1. Introduction
The intensive use of agricultural land worldwide, along with the large-scale development of the agrochemical industry, has dramatically increased the variety and quantity of agrochemicals in both continental and marine waters. Contaminated water poses risks to human and animal health, and the removal of these contaminants has been a challenge for researchers in recent years [
1].
Technologies known as advanced oxidation processes (AOPs) have emerged as strategies to remediate waters contaminated with agrochemicals [
2,
3]. These technologies allow pesticides to be removed up to mineralization, and include methods such as O
3, O
3/UV-B, H
2O
2/UV-C, Fenton, photo-Fenton, TiO
2-ZnO/UV-A, and TiO
2-ZnO/UV-B [
4,
5,
6]. These methods share a common factor: all of them generate hydroxyl radicals (OH
•).
Zinc oxide (ZnO) is a semiconducting species with a band gap similar to the band gap of TiO2, but it offers a major advantage: ZnO absorbs more than one fraction of the UV spectrum, and has a threshold of 425 nm.
It is importate remark also that the UV emission corresponds to the near band edge (NBE) emission assigned to to the radiative annihilation of excitons and the visible emission is commonly referred to as a deep-level or trap-state emission [
7,
8,
9]. The relative strength of NBE to deep level defect emissions exhibits a dramatic threshold dependence on surface roughness, and this point involves directly the method employed to preare the semicoductor. Surface optical emission efficiency increases over the roughness decreases to unit cell dimensions, highlighting the coupled role of surface morphology and near-surface defects for high efficiency ZnO emitters [
7,
8,
9].
In addition, compared to TiO2, materials containing the Zn(II) ion constitute more effective photocatalysts to remediate water, because they generate H
2O
2 more efficiently [
7,
8,
9], afford higher mineralization rates, contain a larger number of active sites for photocatalysis, and exhibit greater surface reactivity.
Incorporation of photoactive elements into molecular sieves provides materials with highly dispersed active species and enhanced photocatalytic activity [
10,
11]. In general, the high dispersion of photoactive sites in the network of the molecular sieves, along with the effective separation between electrons and holes, improves the photocatalytic activity of molecular sieves containing metallic transition elements. Photoactive elements (e.g., Zr and Ti) incorporated into the zeolite MCM-41 network cleave H
2O at least 80 times more effectively than ZrO
2 [
12,
13,
14]. Hence, isomorphous substitution of Zn(II) ions into aluminophosphates seems to be a good strategy to make this semiconductor more efficient, and to avoid the corrosion that free Zn(II) species commonly experience under electrochemical conditions, acidic media, and UV light [
15].
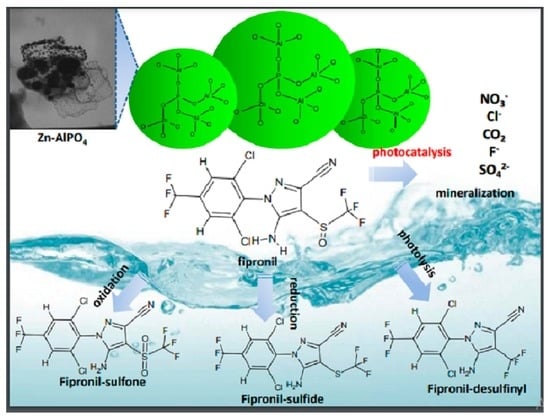
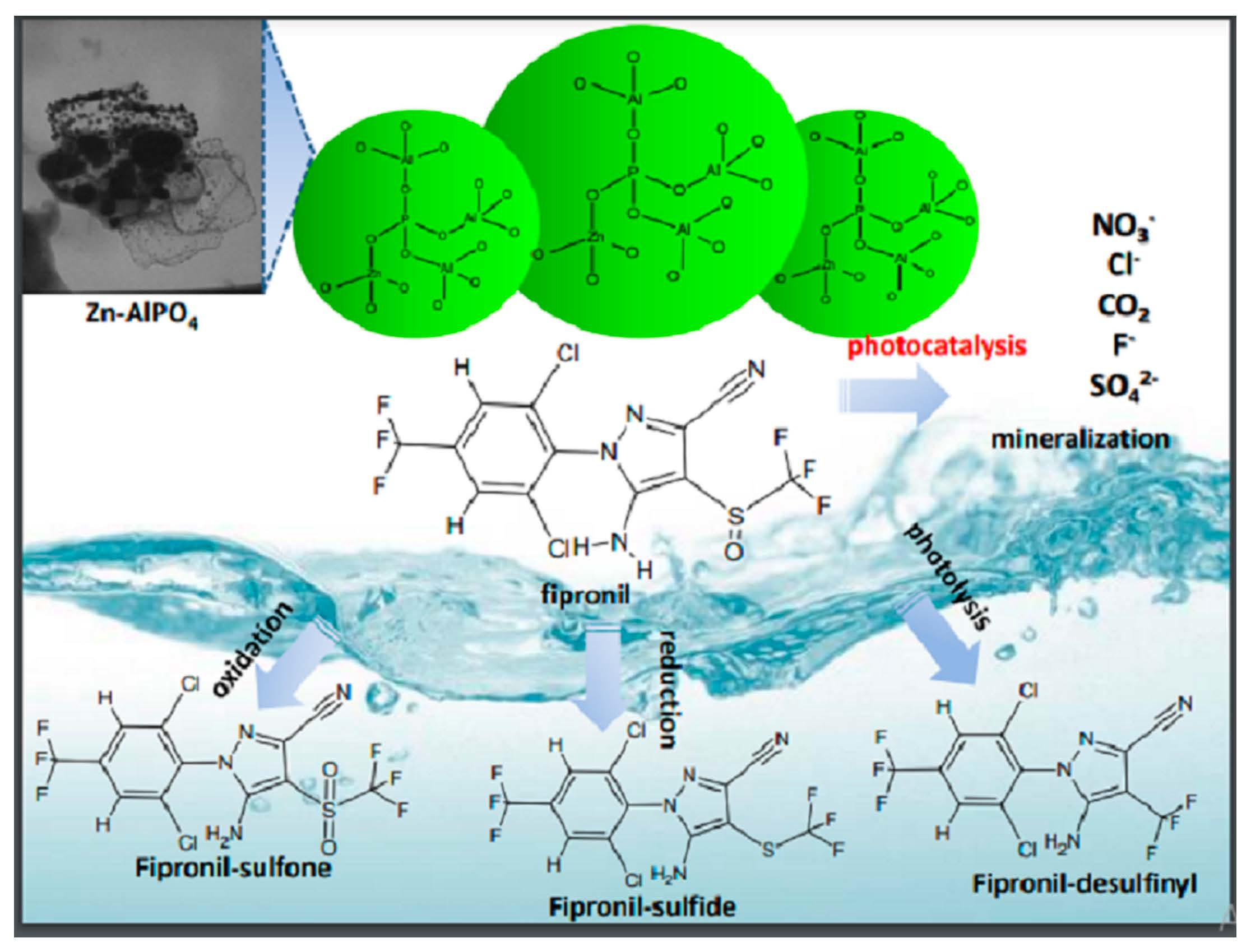
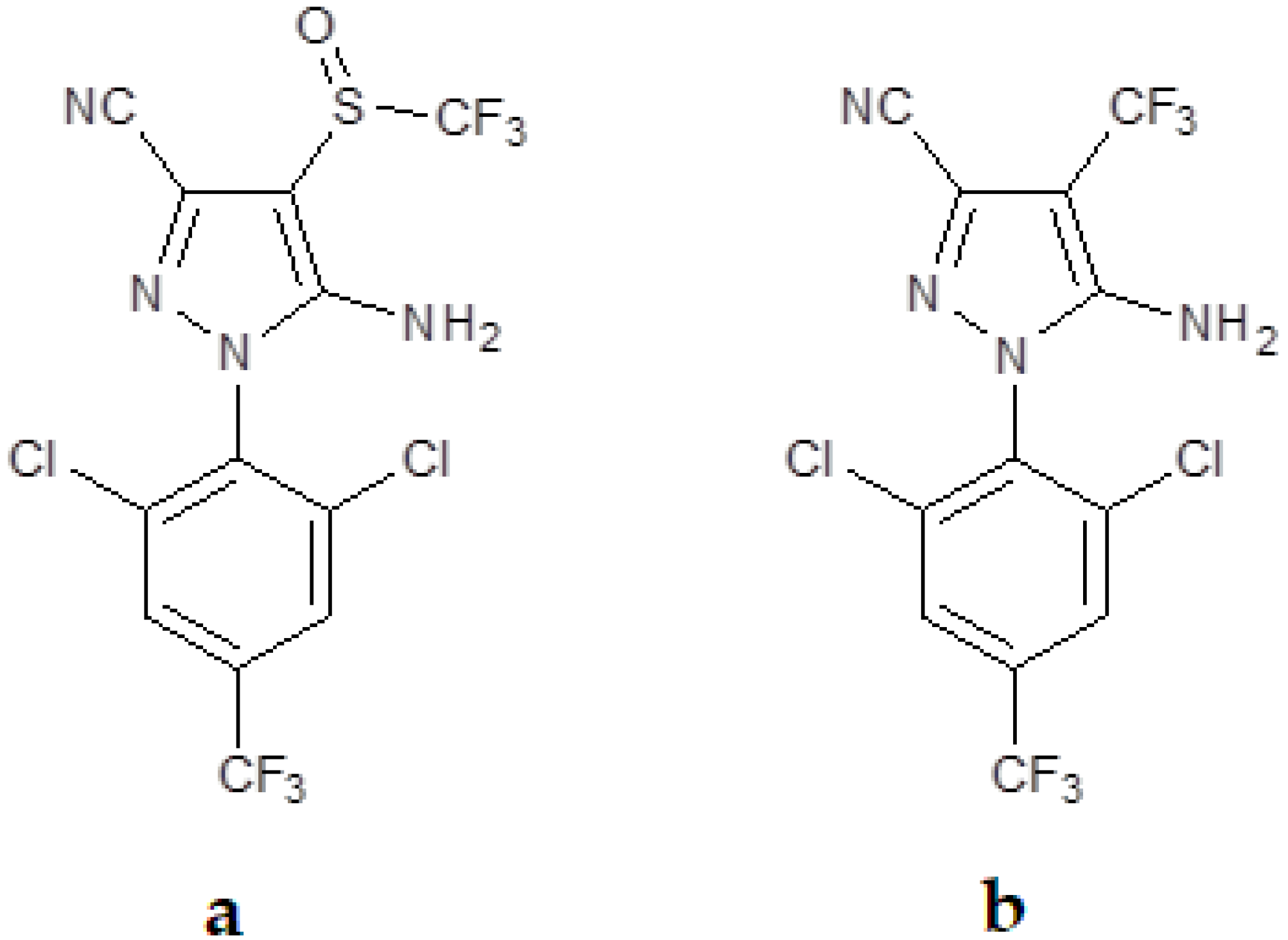
Fipronil, with the minimum formula C
12H
4Cl
2F
6N
4OS (
Figure 1a, is an insecticide of the phenylpyrazole series used to control pests of corn, cotton, and rice in several parts of the world [
16]. It is effective against insects with resistance to pyrethroids, cyclodiene, organophosphorus, and carbamate insecticides [
17]. Its mechanism of action involves the selective blocking of the passage of chloride ions through the γ-aminobutyric acid (GABA)-regulated chloride channel, which disrupts the activity of the central nervous system and, at sufficient doses, kills insects with a favorable safety factor between insects and mammals [
18]. In aquatic environments, fipronil has been found at levels between 0.5 and 9 μg L
−1 in surface waters and downstream of treated rice-cultivation fields, and up to 12.6 μg L
−1 in residential areas [
16]. In the Brazilian southeast region, concentrations between 6 and 465 μg L
−1 were found [
16].
In this context, the sol–gel process, which involves the hydrolysis and polycondensation of molecular precursors and formation of oxo bridges, is an important methodology to design and synthesize materials that are potentially applicable in catalysis and photocatalysis [
19,
20,
21]. The sol–gel chemistry has the following advantages: it provides materials with (i) a high degree of purity; (ii) strictly controlled composition, structure, and homogeneity at the molecular level; and (iii) regulated texture. In recent years, authors have suggested many modifications to the sol–gel routes. In the modified methodology, the oxo bonds originate from other oxygen donors other than water—for example, alkoxides, ethers, or alcohols—giving rise to the term “non-hydrolytic sol-gel” route coined by Acosta et al. in 1994 [
22].
This work aimed to synthesize new materials to treat sewage and wastewater from agrochemical activities. More specifically, it describes the synthesis of zinc aluminophosphate matrices via the non-hydrolytic sol–gel process, and their application in the photocatalytic degradation of fipronil—an insecticide that is widely used worldwide. Techniques such as X-ray powder diffraction, infrared spectroscopy, thermal analysis, and scanning electron microscopy aided the characterization of the new materials, calcined at different temperatures.
2. Materials and Methods
2.1. Reagents
All of the reagents were analytical grade, and were purchased from Aldrich, Sigma, or Merck, and treated when necessary. Fipronil was acquired from Sigma-Aldrich (St. Louis, MO, USA).
