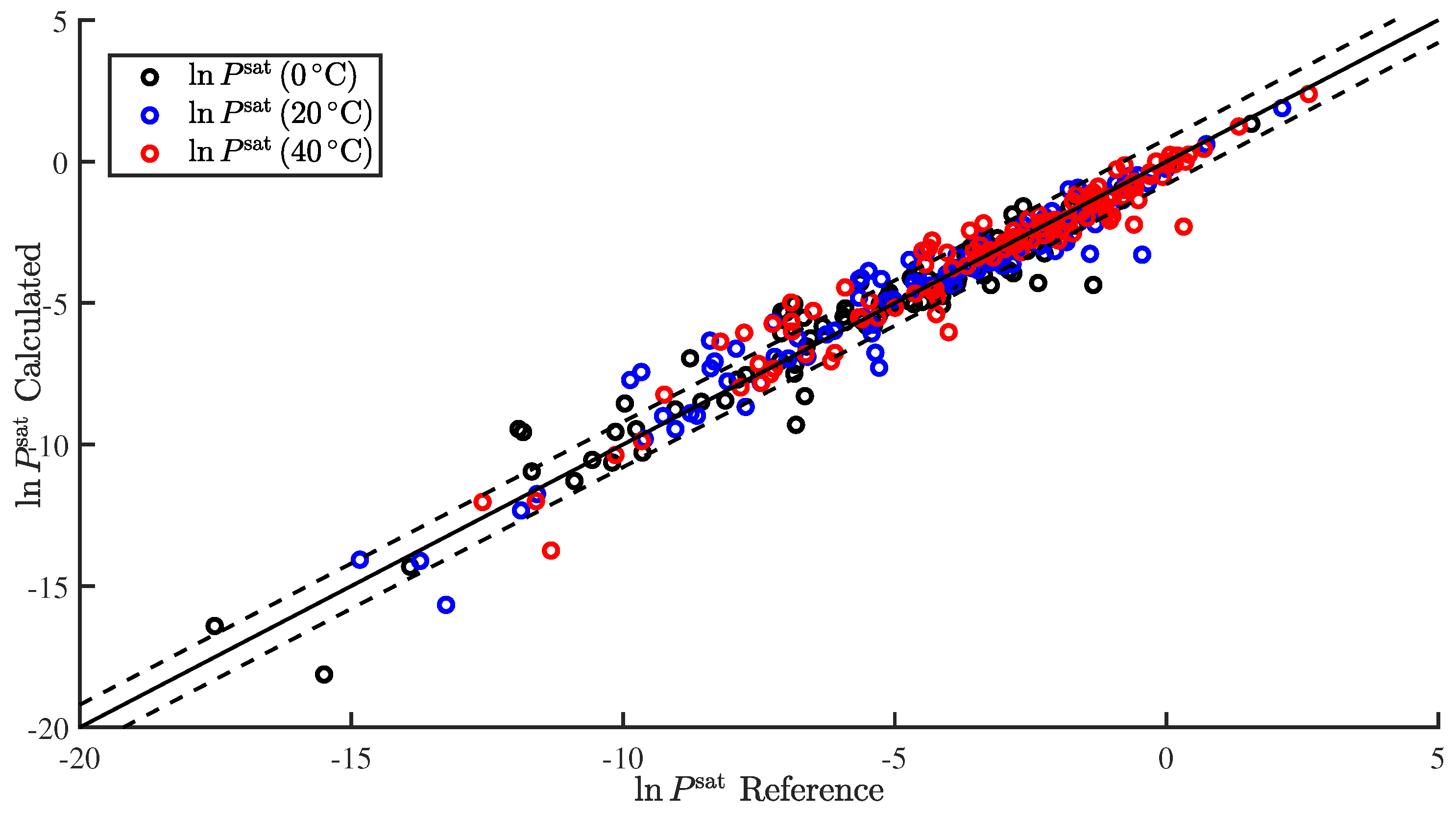
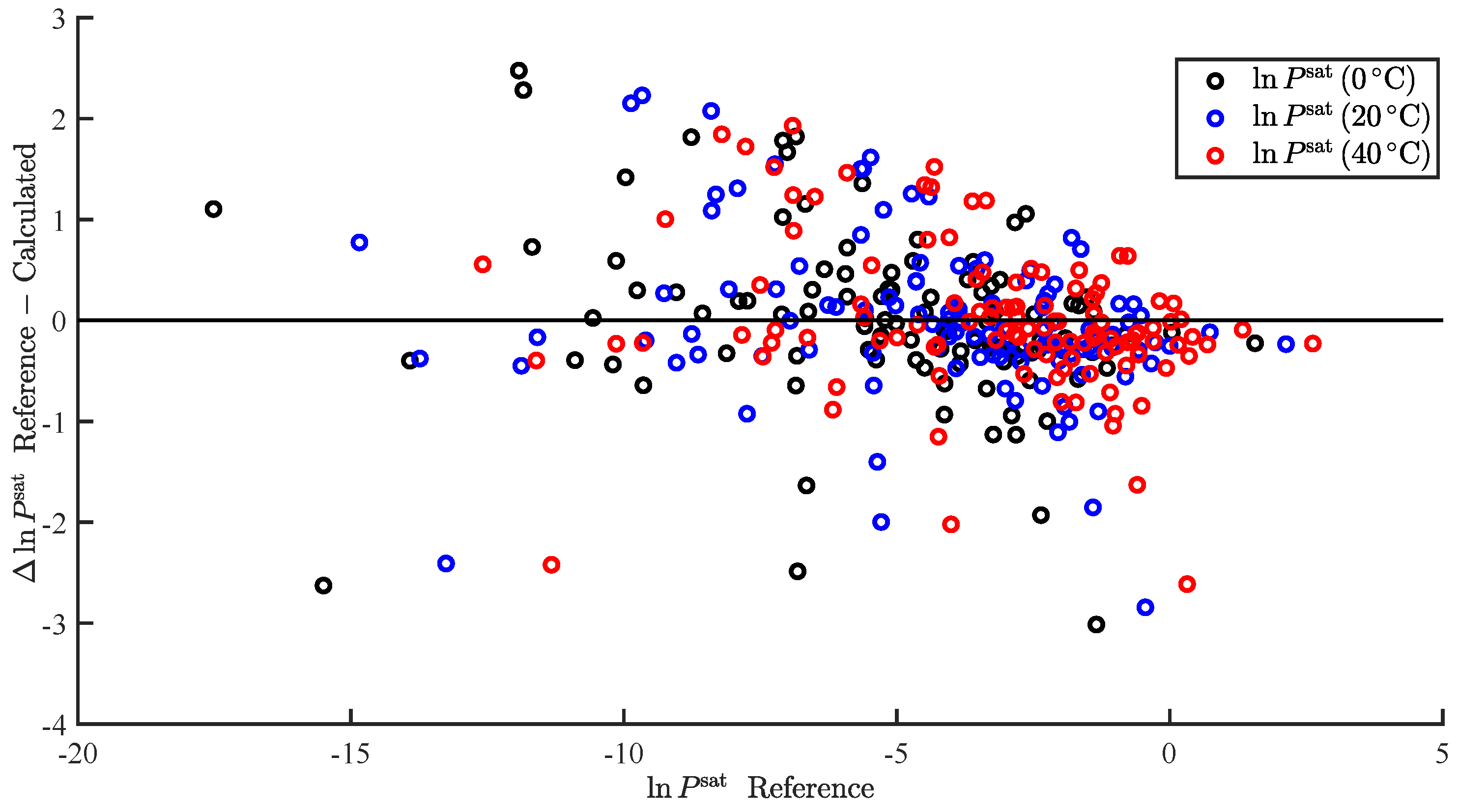
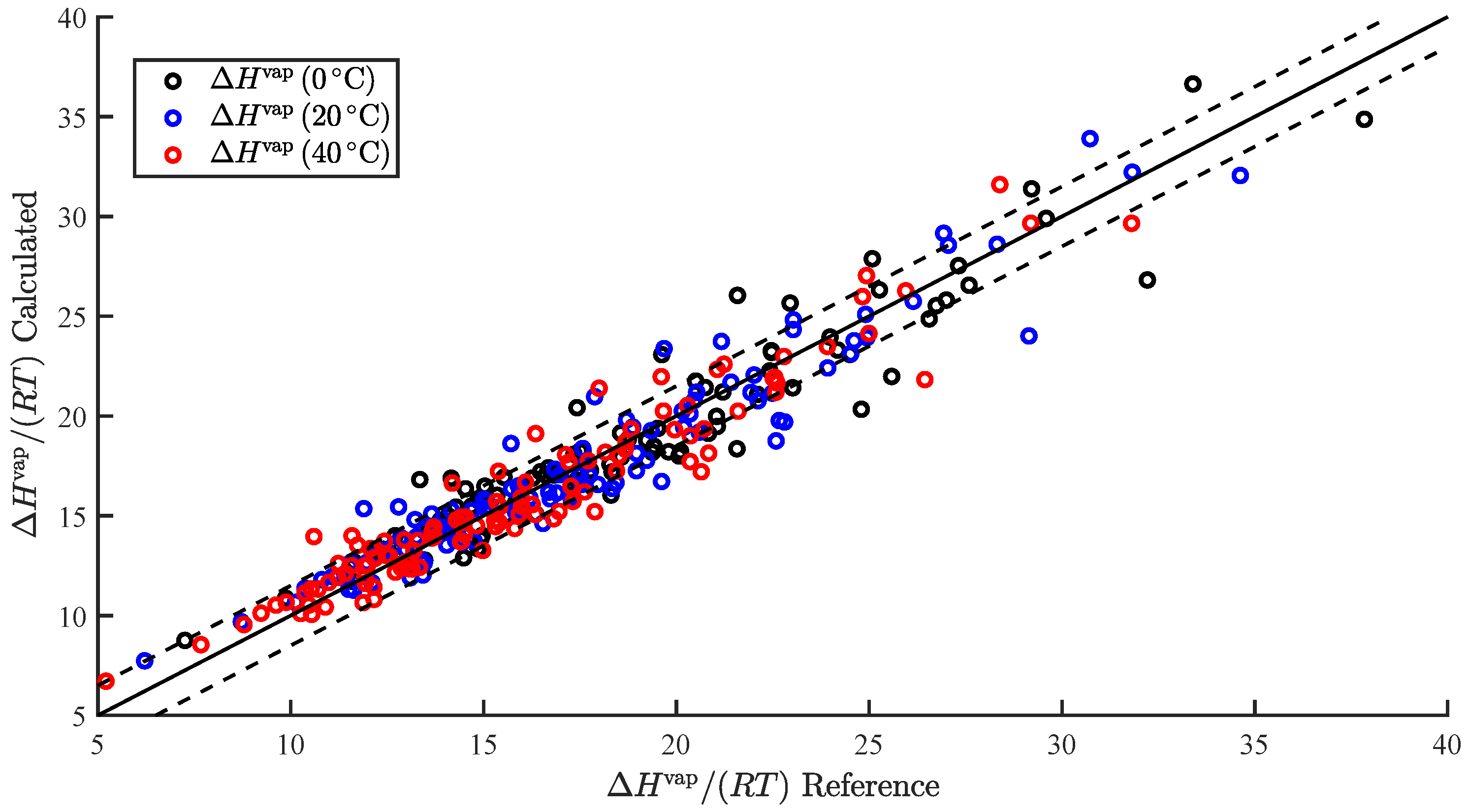
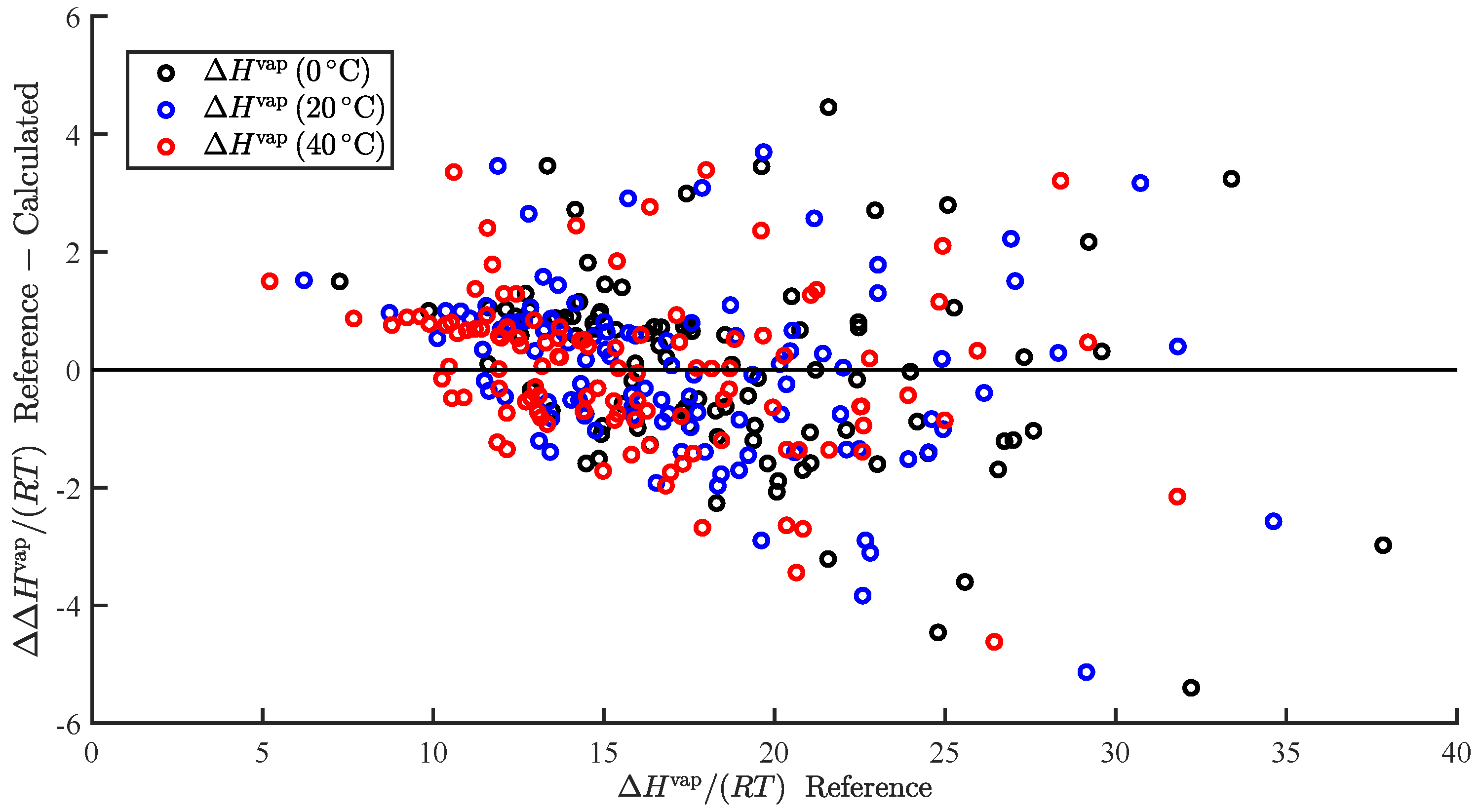
3.1. Regression
3.2. Temperature-Dependent
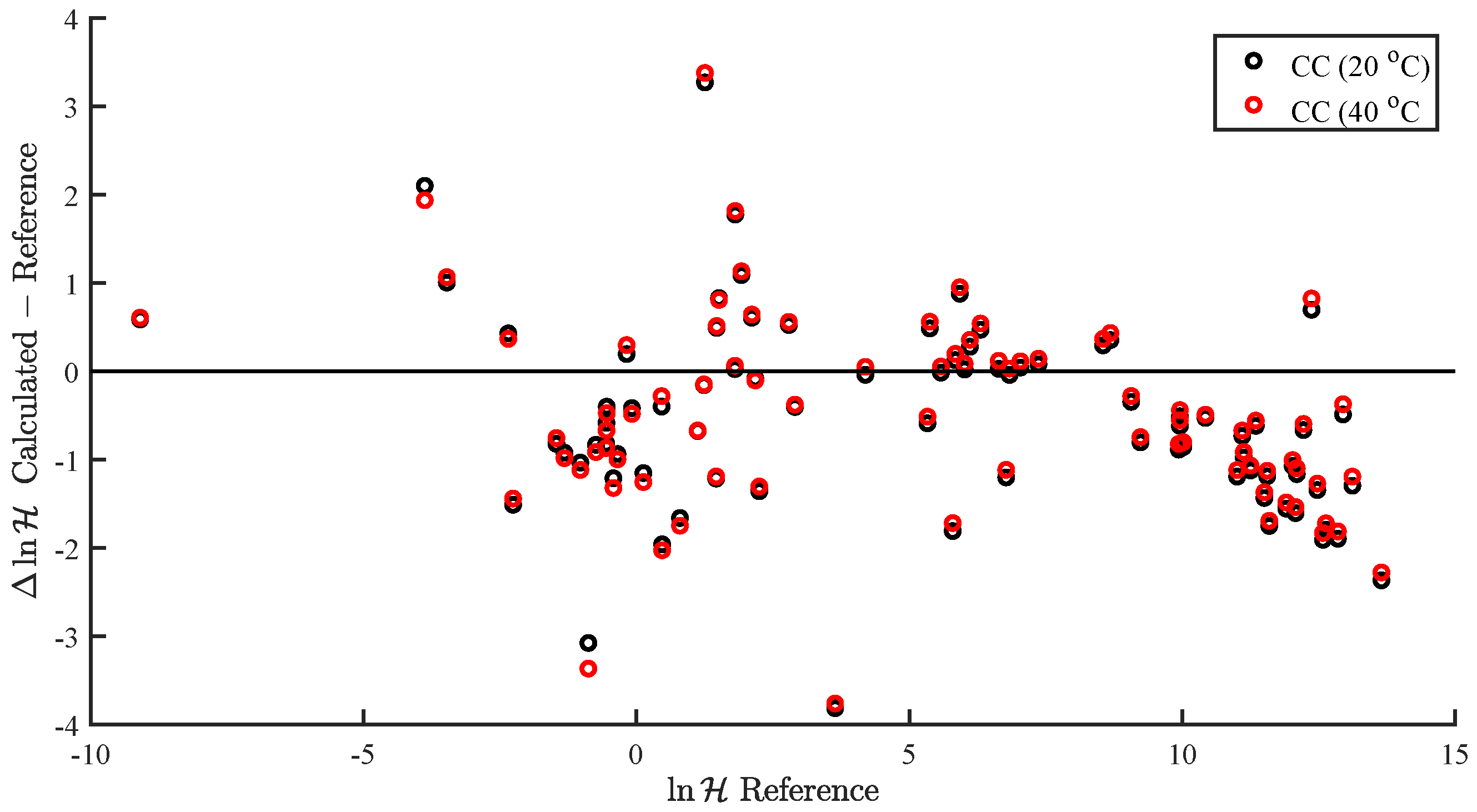
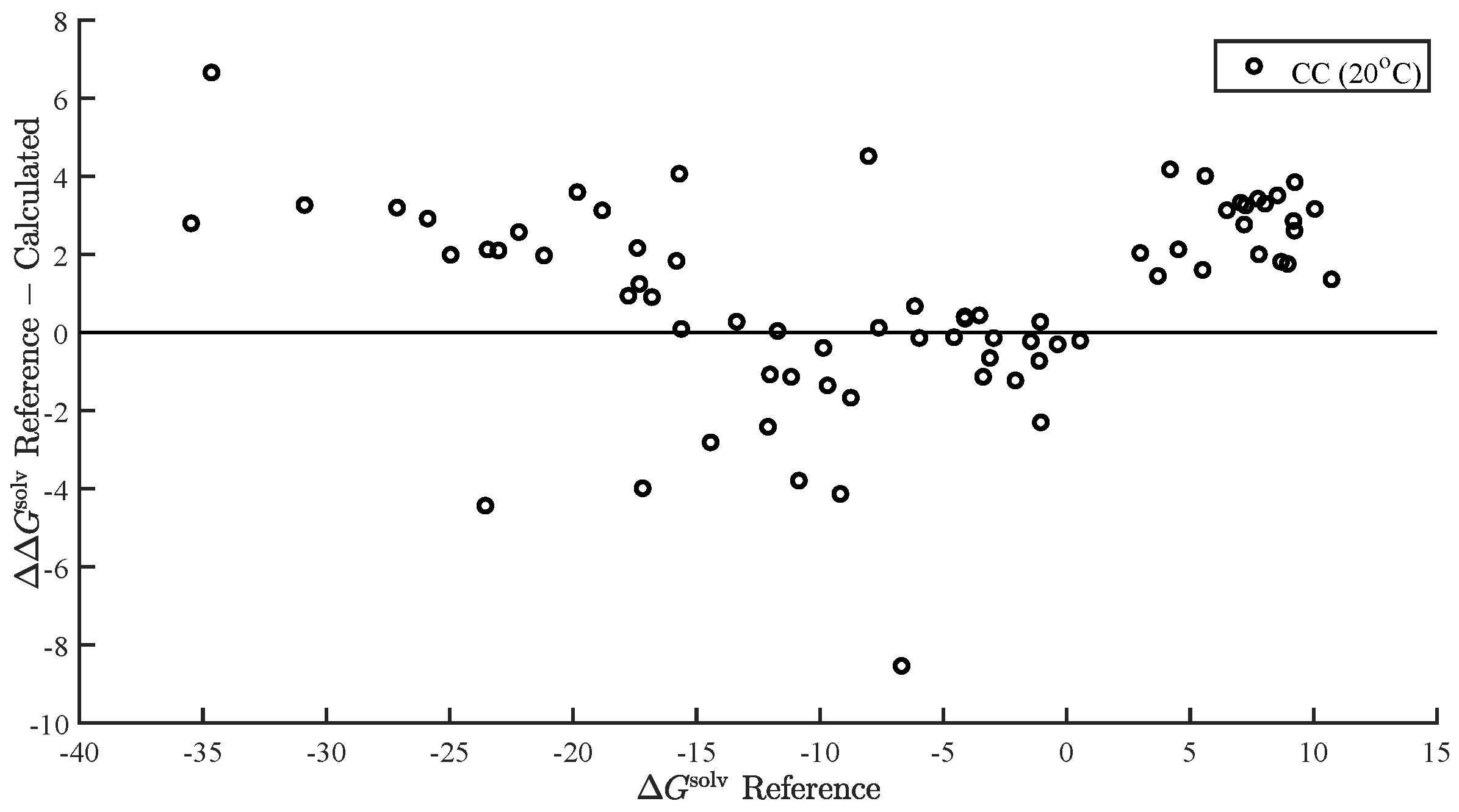
3.3. and in Water
3.4. Other Correlations
4. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wankat, P.C. Separation Process Engineering: Includes Mass Transfer, 3rd ed.; Pearson Education, Inc.: Upper Saddle River, NJ, USA, 2012. [Google Scholar]
- Gmehling, J.; Menke, J.; Krafcyzk, J.; Fischer, K. Azeotropic Data, Part 1, 2nd ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2004. [Google Scholar]
- Materials for Separation Technologies: Energy and Emission Reduction Opportunities; Technical Report; U.S. Department of Energy: Washington, DC, USA, 2005.