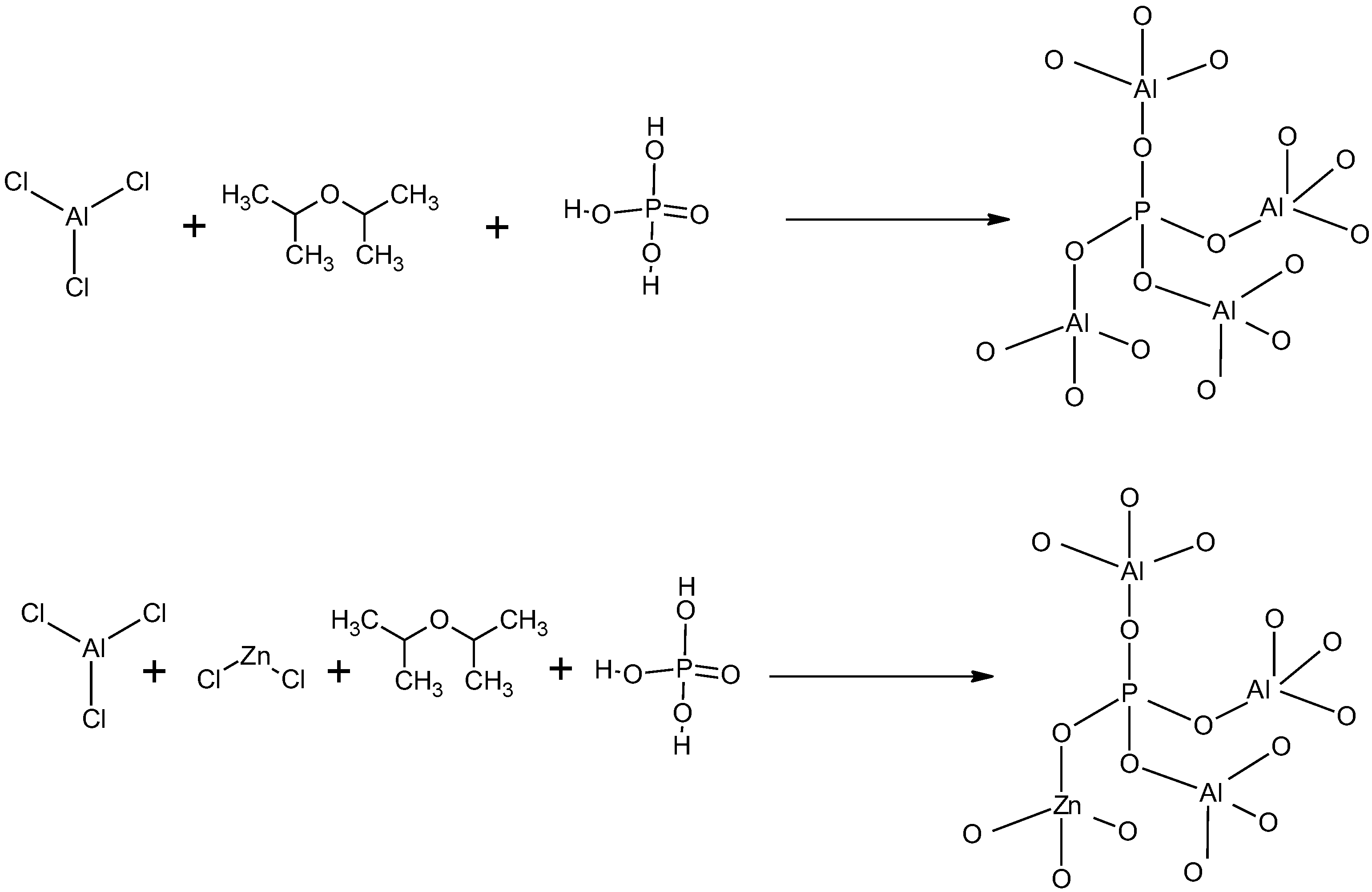
2.2. Preparation of Zn-AlPO4 Matrices via the Non-Hydrolytic Sol–Gel Route (NHG)
The material was synthesized by modifying the methods described by Acosta et al. [
22], Bourget et al. [
23], and de Lima et al. [
19]. In a two-necked round-bottomed flask, aluminum chloride (9.4 × 10
−2 mol), isopropyl ether (iPr
2O, 2.8 mol), and zinc(II) chloride (2.7 × 10
−2 mol) were mixed and refluxed at 110 °C, for 4 h, under an argon 5.0 quality (≥99.999%) atmosphere. The condenser was adapted to a thermostatic bath and kept at −8 °C. After the gel merged, H
3PO
4 (1.5 × 10
−1 mol) was added to the reaction system. After reflux, the system was cooled to room temperature and aged overnight in the mother liquor. The solvent was eliminated in a rotary evaporator, and the solid material was washed with different solvents, in the following order: dichloromethane, acetonitrile, and methanol. This procedure generated a non-hydrolytic gel (NHG) during the process, namely, an aluminum halide condensed with isopropyl ether upon cleavage of the O-R bond, which later produced an alkyl halide [
22,
24]. In the presence of aluminum alkoxides or chloroalkoxides, aluminum chloride exhibits stretching of structures of the μ-Cl and μ-OR type, which elicits two types of reaction: (a) nucleophilic attack of chlorine and electrophilic attack of aluminum, and (b) formation of Al-O-Al, Al-O-PO
4, and -P-O-Zn-OH-Al-O bonds due to nucleophilic attack of chlorine and electrophilic attack of carbon [
25,
26].
Figure 2 depicts a schematic representation of the prepared material, designated Zn-AlPO
4. Contact of the solid with the mother liquor is an important step during aging of the gel. Indeed, a previous work demonstrated [
19] that longer aging periods give better yields, and promote anchoring of a larger amount of metallic ions onto the matrix. Up to this step, the network is flexible; additional condensation reactions and new crosslinkages occur during aging in the mother liquor and solvent removal.
Parts of the solid material were thermally treated at 260, 400, 750, or 1000 °C, which afforded the samples named Zn-AlPO4-260, Zn-AlPO4-400, Zn-AlPO4-750, and Zn-AlPO4-1000, respectively. As a reaction control (blank), heat-treated materials were prepared under the same conditions, but in the absence of ZnCl2, which yielded the control samples AlPO4-260, AlPO4-400, AlPO4-750, and AlPO4-1000, respectively.
2.3. Characterizations
Thermogravimetric and differential thermal data (TG/DTA) were acquired using a thermal analyzer from TA Instruments (SDT Q600). Simultaneous DTA–TGA was carried out between ~25 and 1000 °C, at a heating rate of 20 °C min−1 and air flow of 100 mL min−1.
The X-ray diffractograms were registered on a Rigaku MiniFlex II DESKTOP X-ray diffractometer, at room temperature; CuKα radiation (λ = 1.54 Å) was used. A 0.04° s−1 path was employed for 2θ values ranging from 17 to 80°.
Infrared spectroscopy was carried out on a Fourier-transform infrared spectrometer (Fourier ABB, Bomem, model ME 100), using KBr pellets prepared by mixing 2% of the sample in weight with KBr. The spectra were recorded after 20 scans, from 4000 to 400 cm−1.
Specific surface areas were determined according to the BET method [
27], by analyzing the nitrogen adsorption isotherms. A Micrometrics ASAP 2020 physical adsorption analyzer was employed.
Transmission electron microscopy (TEM) was performed using a JEOL JEM CX 100 II microscope.
Scanning electron microscopy (SEM) and energy-dispersive X-ray experiments were conducted using a Philips model XL30 scanning electron microscope, equipped with an EDAX energy-dispersive X-ray detector.
The electronic spectra were recorded on a Hewlett-Packard 8453 diode-array spectrophotometer coupled with an HP KAYAK-XA microcomputer; the program provided by the manufacturer was employed.
2.4. Photocatalytic Reactions
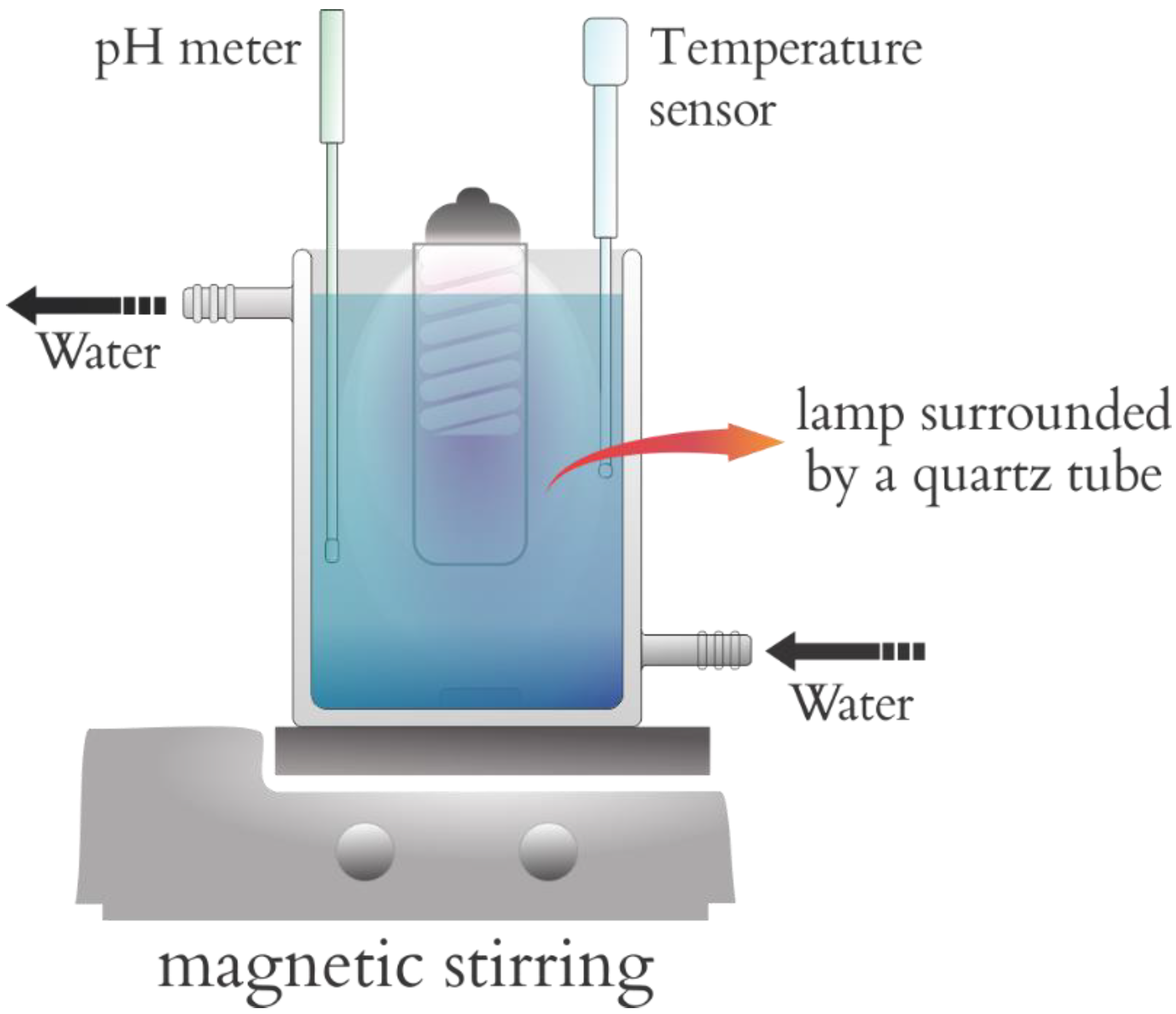
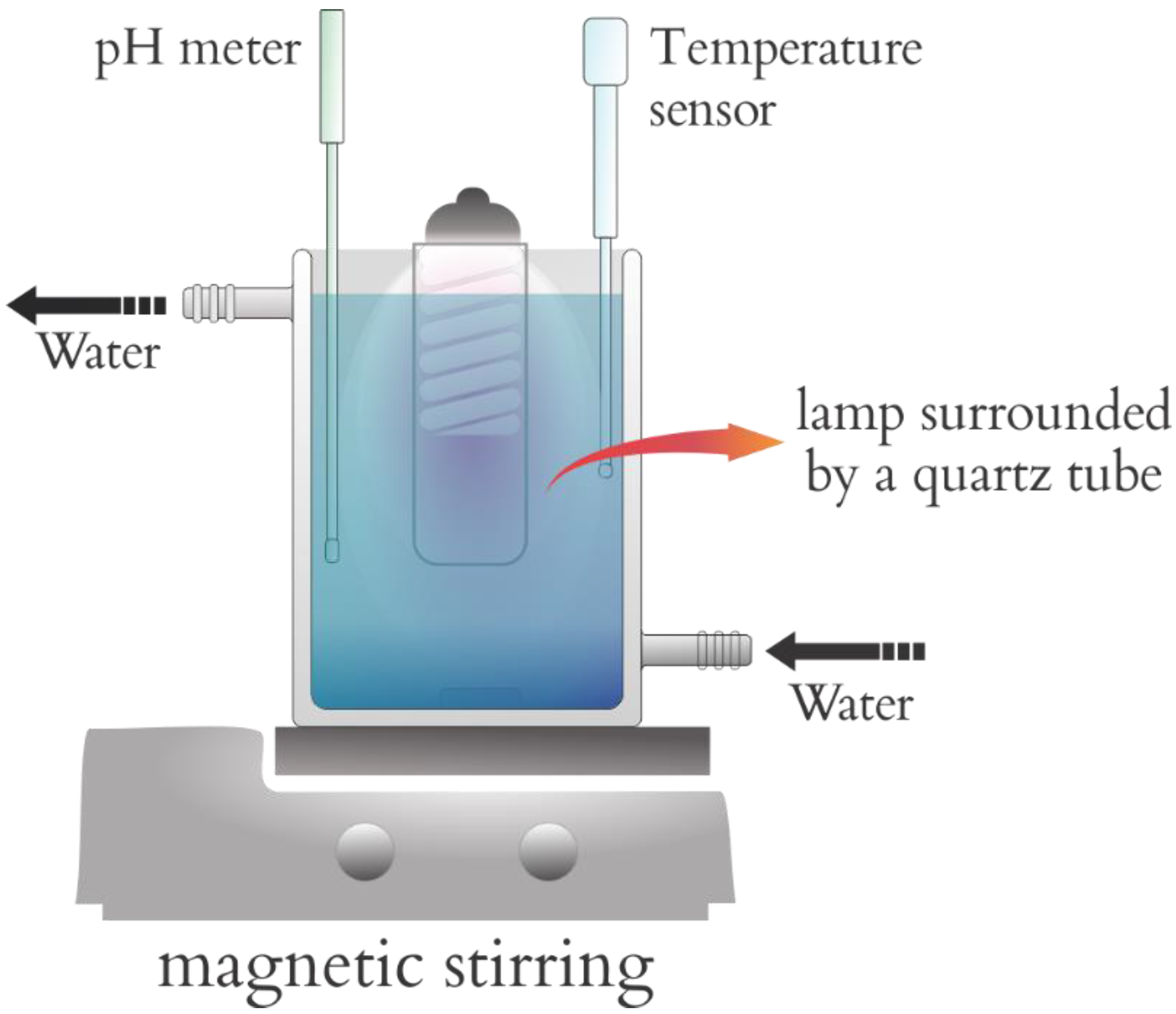
The photocatalytic reactions involving fipronil as a substrate were conducted in a sealed chamber containing a 95-watt lamp emitting short-wavelength UV-C radiation with a peak at 253.7 nm and germicidal action. This chamber was designed on the basis of other models described in the literature (
Figure 3) [
28,
29], with a total volume of 2000 cm
3. A device that allowed us to accomplish simple and cost-effective photodegradation reactions was utilized. The lamp was surrounded by a quartz tube, which enabled us to use it inside the solution containing the substance to be degraded. The chamber also relied on a system mounted to disperse the solution and the photocatalyst, on a temperature sensor, and on an electrode coupled with a potentiometer (Hanna Instruments HI 9321) placed outside the chamber to measure pH (Orion
® semi-micro pH 911600). An aqueous solution of fipronil with concentrations ranging from 2830 to 3370 µg L
−1 was employed. The influence of exposure time and the mass of the photocatalysts was studied herein, with 0.100 or 0.153 g of photocatalyst in 1000 cm
3 of fipronil solution at two different concentrations—2830 or 3370 µg L
−1 (quantified by calibration curves from UV–Vis spectroscopy)—and exposed to artificial ultraviolet radiation under constant stirring and placed in a thermostatic bath maintained at 25 ± 1 °C.