- Prausnitz, J.M.; Lichtenthaler, R.N.; de Azevedo, E.G. Molecular Thermodynamics of Fluid-phase Equilibria, 2nd ed.; Prentice-Hall, Inc.: Englewood Cliffs, NJ, USA, 1986. [Google Scholar]
- Hildebrand, J.H.; Prausnitz, J.M.; Scott, R.L. Regular and Related Solutions; Van Nostrand Reinhold Company: New York, NY, USA, 1970. [Google Scholar]
- Hansen, C.M. The Universality of the Solubility Parameter. Ind. Eng. Chem. Prod. Res. Dev. 1969, 8, 2–11. [Google Scholar] [CrossRef]
- Panayiotou, C. Redefining solubility parameters: The partial solvation parameters. Phys. Chem. Chem. Phys. 2012, 14, 3882–3908. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, E.; Panayiotou, C. A new expanded solubility parameter approach. Int. J. Pharm. 2012, 426, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Beerbower, A.; Wu, P.L.; Martin, A. Expanded Solubility Parameter Approach I: Naphthalene and Benzoic Acid in Individual Solvents. J. Pharm. Sci. 1984, 73, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Louwerse, M.J.; Maldonado, A.; Rousseau, S.; Moreau-Masselon, C.; Roux, B.; Rothenberg, G. Revisiting Hansen Solubility Parameters by Including Thermodynamics. ChemPhysChem 2017, 18, 2999–3006. [Google Scholar] [CrossRef]
- Blanks, R.F.; Prausnitz, J.M. Thermodynamics of Polymer Solubility in Polar and Nonpolar Systems. Ind. Eng. Chem. Fundam. 1964, 3, 1–8. [Google Scholar] [CrossRef]
- Tijssen, R.; Billiet, H.A.H.; Schoenmakers, P.J. Use of the solubility parameter for predicting selectivity and retention in chromatography. J. Chromatogr. A 1976, 122, 185–203. [Google Scholar] [CrossRef]
- Krooshof, G.J.P.; Tuinier, R.; de With, G. Generalization of Guggenheim’s combinatorial activity coefficient equation. J. Mol. Liq. 2018, 266, 467–471. [Google Scholar] [CrossRef]
- Krooshof, G.J.P.; Tuinier, R.; de With, G. On the calculation of nearest neighbors in activity coefficient models. Fluid Phase Equilib. 2018, 465, 10–23. [Google Scholar] [CrossRef]
- Thomas, E.R.; Eckert, C.A. Prediction of limiting activity coefficients by a modified separation of cohesive energy density model and UNIFAC. Ind. Eng. Chem. Proc. Des. Dev. 1984, 23, 194–209. [Google Scholar] [CrossRef]
- Park, J.H.; Carr, P.W. Predictive Ability of the MOSCED and UNIFAC Activity Coefficient Estimation Methods. Anal. Chem. 1987, 59, 2596–2602. [Google Scholar] [CrossRef]
- Howell, W.J.; Karachewski, A.M.; Stephenson, K.M.; Eckert, C.A.; Park, J.H.; Carr, P.W.; Rutan, S.C. An Improved MOSCED Equation for the Prediction and Application of Infinite Dilution Activity Coefficients. Fluid Phase Equilib. 1989, 52, 151–160. [Google Scholar] [CrossRef]
- Hait, M.J.; Liotta, C.L.; Eckert, C.A.; Bergmann, D.L.; Karachewski, A.M.; Dallas, A.J.; Eikens, D.I.; Li, J.J.; Carr, P.W.; Poe, R.B.; et al. Space Predictor for Infinite Dilution Activity Coefficients. Ind. Eng. Chem. Res. 1993, 32, 2905–2914. [Google Scholar] [CrossRef]
- Castells, C.B.; Carr, P.W.; Eikens, D.I.; Bush, D.; Eckert, C.A. Comparative Study of Semitheoretical Models for Predicting Infinite Dilution Activity Coefficients of Alkanes in Organic Solvents. Ind. Eng. Chem. Res. 1999, 38, 4104–4109. [Google Scholar] [CrossRef]
- Lazzaroni, M.J.; Bush, D.; Eckert, C.A.; Frank, T.C.; Gupta, S.; Olson, J.D. Revision of MOSCED Parameters and Extension to Solid Solubility Calculations. Ind. Eng. Chem. Res. 2005, 44, 4075–4083. [Google Scholar] [CrossRef]
- Draucker, L.C.; Janakat, M.; Lazzaroni, M.J.; Bush, D.; Eckert, C.A.; Frank, T.C.; Olson, J.D. Experimental determination and model prediction of solid solubility of multifunctional compounds in pure and mixed nonelectrolyte solvents. Ind. Eng. Chem. Res. 2007, 46, 2198–2204. [Google Scholar] [CrossRef]
- Frank, T.C.; Anderson, J.J.; Olson, J.D.; Eckert, C.A. Application of MOSCED and UNIFAC to screen hydrophobic solvents for extraction of hydrogen-bonding organics from aqueous solution. Ind. Eng. Chem. Res. 2007, 46, 4621–4625. [Google Scholar] [CrossRef]
- Widenski, D.J.; Abbas, A.; Romagnoli, J.A. Use of Predictive Solubility Models for Isothermal Antisolvent Crystallization Modeling and Optimization. Ind. Eng. Chem. Res. 2011, 50, 8304–8313. [Google Scholar] [CrossRef]
- Dhakal, P.; Roese, S.N.; Stalcup, E.M.; Paluch, A.S. Application of MOSCED to Predict Hydration Free Energies, Henry’s Constants, Octanol/Water Partition Coefficients, and Isobaric Azeotropic Vapor-Liquid Equilibrium. J. Chem. Eng. Data 2018, 63, 352–364. [Google Scholar] [CrossRef]
- Eckert, C.A.; Newman, B.A.; Nicolaides, G.L.; Long, T.C. Measurement and Application of Limiting Activity Coefficients. AIChE J. 1981, 27, 33–40. [Google Scholar] [CrossRef]
- Missen, R.W. On Criteria for Occurence of Azeotropes in Isothermal and Isobaric Binary Systems. Can. J. Chem. Eng. 2005, 83, 667–674. [Google Scholar] [CrossRef]
- Brandani, V. Use of Infinite-Dilution Activity Coefficients for Predicting Azeotrope Formation at Constant Temperature and Partial Miscibility in Binary Liquid Mixtures. Ind. Eng. Chem. Fundam. 1974, 13, 154–156. [Google Scholar] [CrossRef]
- Schreiber, L.B.; Eckert, C.A. Use of Infinite Dilution Activity Coefficients with Wilson’s Equation. Ind. Eng. Chem. Process Des. Develop. 1971, 10, 572–576. [Google Scholar] [CrossRef]
- Dhakal, P.; Roese, S.N.; Stalcup, E.M.; Paluch, A.S. GC-MOSCED: A Group Contribution Method for Predicting MOSCED Parameters with Application to Limiting Activity Coefficients in Water and Octanol/Water Partition Coefficients. Fluid Phase Equilib. 2018, 470, 232–240. [Google Scholar] [CrossRef]
- Gnap, M.; Elliott, J.R. Estimation of MOSCED parameters from the COSMO-SAC database. Fluid Phase Equilib. 2018, 470, 241–248. [Google Scholar] [CrossRef]
- Churchill, B.; Acree, E., Jr.; Abraham, M.H. Development of Abraham model expressions for predicting the standard molar enthalpies of vaporization of organic compounds at 298.15 K. Thermochim. Acta 2019, 681, 178372. [Google Scholar] [CrossRef]
- Quina, F.H.; Carroll, F.A.; Cheuy, D.H. A Linear Solvation Energy Relationship to Predict Vapor Pressure from Molecular Structure. J. Braz. Chem. Soc. 2005, 16, 1010–1016. [Google Scholar] [CrossRef]
- Vitha, M.; Carr, P.W. The chemical interpretation and practice of linear solvation energy relationships in chromatography. J. Chromatogr. A 2006, 1126, 143–194. [Google Scholar] [CrossRef]
- Dhakal, P.; Ouimet, J.A.; Roese, S.N.; Paluch, A.S. MOSCED parameters for 1-n-alkyl-3-methylimidazolium-based ionic liquids: Application to limiting activity coefficients and intuitive entrainer selection for extractive distillation processes. J. Mol. Liq. 2019, 293, 111552. [Google Scholar] [CrossRef]
- Dhakal, P.; Weise, A.R.; Fritsch, M.C.; O’Dell, C.M.; Paluch, A.S. Expanding the Solubility Parameter Method MOSCED to Pyridinium, Quinolinium, Pyrrolidinium, Piperidinium, Bicyclic, Morpholinium, Ammonium, Phosphonium, and Sulfonium Based Ionic Liquids. ACS Omega 2020, 5, 3863–3877. [Google Scholar] [CrossRef]
- Dhakal, P.; Roese, S.N.; Lucas, M.A.; Paluch, A.S. Predicting Limiting Activity Coefficients and Phase Behavior from Molecular Structure: Expanding MOSCED to Alkanediols Using Group Contribution Methods and Electronic Structure Calculations. J. Chem. Eng. Data 2018, 63, 2586–2598. [Google Scholar] [CrossRef]
- Ley, R.T.; Fuerst, G.B.; Redeker, B.N.; Paluch, A.S. Developing a Predictive Form of MOSCED for Nonelectrolyte Solids Using Molecular Simulation: Application to Acetanilide, Acetaminophen, and Phenacetin. Ind. Eng. Chem. Res. 2016, 55, 5415–5430. [Google Scholar] [CrossRef]
- Phifer, J.R.; Solomon, K.J.; Young, K.L.; Paluch, A.S. Computing MOSCED parameters of nonelectrolyte solids with electronic structure methods in SMD and SM8 continuum solvents. AIChE J. 2017, 63, 781–791. [Google Scholar] [CrossRef]
- Phifer, J.R.; Cox, C.E.; da Silva, L.F.; Nogueira, G.G.; Barbosa, A.K.P.; Ley, R.T.; Bozada, S.M.; O’Loughlin, E.J.; Paluch, A.S. Predicting the equilibrium solubility of solid polycyclic aromatic hydrocarbons and dibenzothiophene using a combination of MOSCED plus molecular simulation or electronic structure calculations. Mol. Phys. 2017, 115, 1286–1300. [Google Scholar] [CrossRef]