Measurement of light intensity: The amount of light emission under artificial light and sunlight was quantified by a INSTRUTEMP model LM801 luximeter (Instrutemp, São Paulo, Brazil) with a 0 to 500,000 lux range, and UV light was quantified by an Instrutherm model MRU-201 UV light meter (Instrutemp, São Paulo, Brazil), from 290 nm to 390 nm, and with a quantification range from 4000 to 19,990 µW/cm2 (distance varying from 5–38 cm).
Extraction protocol for GC–MS analysis: Photodegradation products were extracted using a typical extraction method. Briefly, 150 mL of the supernatant was placed into conical flasks and extracted with an organic solvent (150 mL of ethyl acetate). The samples were filtered into bottles and allowed to settle for 24 h. The filtrates from each photocatalytic reaction were then concentrated in vacuo at 40 °C using a rotary evaporator, and proceeded to analysis under GC–MS.
The photocatalyzed reactions were monitored by UV–Vis spectroscopy and gas chromatography–mass spectrometry (GC–MS) analyses. For the UV–Vis analysis, a 2000 μL aliquot was removed from the reaction medium and centrifuged at 3500 rpm for 15 min, followed by analysis on a Hewlett-Packard 8453 diode-array UV–Vis spectrometer, without previous treatment. For the GC–MS analysis, the reaction medium was extracted with ethyl acetate and analyzed using a Shimadzu.
A GCMS 2010 Plus (Shimadzu Corporation, Kyoto, Japan) system equipped with an AOC-20i autosampler was utilized; the column consisted of an Rtx-5MS (Restek Co., Bellefonte, PA, USA) fused silica capillary column (30 m length × 0.25 mm i.d. × 0.25 μm film thickness). The electron ionization mode was used at 70 eV. Helium (99.999%), with positive ionization, was employed as the carrier gas at a constant flow of 1.0 mL/min. The injection volume was 0.1 μL (split ratio of 1:10). The injector and the ion-source temperatures were set at 240 and 280 °C, respectively. The column temperature was programmed to rise from 80 to 240 °C at 3 °C/min, and was then held at 240 °C for 5 min. Mass spectra were taken with a scan interval of 0.5 s, in the mass range from 40 to 600 Da. The products from the photodegradation of fipronil were identified in the single-ion mode (SIM) chromatogram on the basis of retention times (R
t); the MS data were compared with the literature results [
17,
30].
3. Results
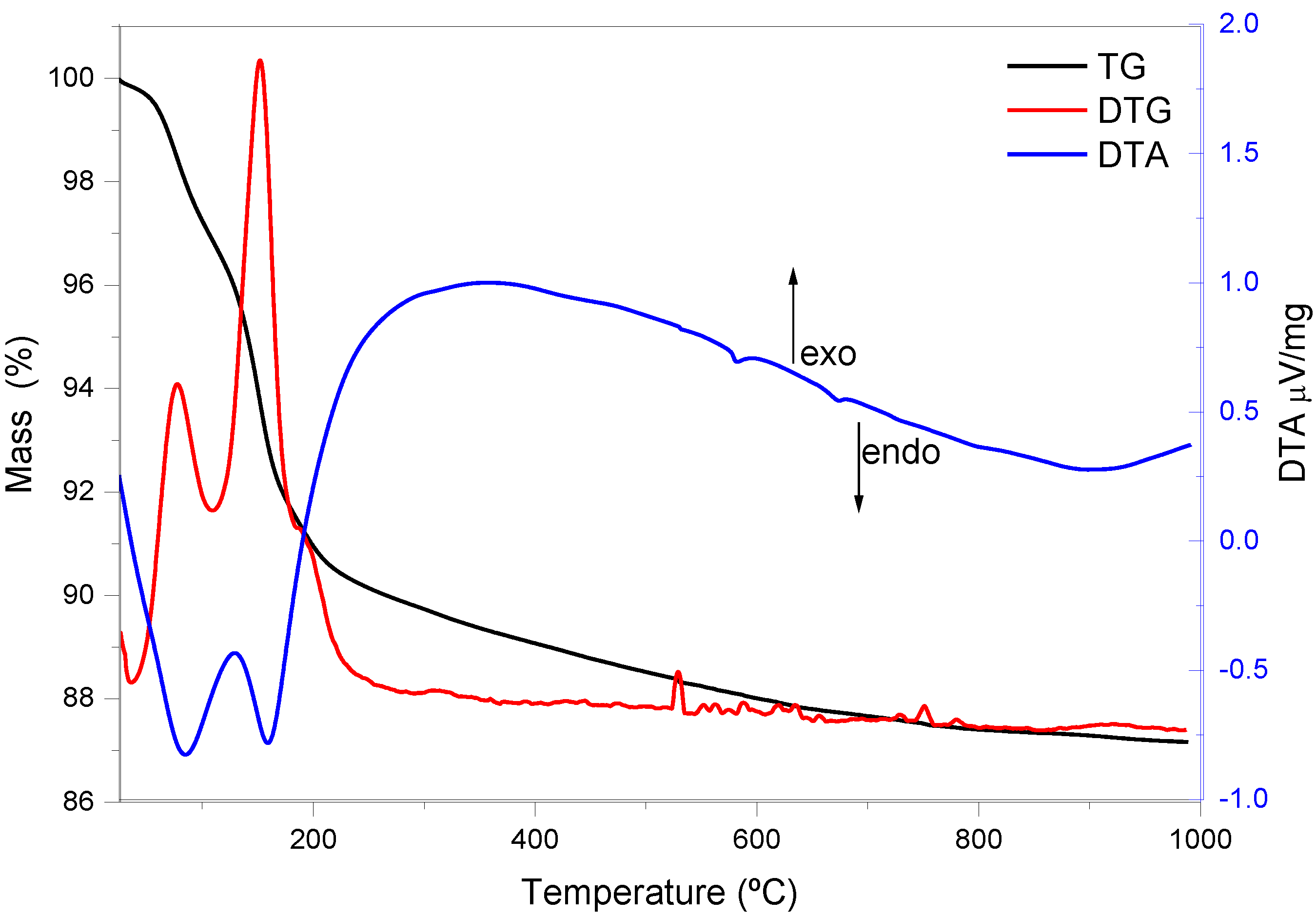
Figure 4 illustrates the thermogravimetric (TG/DTG and DTA) curves recorded for aluminophosphate samples synthesized in the presence of Zn(II) ions.
Treatment of the samples up to 260 °C removed the liquid retained in the material after the synthesis. However, the AlPO
4 sample underwent successive mass losses from the onset of heating at 25 °C until 200 °C, assigned to volatile solvents employed on washing steps and adsorbed water that remained in the product. A similar phenomenon has already been verified for alumina matrices prepared via the same route, and it was attributed to the contact of the material with the room atmosphere after the synthetic procedure [
19,
31].
The mass losses taking place from 200 to 400 °C (around 12% wt) referred to loss of residual groups of alkyl halides from the precursors employed during the synthesis. In the DTA curve, endothermic peaks arose at 523, 589, 671, and 870 °C. These peaks were related to small mass losses, and indicated that the sample underwent successive structural arrangements and rearrangements under rising temperatures.
Regarding the Zn-AlPO
4 samples, they lost weakly bound or weakly adsorbed water between 63 and 200 °C (8.0% wt). As explained above for AlPO
4, these water molecules were adsorbed onto the material after the synthesis. Further mass loss took place between 200 and 600 °C, due to pyrolysis and oxidation of residual alkyl halide groups [
32]. The DTA curve also displayed two endothermic peaks, at 578 and 670 °C—a region where mass loss did not occur; in fact, this region indicated a phase transition in the network.
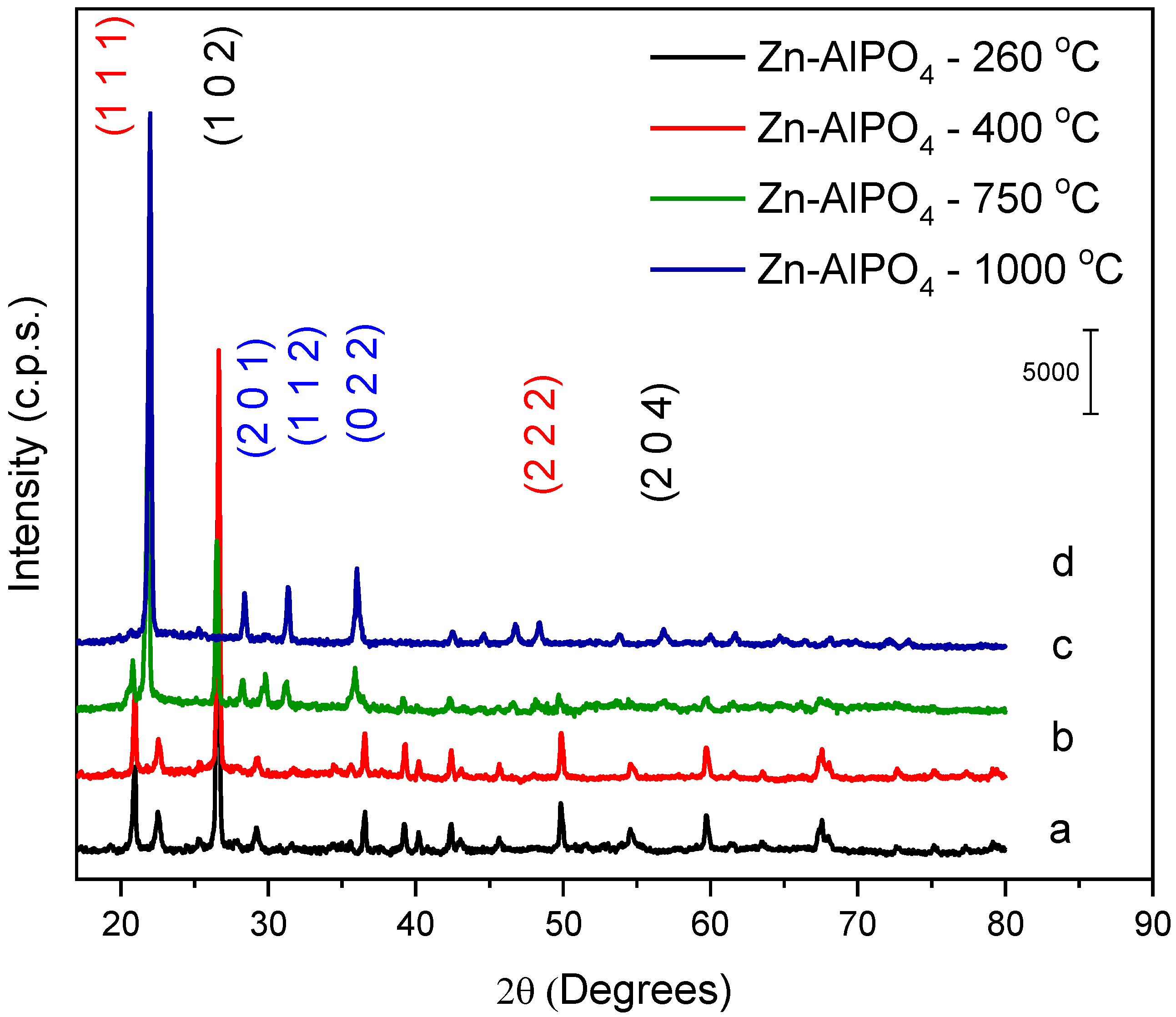
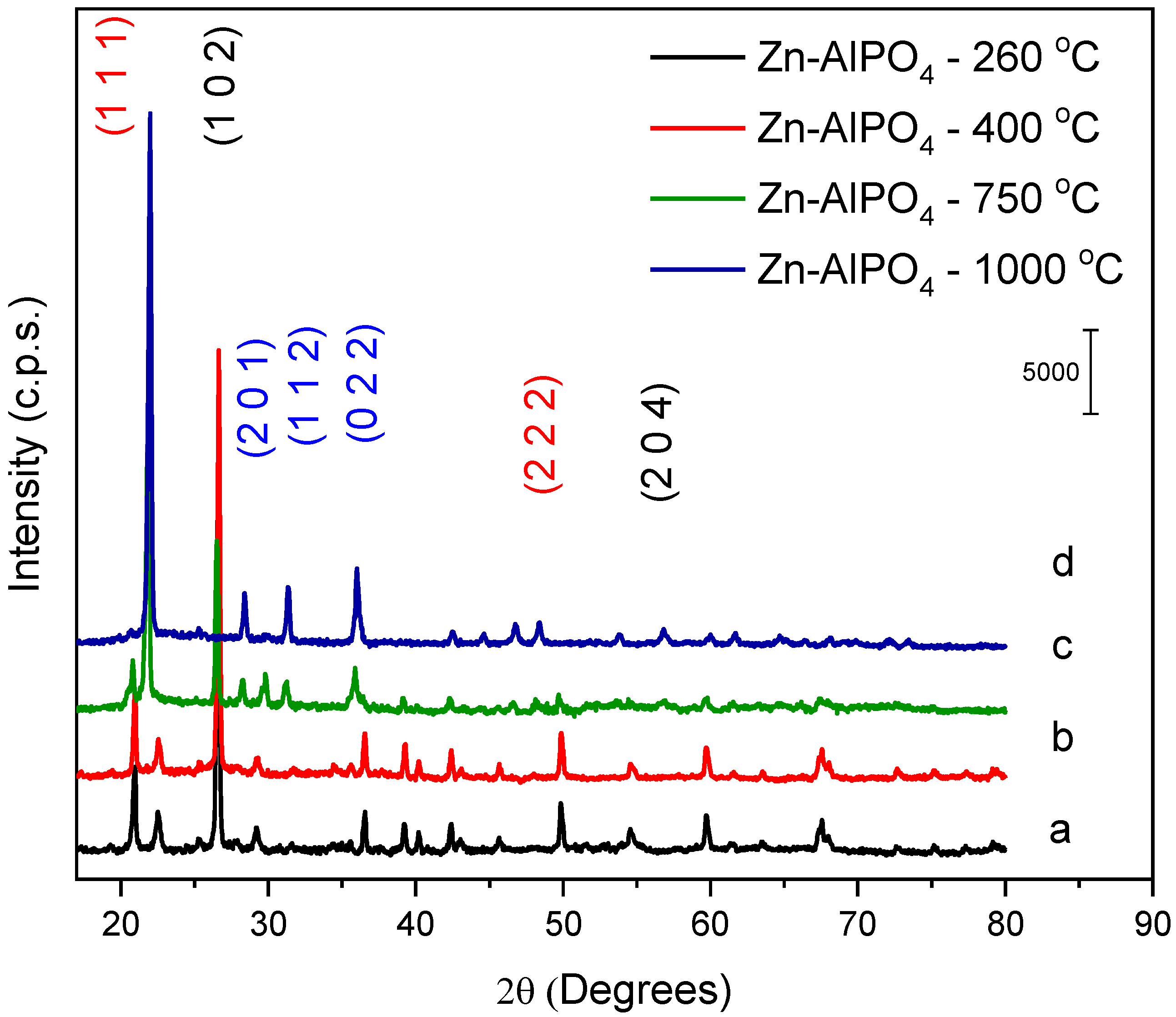
Identification of the phases in Zn-AlPO
4 (
Figure 5) relied on comparison of the peak positions and diffraction planes, as well as their intensities, with the corresponding files of the National Bureau Standards (NBSCAA-00-010-0423) for pure crystalline compounds.
The diffractograms of the Zn-AlPO4 samples heat-treated at 260 and 400 °C demonstrated that the berlinite phase—a hexagonal system—predominated in these products. The materials heat-treated at 750 and 1000 °C exhibited different diffraction planes, suggesting that they had distinct configurations from those of the samples treated at lower temperatures. The sample heated at 750 °C had a trigonal phase, ascribed to AlZnP2O8 (ACHSE7-00-052-1506); the material treated at 1000 °C presented an orthorhombic phase, assigned to AlPO4. This might have resulted from ZnO migration to regions outside the aluminophosphate network, corroborating formation of the AlPO4 phase.
The hexagonal structure of the berlinite type prevailed in AlPO
4 (2θ = 26.43° and 54.34°), with diffraction planes typical of this phase: (hkl = 1,0,2) and (2,0,4) (NBSCAA-00-010-0423). The AlPO
4 berlinite structure presents the same atomic structure as quartz. It is obtained by alternately substituting Si for Al and P [
33]. The presence of diffraction peaks due to the planes (1,0,0), (1,1,0), and (2,0,0) at larger angles also indicated that a hexagonal phase of the wurtzite type existed in the sample, suggesting that it was a polycrystalline material.
The AlPO4 samples heat-treated at 260, 400, and 750 °C displayed the same diffraction planes as the material that did not undergo heat treatment; therefore, the berlinite phase prevailed in these cases as well. The AlPO4 sample heat-treated at 1000 °C also contained berlinite, but structural defects gave rise to another non-identified phase.
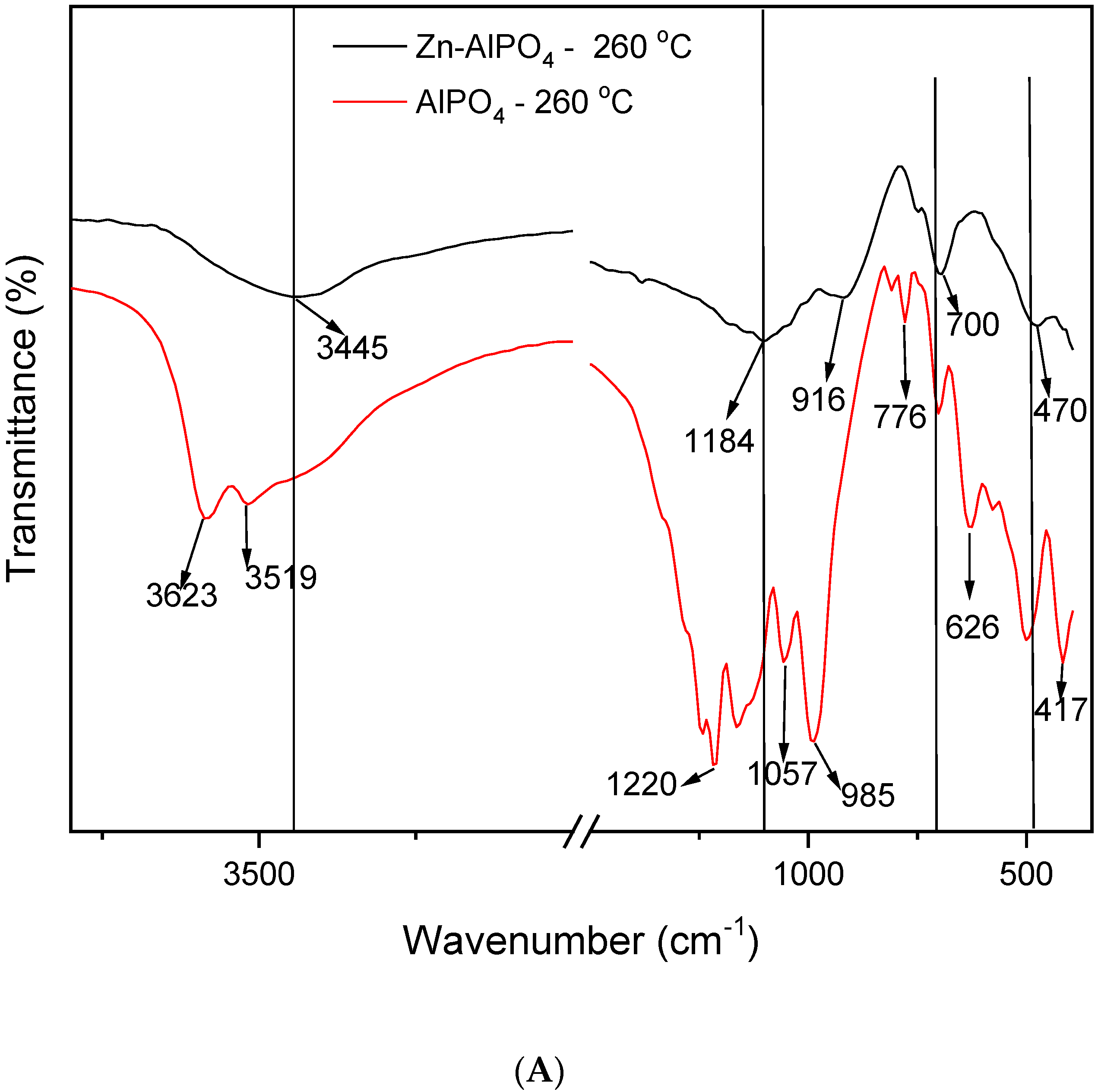
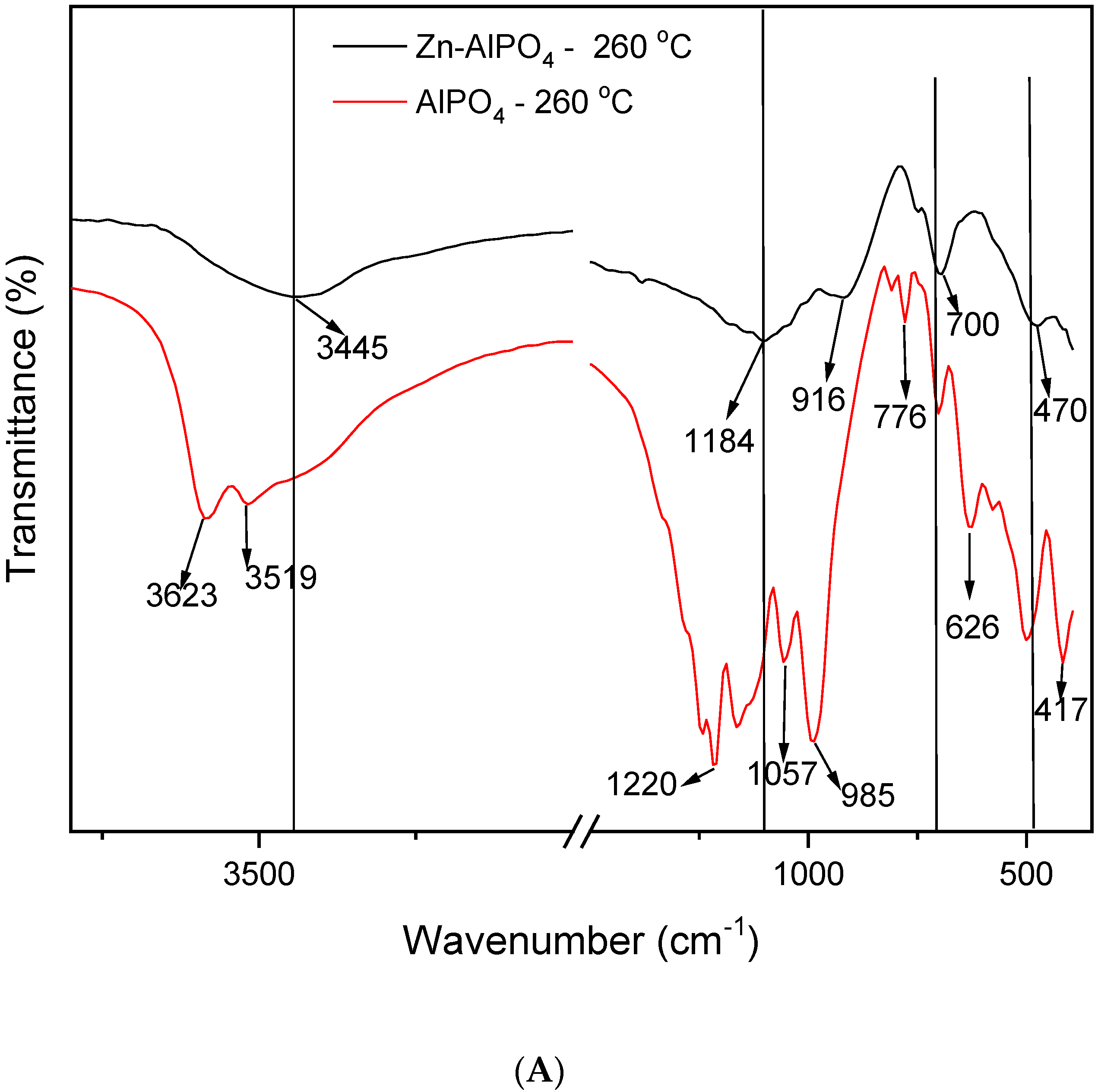
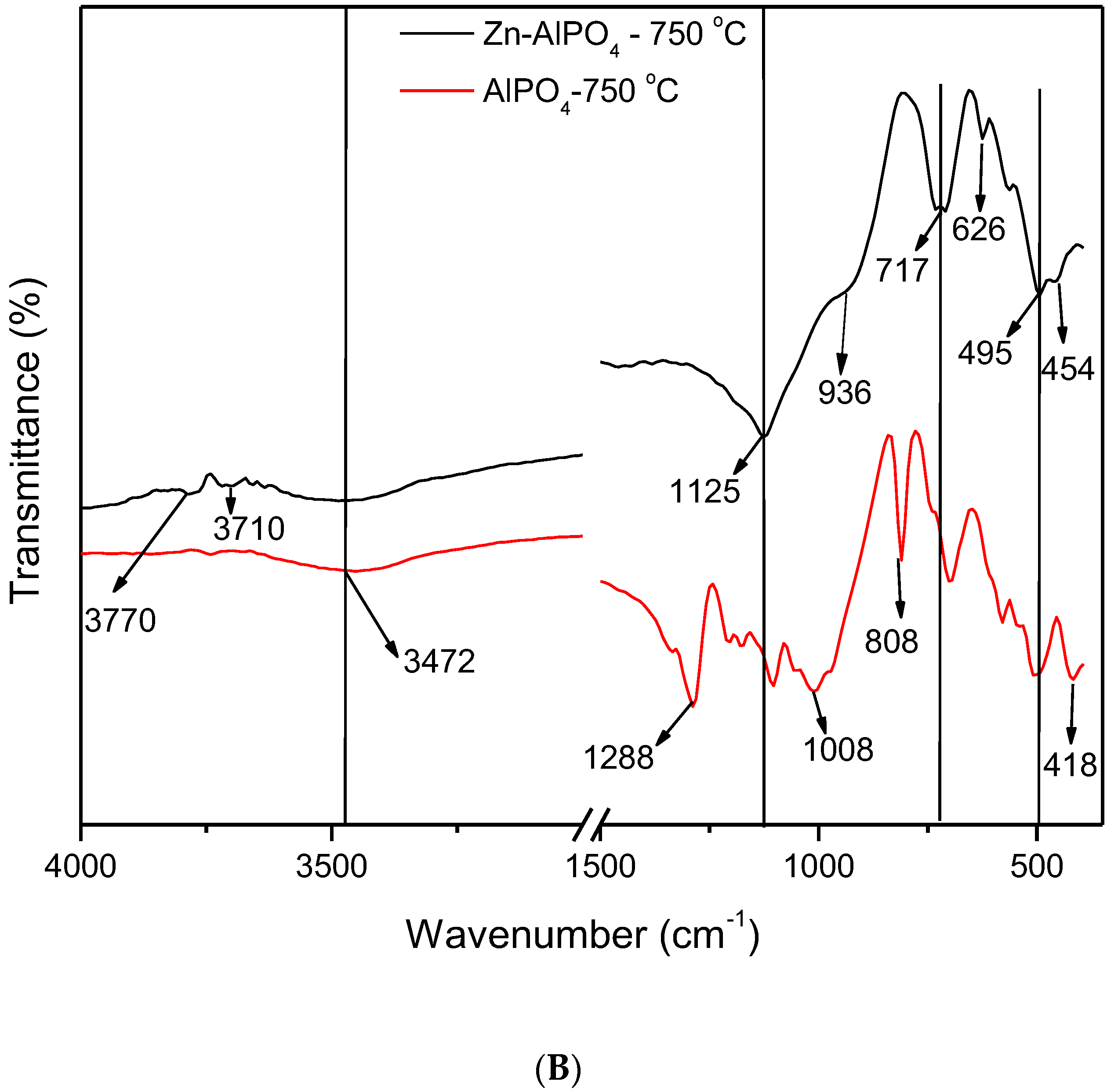
The IR spectra (
Figure 6) enabled us to compare the AlPO
4 and Zn-AlPO
4 samples heat-treated at different temperatures. The spectra of the AlPO
4 samples contained the typical vibrations of the berlinite structure [
34,
35,
36], the bands associated with vibrations of the berlinite unit at 1220, 1096, 504, 468, and 418 cm
−1, and the bands related to the vibrations of the Al pseudo-network at 700, 690, 626, and 580 cm
−1. A band also appeared at 735 cm
−1, which corresponded to defects of the Al subnetwork in berlinite [
35].