- Marrero, J.; Gani, R. Group-contribution based estimation of pure component properties. Fluid Phase Equilib. 2001, 183–184, 183–208. [Google Scholar] [CrossRef]
- Hukkerikar, A.S.; Sarup, B.; Kate, A.T.; Abildskov, J.; Sin, G.; Gani, R. Group-contribution+ (GC+) based estimation of properties of pure components: Improved property estimation and uncertainty analysis. Fluid Phase Equilib. 2012, 321, 25–43. [Google Scholar] [CrossRef]
- Gharagheizi, F.; Eslamimanesh, A.; Ilani-Kashkouli, P.; Mohammadi, A.H.; Richon, D. Determination of Vapor Pressure of Chemical Compounds: A Group Contribution Model for an Extremely Large Database. Ind. Eng. Chem. Res. 2012, 51, 7119–7125. [Google Scholar] [CrossRef]
- Poe, R.B.; Rutan, S.C.; Hait, M.J.; Eckert, C.A.; Carr, P.W. Developing Models for Infinite Dilution Activity-Coefficients Using Factor-Analysis Methods. Anal. Chim. Acta 1993, 277, 223–238. [Google Scholar] [CrossRef]
- Watson, K.M. Thermodynamics of the Liquid State. Ind. Eng. Chem. 1943, 35, 398–406. [Google Scholar] [CrossRef]
- Yaws, C.L. Yaws’ Handbook of Thermodynamic and Physical Properties of Chemical Compounds; Knovel: New York, NY, USA, 2003; ISBN 978-1-59124-444-8. [Google Scholar]
- Verevkin, S.P. Measurement and Prediction of the Monocarboxylic Acids Thermochemical Properties. J. Chem. Eng. Data 2000, 45, 953–960. [Google Scholar] [CrossRef]
- MATLAB. R2019a; The MathWorks Inc.: Natick, MA, USA, 2019. [Google Scholar]
- Lay, D.C. Linear Algebra and Its Applications, 3rd ed.; Addison Wesley, Inc.: Boston, MA, USA, 2003. [Google Scholar]
- Yaws, C.L. Yaws’ Handbook of Properties for Aqueous Systems; Knovel: New York, NY, USA, 2012; ISBN 978-1-62198-034-6. [Google Scholar]
- Mobley, D.L.; Guthrie, J.P. FreeSolv: A database of experimental and calculated hydration free energies, with input files. J. Comput.-Aided Mol. Des. 2014, 28, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Matos, G.D.R.; Kyu, D.Y.; Loeffler, H.H.; Chodera, J.D.; Shirts, M.R.; Mobley, D.L. Approaches for Calculating Solvation Free Energies and Enthalpies Demonstrated with an Update of the FreeSolv Database. J. Chem. Eng. Data 2017, 62, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- FreeSolv: Experimental and Calculated Small Molecule Hydration Free Energies, Version 0.51. Available online: https://github.com/MobleyLab/FreeSolv (accessed on 11 June 2017).
- Poling, B.E.; Prausnitz, J.M.; O’Connell, J.P. The Properties of Gases and Liquids, 5th ed.; The McGraw-Hill Companies, Inc.: New York, NJ, USA, 2001. [Google Scholar]
- Weidlich, U.; Gmehling, J. A Modified UNIFAC Model. 1. Prediction of VLE, hE, and γ∞. Ind. Eng. Chem. Res. 1987, 26, 1372–1381. [Google Scholar] [CrossRef]
- Gmehling, J.; Li, J.; Schiller, M. A Modified UNIFAC Model. 2. Present Parameter Matrix and Results for Different Thermodynamic Properties. Ind. Eng. Chem. Res. 1993, 32, 178–193. [Google Scholar] [CrossRef]
- Gmehling, J.; Lohmann, J.; Jakob, A.; Li, J.; Joh, R. A Modified UNIFAC (Dortmund) Model. 3. Revision and Extension. Ind. Eng. Chem. Res. 1998, 37, 4876–4882. [Google Scholar] [CrossRef]
- Gmehling, J.; Wittig, R.; Lohmann, J.; Joh, R. A Modified UNIFAC (Dortmund) Model. 4. Revision and Extension. Ind. Eng. Chem. Res. 2002, 41, 1678–1688. [Google Scholar] [CrossRef]
- Jakob, A.; Grensemann, H.; Lohmann, J.; Gmehling, J. Further Development of Modified UNIFAC (Dortmund): Revision and Extension 5. Ind. Eng. Chem. Res. 2006, 45, 7924–7933. [Google Scholar] [CrossRef]
- Constantinescu, D.; Gmehling, J. Further Development of Modified UNIFAC (Dortmund): Revision and Extension 6. J. Chem. Eng. Data 2016, 61, 2738–2748. [Google Scholar] [CrossRef]
- Koenhen, D.M.; Smolders, C.M. The Determination of Solubility Parameters of Solvents and Polymers by Means of Correlations with Other Physical Quantities. J. Appl. Polym. Sci. 1975, 19, 1163–1179. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | RMS | AAPE (%) | N | |||||