The Zn-AlPO
4 samples also displayed the bands detected for AlPO
4, although some of them shifted to lower wavenumbers as a result of the presence of Zn(II) in the berlinite network. Two additional vibrations appeared at 1009 and 936 cm
−1, as a result of Zn-OP vibrations [
37]. ZnO IR vibrations usually occur below 600 cm
−1; in fact, a broad band in the 400–550 cm
−1 region has been reported, constituting further evidence of Zn(II)’s incorporation into the network of berlinite [
37]. The altered vibrations between 450 and 1290 cm
−1, along with an asymmetric band centered at 1125 cm
−1, clearly attested to the isomorphous substitution of Al(III) for Zn(II); the latter band intensified as the heat treatment temperature increased. According to the X-ray diffractometry results, this may have stemmed from formation of AlZnP
2O
8. Recent studies have described that both the activity and selectivity within a series of zeolites and molecular sieves—such as aluminophosphates—depend on total acidity. Apart from hydroxyl groups, other types of acid sites (for example, Lewis centers) contribute to the acidity of molecular sieves [
26].
All of the Zn-AlPO4 and AlPO4 samples presented a broad band in the region of 3650–2900 cm−1 (centered at 3500 cm−1), irrespective of the heat treatment temperature. Various authors have attributed this band to hydroxyl groups on the surface of free phosphorus; indeed, a hydrogen bond of the hydroxyl group on the surface of the compound disturbs phosphorus.
Hydroxyl vibrations appeared between 3000 and 4000 cm
−1. These bands allow for the detection of different types of OH groups, such as Al-OH, P-OH, and Zn-OH, which result from crystal defects and bridge hydroxyls, and confer Bronsted acidity to the material [
32]. They indicate the type of metal incorporation into MeALPO and MeSAPO; that is, whether the metal isomorphically substitutes Al, P, or Si, or if only ion exchange occurs. Isomorphous substitution generates new OH groups that bind the metal to another tetrahedral ion in the structure of the zeolite or the molecular sieve. In contrast, ion exchange does not alter the typical structural vibrations. Hence, isomorphous substitution did take place in the synthesized materials.
Irrespective of the presence of Zn(II) in the matrix and the heat treatment temperature, all of the samples displayed bands between 3445 and 3500 cm
−1, characteristic of OH groups. These groups could correspond to water physisorbed onto the KBr pellet, or even onto the sample exposed to the environment after heat treatment. Indeed, thermogravimetry detected the presence of water due to handling of the material, as discussed earlier in this paper. The bands at 3583, 3623, 3710, and 3748 cm
−1 corresponded to terminal -Al-OH and -P-OH groups [
38]. The Zn-AlPO
4 samples may also present interactions of the -P-OH + Zn(OH)
2 type [
38,
39].
Table 1 lists data on the textural properties of the samples thermally treated at different temperatures, as determined by N
2 adsorption/desorption isotherms [
27].
The Zn-AlPO4 samples treated at 750 and 1000 °C had larger pore sizes. Increased heat treatment temperature rearranged the constituents in the matrix network, and subsequently eliminated organic residues, producing a more ordered crystalline structure. It is noteworthy that all of the AlPO4 and Zn-AlPO4 samples presented a lower surface area than those reported in the literature. This happened because the syntheses occurred in the absence of surfactants or any other molecular guides, to afford extremely dense materials.
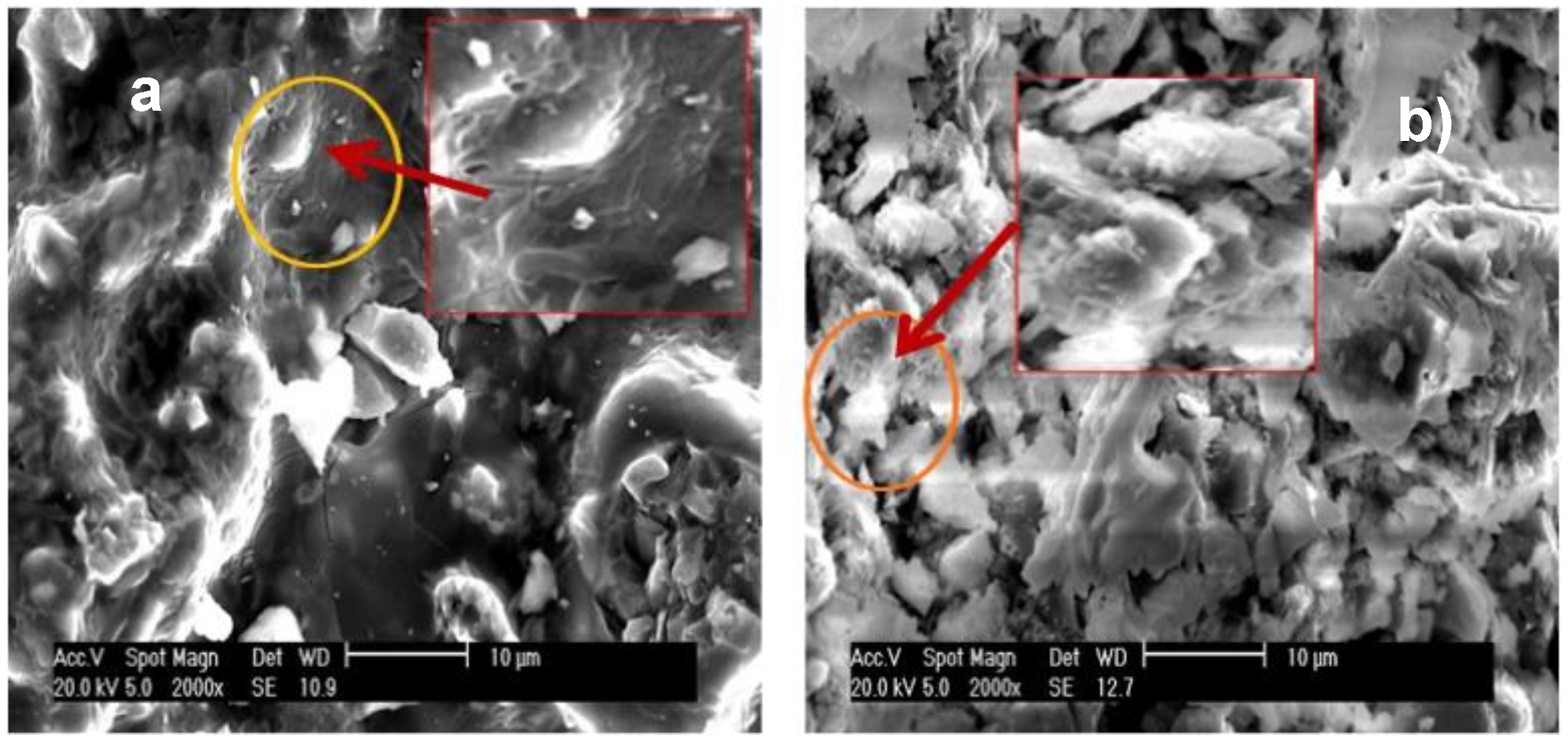
The micrographs depicted in
Figure 7A,B demonstrate that the samples were arranged as plaques, which generated a dense material and made it difficult for the electron beams to cross the structure. This reduced the TEM resolution, as observed for all of the AlPO
4 and Zn-AlPO
4 samples.
AlPO
4 treated at 400 °C contained cavities, whereas Zn-AlPO
4 (
Figure 7A,B), with high electron density, did not. In Zn-AlPO
4, Zn(II) may have been homogeneously dispersed in the aluminophosphate matrix, which justified the absence of cavities in the sample, and indicated that Zn(II) isomorphically substituted the Al(III) ions.
Zn-AlPO
4 thermally treated at 1000 °C (
Figure 8B) bore agglomerates on the surface, which evidenced that the metal migrated to the surface at higher temperatures. As discussed earlier in the X-ray results confirmed the presence of ZnO and AlPO
4, which corroborated this migration.
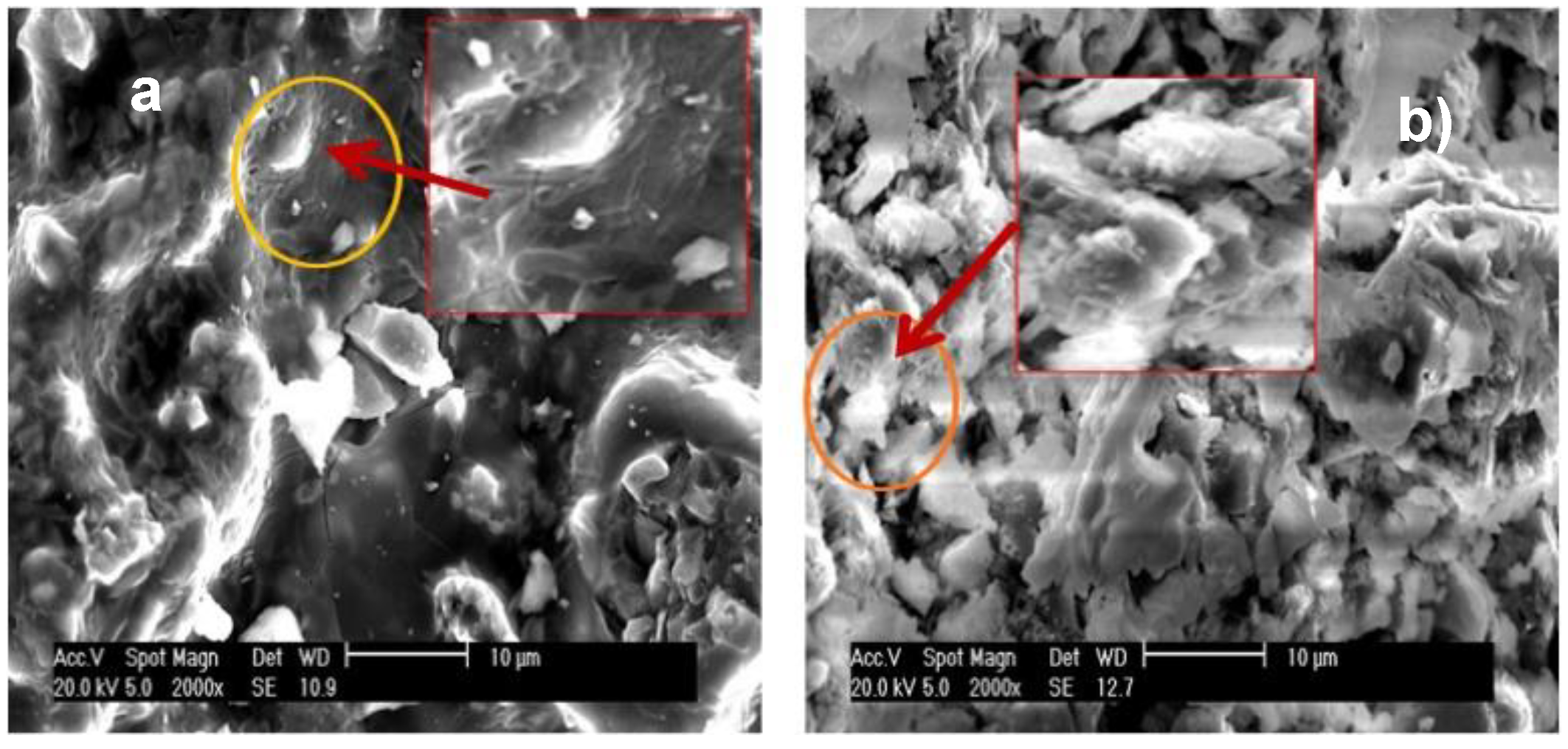
The SEM images depicted in
Figure 9 and
Figure 10 show that the Zn(II) ions elicited changes in the features of the aluminophosphate matrix. Rising heat treatment temperature accentuated the differences between Zn-AlPO
4 and AlPO
4. Zn-AlPO
4 thermally treated at 400 °C had a smoother surface than AlPO
4; the latter material presented a more disordered structure, and granules of varied sizes.
The differences between Zn-AlPO4 and AlPO4 became more pronounced upon heat treatment of the materials. The surface of the Zn-AlPO4 sample contained some holes that resembled volcanoes, which may have originated during rising temperatures and the subsequent elimination of volatile compounds. Cracks also existed on the surface of Zn-AlPO4. As for AlPO4, the sample became whiter upon the removal of impurities. In the signaled area, and juxtaposed hexagonal plaques arose, which confirmed the X-ray and TEM results.
As for the Zn-AlPO4 and AlPO4 samples thermally treated at 1000 °C, Zn-AlPO4 displayed a rough surface because the structure collapsed upon increasing heat treatment temperature. The AlPO4 sample exhibited larger agglomerates and, according to the TEM results, this material consisted of juxtaposed plaques with extremities of lower thickness.
The surface was irregular. Synthetization seemed to culminate in low viscosity. The AlPO4 surface presented larger agglomerates, and the particles resembled loose scales. These scales or plaques may also have originated from the collapse of the structures upon rising heat treatment temperature. The larger block was rough, and contained cracks along the surface.
To conduct a more detailed study on the composition of the samples analyzed by SEM, EDX analysis was carried out.
Table 2 summarizes the percentage mass values of aluminum, phosphorus, zinc, oxygen, and carbon in the samples.
The amount of Zn(II) in the material decreased with the heat treatment temperature, because ZnO agglomerates arose, as verified by TEM. This provided further evidence that the ZnO agglomerates migrated to the surface of the polymeric matrix. Therefore, the samples heat-treated at higher temperatures should more efficient as photodegradation catalysts—the presence of these ZnO agglomerates on the surface of the matrix or around it should improve the quantum efficiency, consequently increasing the photogeneration of radicals that oxidize the target pesticide. As for AlPO4, the ratios between the matrix constituents remained the same even after treatment at high temperatures. The exception was the sample treated at 1000 °C, which had better crystalline organization.
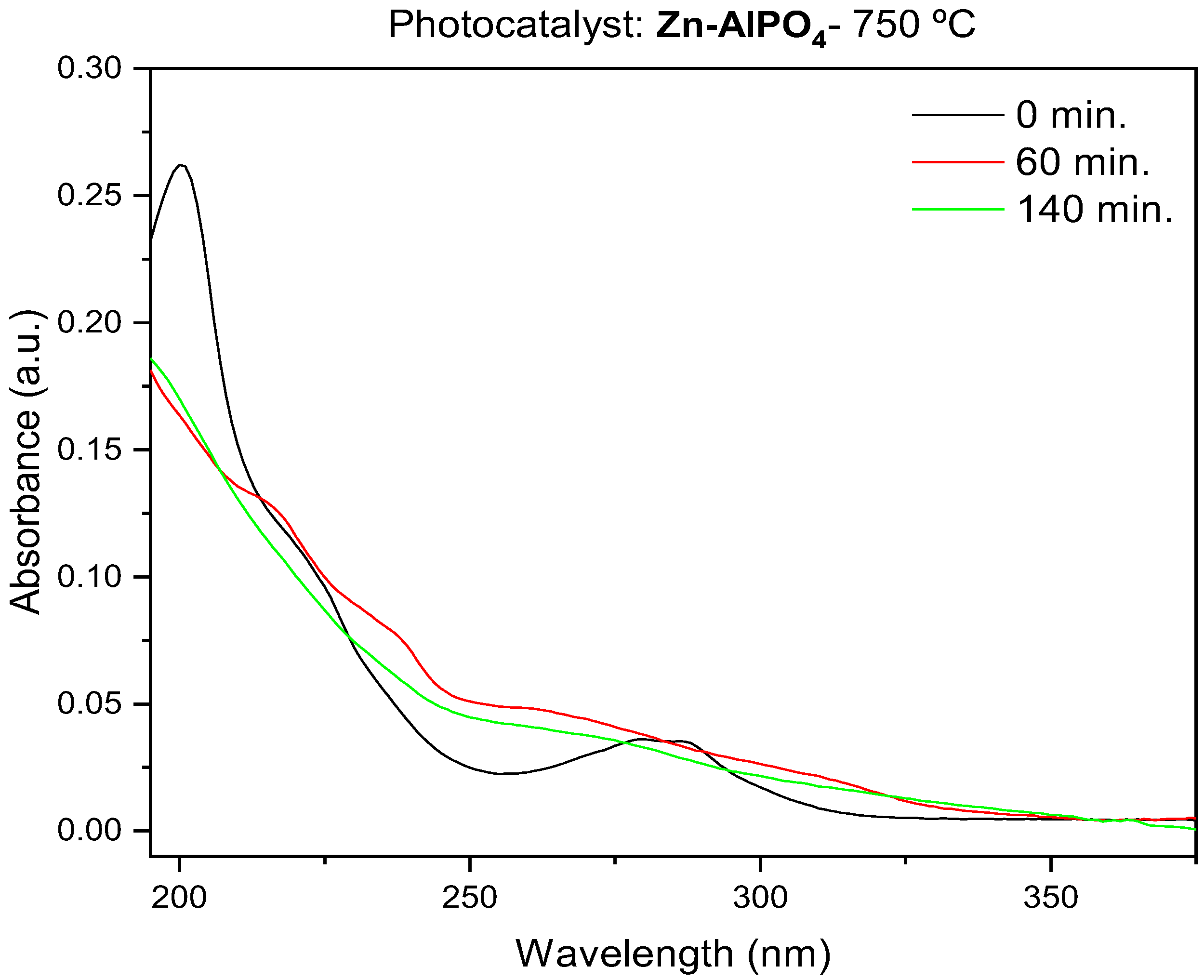
3.1. Photodegradation Studies
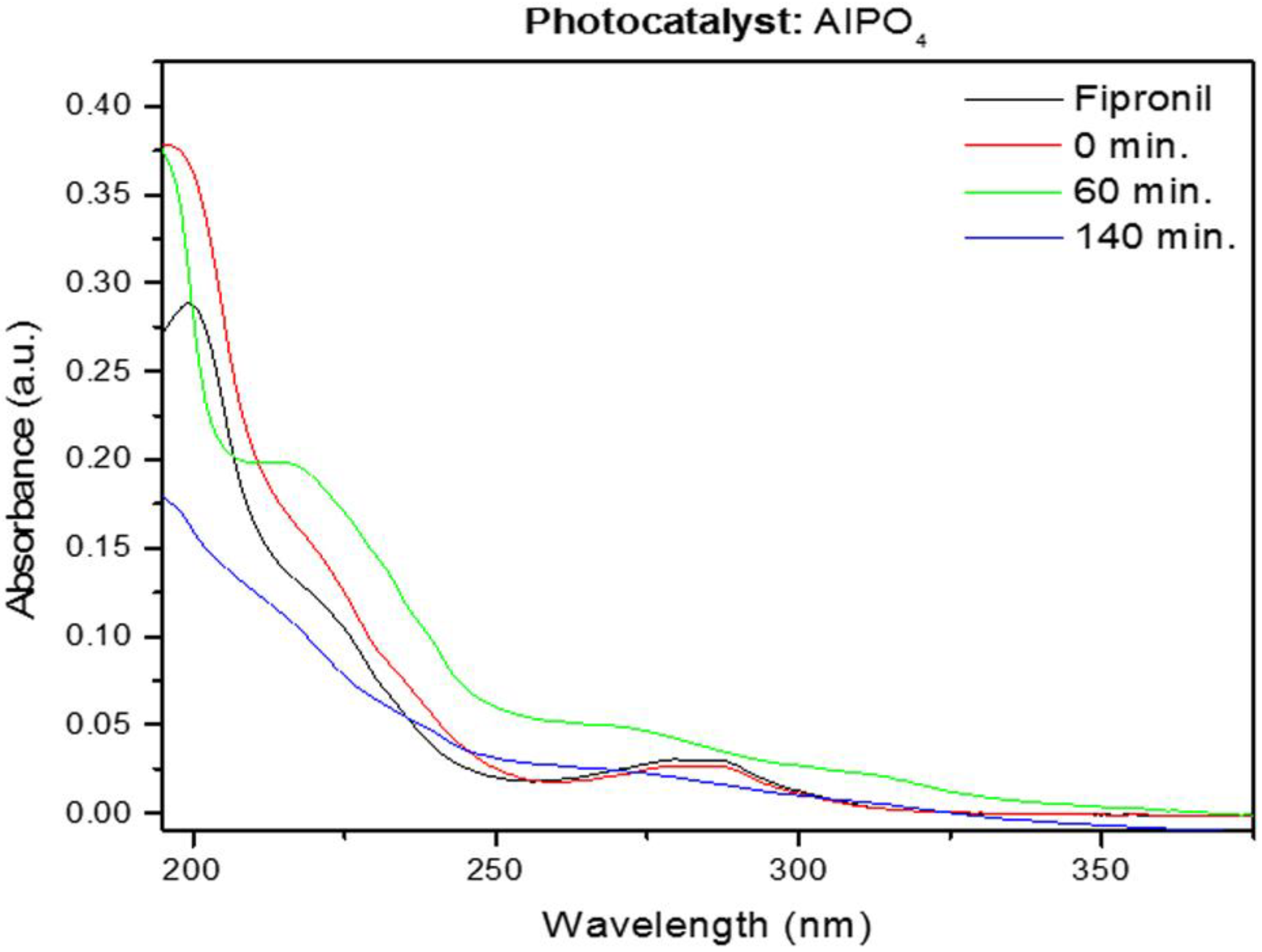
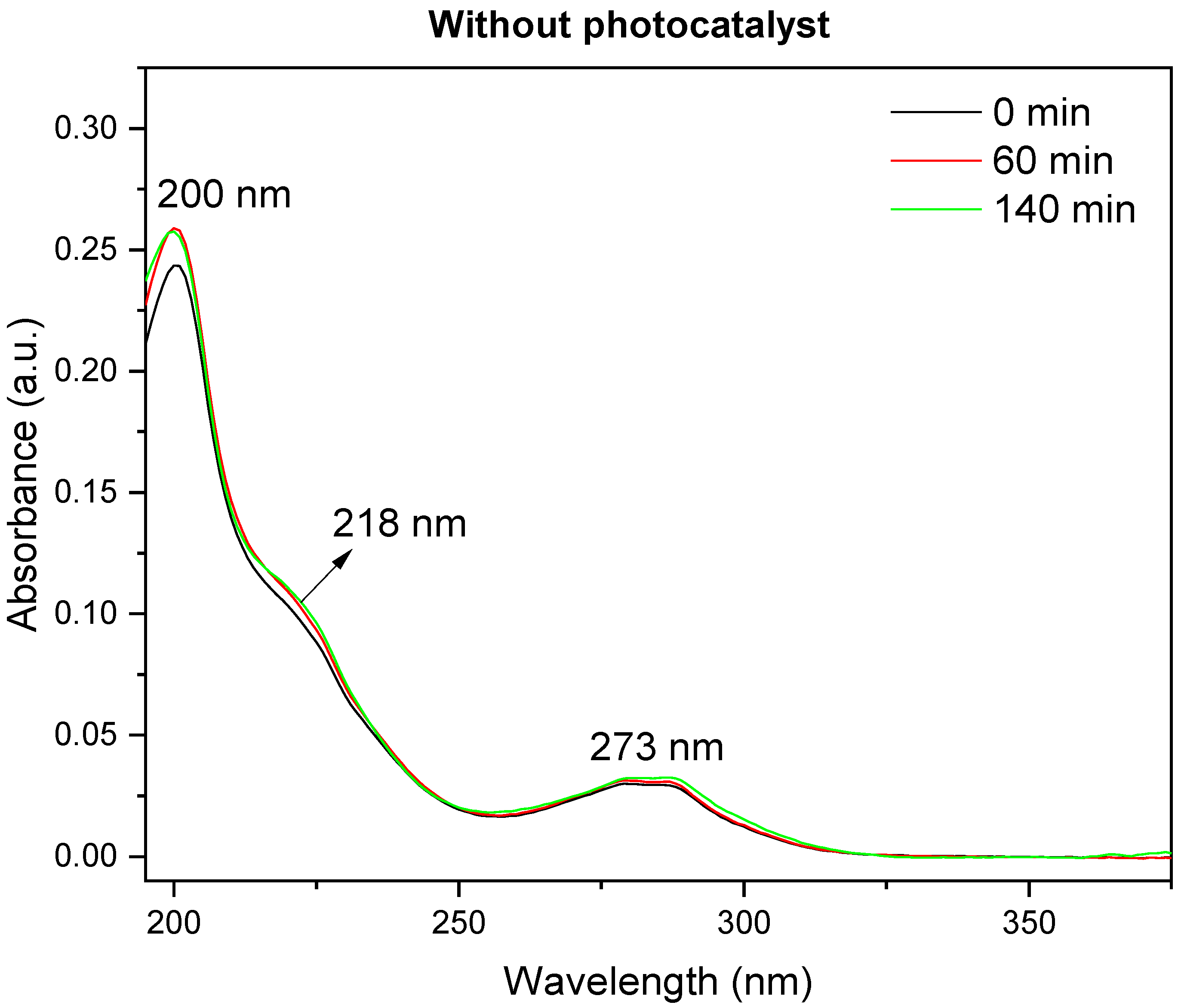
The photodegradation reactions lasted between 60 and 140 min, as monitored by UV spectroscopy (
Figure 11 and
Figure 12). The typical bands of fipronil at 196, 219, and 284 nm disappeared as a function of the time that the sample was exposed to UV light; the temperature and pH values were kept constant. The influence of temperature and pH on photodegradation was also investigated. Finally, the optimized conditions were combined, and GC–MS was used to study how AlPO
4 and Zn-AlPO
4 affected the photodegradation of fipronil.