|---|---|---|---|---|---|---|---|---|
| 4.94 | −0.63 | −1.65 | −1.48 | 0.93 | 0.88 | 18.14 | 102 | |
| 5.14 | −0.61 | −1.61 | −1.38 | 0.94 | 0.80 | 90.14 | 113 | |
| 5.34 | −0.59 | −1.59 | −1.32 | 0.93 | 0.74 | 40.79 | 116 | |
| 3.38 | 0.94 | 2.20 | 2.92 | 0.91 | 1.65 | 6.86 | 95 | |
| 2.76 | 0.93 | 2.27 | 2.88 | 0.92 | 1.46 | 6.59 | 107 | |
| 2.04 | 0.93 | 2.35 | 2.81 | 0.93 | 1.33 | 6.67 | 113 |
| Property | RMS | AAPE (%) | N | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 4.94 | −0.63 | −1.64 | −1.59 | 0.11 | 0.00 | 0.93 | 0.87 | 18.25 | 102 | |
| 5.13 | −0.61 | −1.64 | −1.34 | −0.04 | 0.01 | 0.94 | 0.80 | 100.47 | 113 | |
| 5.30 | −0.58 | −1.56 | −1.26 | −0.03 | −0.01 | 0.93 | 0.74 | 40.35 | 116 | |
| 3.40 | 0.94 | 2.18 | 3.11 | −0.20 | 0.00 | 0.91 | 1.65 | 6.85 | 95 | |
| 2.78 | 0.93 | 2.28 | 2.82 | 0.04 | 0.00 | 0.92 | 1.45 | 6.54 | 107 | |
| 2.07 | 0.90 | 2.34 | 2.74 | 0.04 | 0.01 | 0.93 | 1.32 | 6.62 | 113 |
| Correlation | Clausius–Clapeyron | |||||||
|---|---|---|---|---|---|---|---|---|
| N | AAPE (%) | RMS | AAPE (%) | RMS | ||||
| 0 | 102 | 0.64 | 18.25 | 0.87 | 20 | 0.70 | 70.80 | 0.90 |
| 40 | 0.64 | 42.40 | 0.83 | |||||
| 20 | 114 | 0.58 | 99.75 | 0.79 | 0 | 0.64 | 18.68 | 0.88 |
| 40 | 0.54 | 41.86 | 0.74 | |||||
| 40 | 117 | 0.54 | 40.10 | 0.74 | 0 | 0.67 | 14.55 | 0.88 |
| 20 | 0.79 | 337.73 | 1.48 | |||||
| Calc | RMS | AAPE (%) | AAE | N | ||
|---|---|---|---|---|---|---|
| CC (20 C) | 1.08 | 1.20 | 64.72 | 0.94 | 0.96 | 81 |
| CC (40 C) | 1.10 | 1.20 | 68.98 | 0.94 | 0.96 | 81 |
| reference | 0.97 | 1.07 | 10.61 | 0.74 | 0.97 | 81 |
| mod-UNIFAC | 2.12 | 3.73 | 27.77 | 3.08 | 0.63 | 60 |
| Calc | RMS | AAPE (%) | AAE | N | ||
|---|---|---|---|---|---|---|
| CC (20 C) | 2.54 | 2.71 | 22.07 | 2.18 | 0.96 | 73 |
| reference | 2.52 | 2.73 | 22.89 | 2.04 | 0.96 | 73 |
| mod-UNIFAC | 4.88 | 9.31 | 229.80 | 7.95 | 0.70 | 60 |
| Property | RMS | AAPE (%) | N | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| −2.23 | 49.65 | 95.21 | 38.17 | 2.15 | −1.23 | 0.82 | 3.28 | 3.03 | 96 | |
| −4.72 | 54.03 | 102.96 | 45.08 | −0.82 | −2.92 | 0.82 | 3.31 | 9.07 | 112 | |
| −6.82 | 57.61 | 105.70 | 45.82 | −0.63 | −2.36 | 0.84 | 3.18 | 9.56 | 118 | |
| −2.65 | 0.15 | 0.51 | 0.62 | 0.05 | −0.03 | 0.81 | 0.42 | 204.22 | 77 | |
| −2.40 | 0.13 | 0.41 | 0.64 | −0.03 | −0.01 | 0.79 | 0.43 | 342.72 | 112 | |
| −0.49 | 0.25 | 0.77 | 1.05 | 0.06 | 0.00 | 0.80 | 6.51 | 96.06 | 117 | |
| 33.80 | 0.45 | 0.65 | 0.40 | 0.06 | 0.02 | 0.56 | 35.27 | 13.95 | 124 | |
| 214.45 | 0.04 | 0.11 | 0.08 | 0.00 | 0.00 | 0.88 | 24.00 | 4.33 | 127 | |
| 0.06 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.61 | 0.09 | 19.98 | 122 | |
| −792.98 | 0.55 | 0.42 | 0.55 | −0.09 | 0.00 | 0.56 | 603.18 | 86.51 | 114 | |
| 4.21 | −0.05 | 0.02 | 0.01 | 0.01 | 0.00 | 0.64 | 0.19 | 4.21 | 121 | |
| 220.99 | 0.97 | 1.44 | 0.82 | 0.01 | 0.02 | 0.85 | 33.86 | 7.91 | 121 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, N.H.; Dhakal, P.; Roese, S.N.; Paluch, A.S. Correlating Pure Component Properties with MOSCED Solubility Parameters: Enthalpy of Vaporization and Vapor Pressure. ChemEngineering 2023, 7, 25. https://doi.org/10.3390/chemengineering7020025
Wong NH, Dhakal P, Roese SN, Paluch AS. Correlating Pure Component Properties with MOSCED Solubility Parameters: Enthalpy of Vaporization and Vapor Pressure. ChemEngineering. 2023; 7(2):25. https://doi.org/10.3390/chemengineering7020025
Chicago/Turabian StyleWong, Nick H., Pratik Dhakal, Sydnee N. Roese, and Andrew S. Paluch. 2023. "Correlating Pure Component Properties with MOSCED Solubility Parameters: Enthalpy of Vaporization and Vapor Pressure" ChemEngineering 7, no. 2: 25. https://doi.org/10.3390/chemengineering7020025
APA StyleWong, N. H., Dhakal, P., Roese, S. N., & Paluch, A. S. (2023). Correlating Pure Component Properties with MOSCED Solubility Parameters: Enthalpy of Vaporization and Vapor Pressure. ChemEngineering, 7(2), 25. https://doi.org/10.3390/chemengineering7020025