According to previous studies, exposure to light prompts conversion of fipronil into its desulfurated derivative desthiofipronil, which has also been reported to act as an insecticide [
16,
40]. However, desthiofipronil is stable on the surface of leaves, which could culminate in persistence and non-target toxicity in the environment.
Analyses performed without a photocatalyst and in the presence of 153 mg of AlPO
4 (
Figure 12)—the designated blank (control) reaction under irradiation—revealed that AlPO
4 samples without the semiconductor (Zn(II) species) photodegraded fipronil. The UV/AlPO
4 system initially formed a novel species, as observed through the new UV bands that emerged at 218 and 273 nm. After 140 min, these species had disappeared, as confirmed by the decreased UV absorption and complete change in the UV spectral profile. Reactive groups (F, N, and Cl) in the structure of fipronil probably favored the formation of reactive sites on the surface of the AlPO
4 matrix, which generated oxidizing radicals even in the absence of the photoconductor. Nevertheless, considering the same period of exposure to radiation, Zn-AlPO
4 materials photodegraded fipronil more efficiently.
As noted by Ngim et al. [
30], photodegradation of fipronil under mechanical stirring and UV light radiation, in the absence of the catalyst, produced intermediate species. However, these intermediates seemed to regenerate the reactants throughout the experiment. These results suggested that photoinduced reactions did not occur simply due to UV light irradiation on the insecticide. Indeed, previous literature data demonstrate that total mineralization of certain pollutants exposed to radiation would require days or even months to occur [
41]. The detailed discussion on the formation of intermediate species is presented in the section on CG–MS.
Experiments conducted in the absence and in the presence of UV radiation helped to evaluate how UV light affected the photodegradation of fipronil. In a typical adsorption experiment accomplished in a dark chamber and under mechanical stirring, the concentration of the pesticide did not change over time, suggesting that both the photocatalyst and light radiation were necessary to promote the photodegradation of fipronil via hydroxyl radicals [
42].
The experiment accomplished in the presence of Zn-AlPO4 but in the absence of UV light did not provide the typical degradation profile of the intermediate species, indicating that Zn-AlPO4 only degraded the pesticide in the presence of UV artificial light source (253.7 nm). Fipronil clearly did not adsorb at the same intensity as it did throughout the exposure, because its concentration did not vary over the duration of the experiment. Indeed, BET textural analyses showed that the synthesized Zn-AlPO4 material hardly adsorbed substances, which may explain why the pesticide did not adsorb onto the matrix containing the photosensitizer.
Because not even temporary modification of the chemical structure of fipronil took place during the experiment conducted in the absence of the photocatalyst, efficient decomposition of fipronil must have depended on the presence of hydroxyl groups generated on the surface of Zn-AlPO4 under UV light, to yield the intermediate reactive species. These intermediate species allowed for complete mineralization of the pesticide.
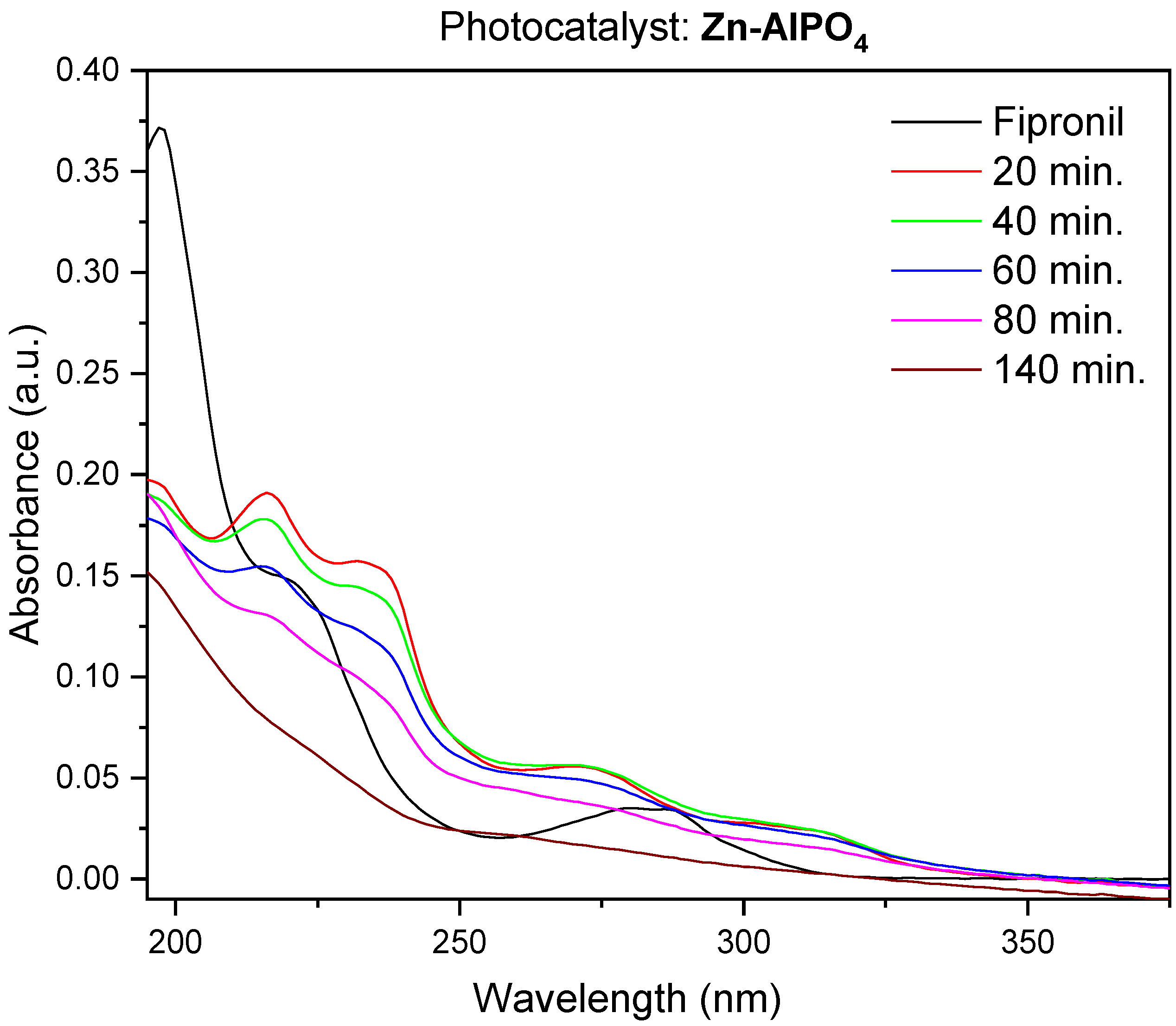
3.1.1. Influence of the Zn-AlPO4 Mass on the Photodegradation of Fipronil
Table 3 shows that photodegradation occurred as a function of the mass of Zn-AlPO
4 employed in the photodegradation experiment. The same conditions of photodegradation and an exposure time of 140 min did not degrade fipronil in the presence of 103 mg of the photocatalyst, regardless of the Zn-AlPO
4 heat treatment temperature. Nevertheless, the concentration of the pesticide decreased, confirming that photodegradation of fipronil occurred in the presence of catalysts treated at 400, 750, and 1000 °C.
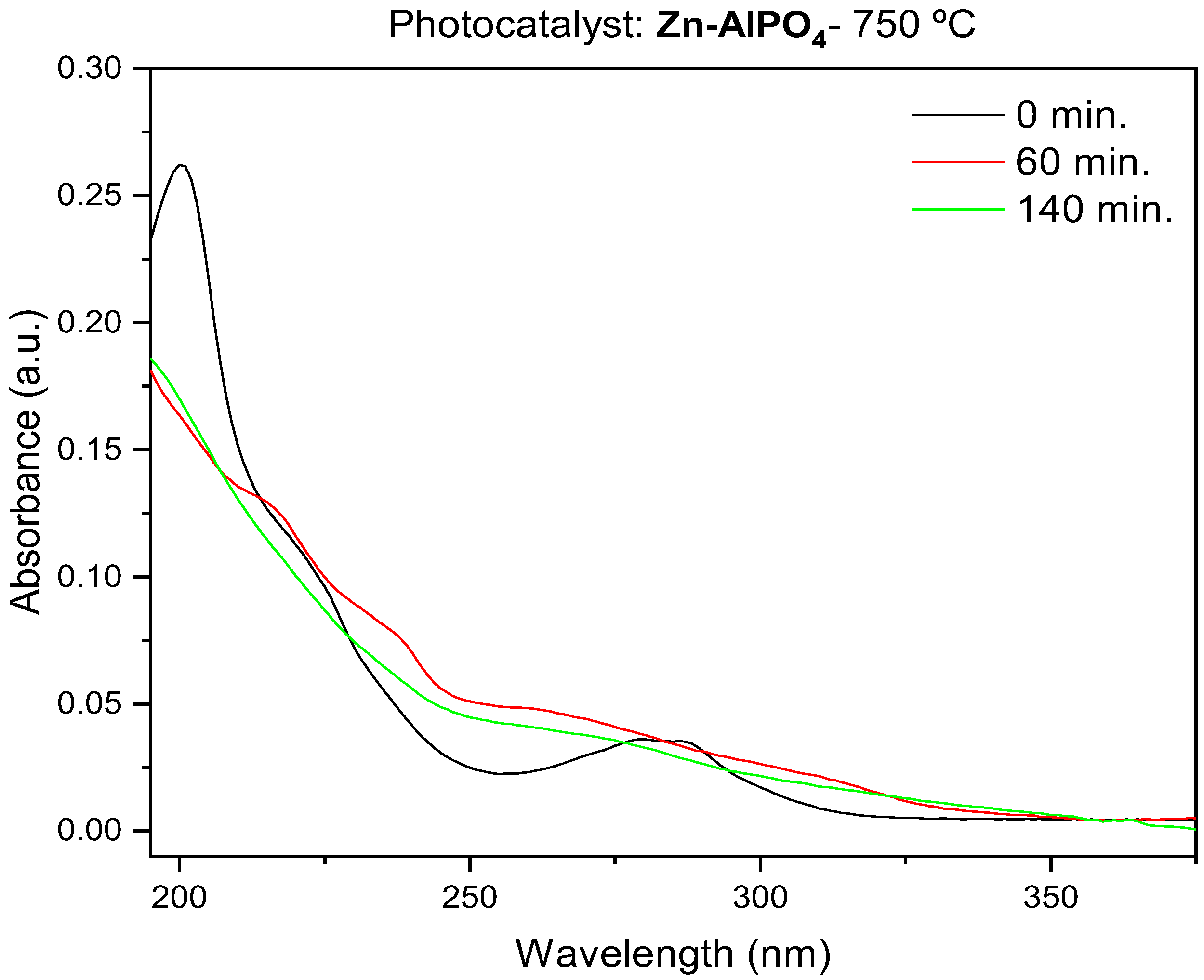
The catalyst calcined at 750 °C produced the best results, as shown in
Figure 13. The sample heated at 750 °C had a trigonal phase, assigned to AlZnP
2O
8. Compared to the hexagonal phase, the trigonal phase probably contributed to more efficient generation of hydroxyl radicals due to the presence of larger amounts of ZnO in the AlZnP
2O
8 phase at the surface of the unit cell. The temperature at which Zn-AlPO
4 was treated also influenced the morphology of the material, as noted from TEM analysis. Indeed, heat treatment was directly related to the formation of particles with features that favored the generation of the electron–gap pair on the surface of the catalyst [
43].
Among other factors, the efficiency of Zn-AlPO
4-750 may have been related to the treatment (higher temperatures) to which it was submitted after the synthesis. This treatment eliminated residual groups (e.g., chloride ions) that could interfere in the adsorption of the compound from which the reactive radicals originated [
44]. The presence of such interfering agents would culminate in an effect known as scavenging—sweeping of the radicals from the surface of the photocatalyst, to favor recombination of the electron–gap pair, with subsequent release of energy as heat. This would harm AOPs, not to mention that these groups could affect adsorption of the substance that generates the radicals on the semiconductor matrix. The experiment depicted revealed that a smaller mass of catalyst (103 mg) and longer exposure time (140 min) decomposed almost 100% of the pesticide. A higher mass of catalyst and longer reaction time (
Figure 13 and
Figure 14) indicated that Zn-AlPO
4 treated at 260 °C degraded the pesticide (Figure not shown).
A calcination temperature of 750 °C afforded the best photodegradation catalyst. All of the Zn-AlPO
4 materials prepared herein performed well during the degradation of fipronil. Degradation increased very quickly within a short time, especially for Zn-AlPO
4-750 and Zn-AlPO
4-1000. Eighty-percent photodegradation was achieved after only 2 h. Longer reaction times favored complete degradation. All of the calcined composites behaved similarly. These results resembled the results reported by Ngim et al. [
30] and Bóbe et al. [
18]. It is important to consider not only the photocatalytic nature of the composites, but also the amount of ZnO in the material when comparing the data obtained herein with the literature results. In some cases, our materials provided even better results than those reported by other authors. The photodegradation of fipronil as a function of time is shown in
Table 4.
Concerning Zn-AlPO
4 heat-treated at 1000 °C, a smaller mass of catalyst afforded better results. A series of studies have indicated that the photocatalytic degradation rate initially rises with larger photocatalyst mass, but later decreases significantly due to light scattering. A higher amount of the solid catalyst also favors agglomeration (particle–particle interaction); hence, lower surface area is available for light absorption, diminishing the rate of photocatalytic degradation. Although the number of active sites in the solution might increase with larger mass of the photocatalyst, the system reaches a point where excess particles no longer allow light to penetrate the catalyst. Therefore, there is an optimal mass that affords the highest efficiency possible. Further increase in the amount of the photocatalyst results in non-uniform distribution of light intensity, reducing the reaction yields despite the larger catalyst mass [
45].
The results showed that the best photocatalytic conditions were obtained using the higher mass of photocatalyst heat-treated at 750 °C; this enhanced photocatalytic activity was assigned to trigonal-phase AlZnP2O8, producing 80% fipronil removal without byproducts; the material heat-treated at 1000 °C presented very low photocatalytic activity under the same conditions—only 10% photodegradation and the presence of byproducts were observed by UV–Vis spectroscopy; this difference could be assigned to the orthorhombic berlinite phase that presented lower photocatalytic activity; the sintering of pores and migration of Zn phases from matrix could also explain this difference.
3.1.2. Investigation into the Photodegradation of Fipronil Catalyzed by Zn-AlPO4
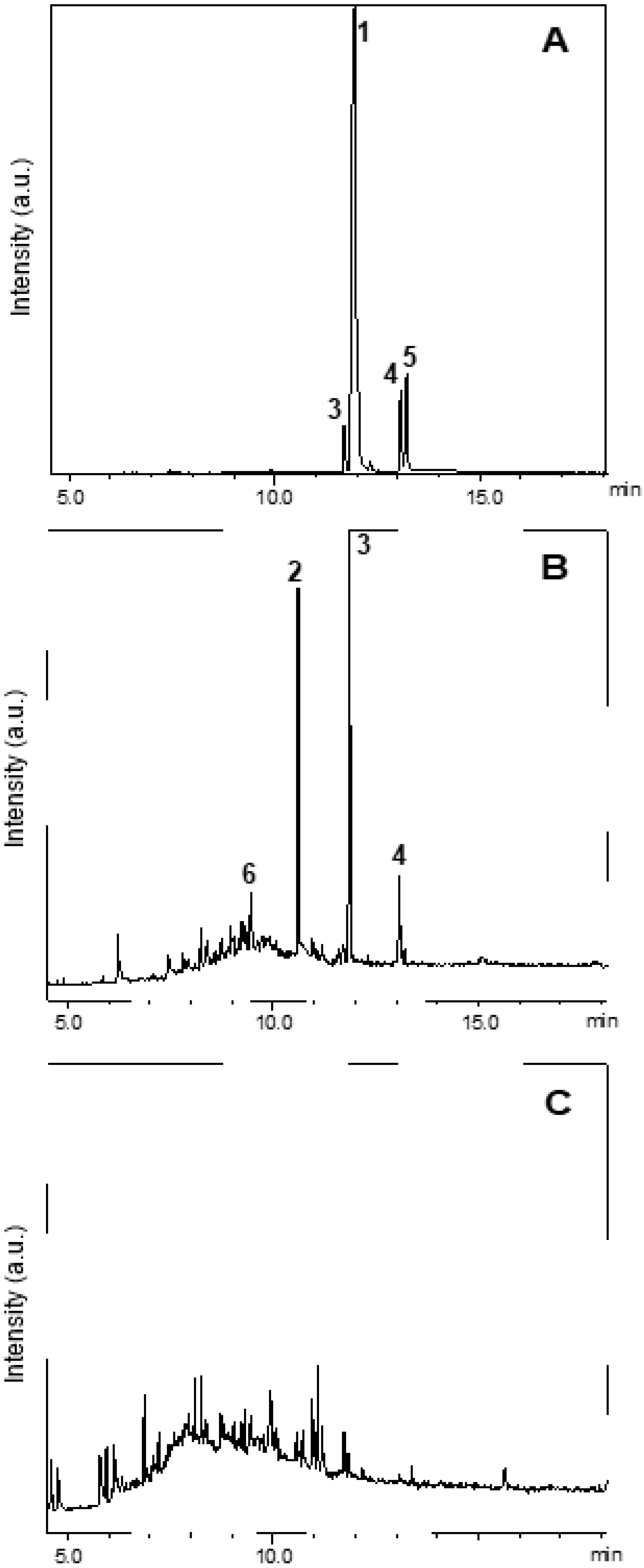
GC–MS helped to investigate the potential of Zn-AlPO
4 to catalyze the photodegradation of fipronil. To this end, an aqueous solution of fipronil (1 μg/mL) was prepared. Then, the solution was placed in a UV chamber in the absence of Zn-AlPO
4 for 2 h, and in the presence of Zn-AlPO
4 for 120 min, and photodegradation was measured as shown in
Figure 15.
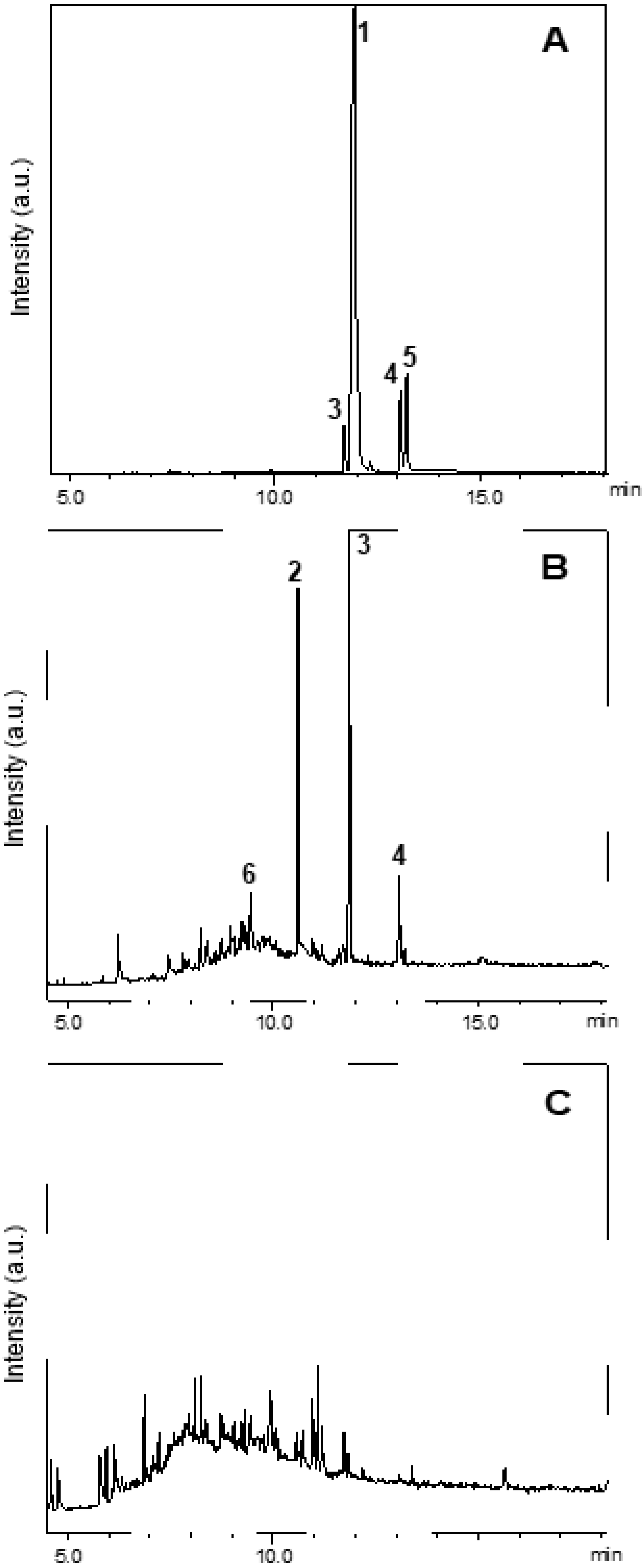
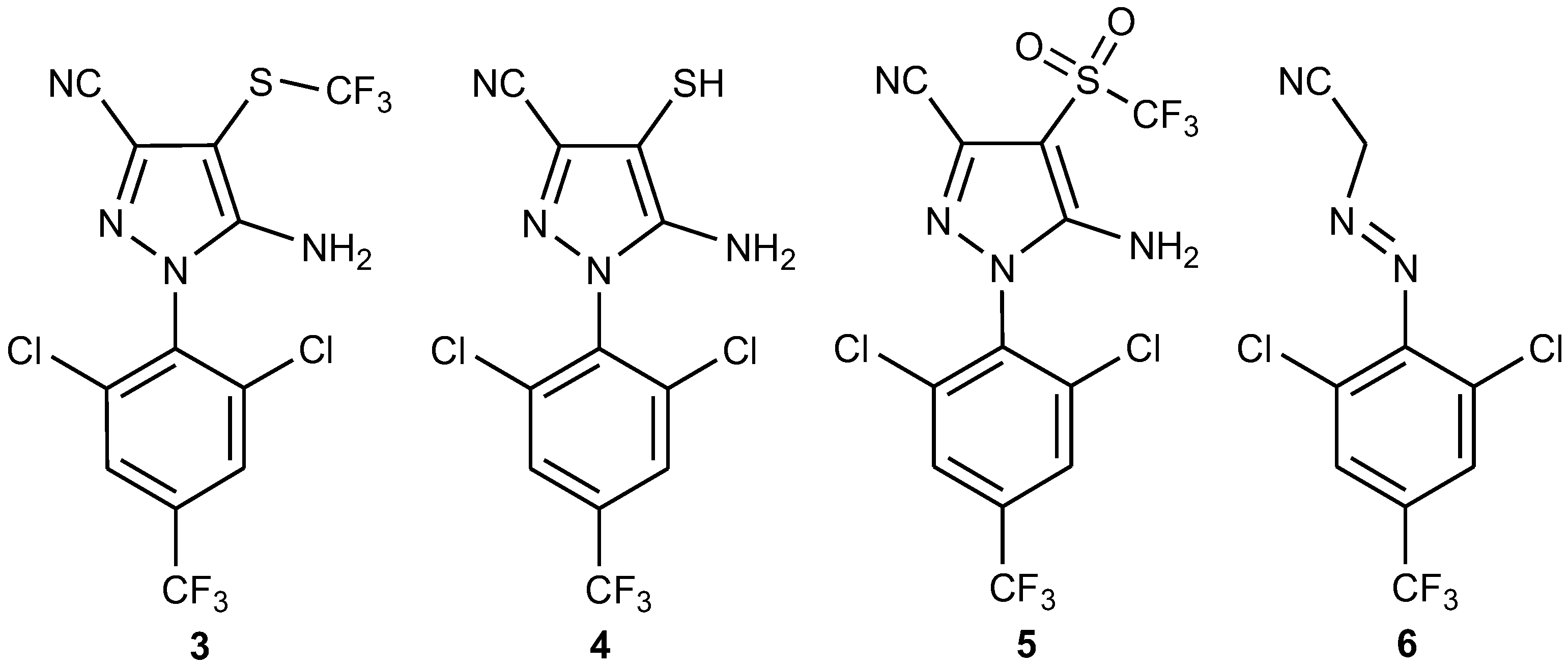
Figure 15A shows the chromatogram of fipronil (
1, R
t = 11.9 min). Peaks with retention times at 11.7 (
m/z 420), 13.1 (
m/z 352), and 13.2 min (
m/z 452) corresponded to fipronil–sulfide (
3), 4-thiol–fipronil (
4), and fipronil–sulfone (
5), respectively (
Figure 16). Compounds
3 and
4 corresponded to products from the photodegradation of fipronil [
32,
33,
46]. Compound
5 referred to an intermediate that emerged during conversion of
1 to desulfinyl–fipronil (
2) [
18].
Table 5 summarizes the MS data of compounds
1 to
6.
Comparison of the GC–MS chromatogram of fipronil with the GC–MS chromatogram obtained from exposure of the aqueous solution of fipronil to UV radiation in the sealed chamber revealed that fipronil generated mainly fipronil–sulfide (
3, R
t = 11.7 min) and desulfinyl–fipronil (
2, R
t = 10.6 min) after 2 h. Compound
6 (R
t = 9.4 min)—a product from the degradation of the pyrazole ring, and a known product from the photodegradation of fipronil—also emerged. Conversion of fipronil to its desthioderivative
2 has been exhaustively investigated in the literature, mainly due to the increased stability of this product as compared to fipronil [
46]. In this study, only traces of fipronil arose after 140 min of exposure of the aqueous solution of fipronil to UV radiation in the sealed chamber without Zn-AlPO
4.
The photodegradation of fipronil by Zn-AlPO
4 at 750 °C is confirmed in
Figure 15C, which shows only lower intensities of the characteristic peaks of fipronil and its byproducts; the peak intensity is quantitatively proportional to the pollutant concentration.
Analysis of the GC–MS chromatogram obtained for the aqueous solution of fipronil after 140 min under exposure to UV light, in the presence of the photocatalyst Zn-AlPO4, revealed that fipronil and its transformation products 2–6 underwent photodegradation. More detailed analysis of the MS data of each peak in this chromatogram evidenced total conversion of fipronil and its most commonly reported transformation products into a complex mixture of other photoproducts. Although it was not possible to identify these transformation products on the basis of GC–MS data alone, these results strongly suggested mineralization of fipronil and its transformation products. In summary, the results obtained in the photodegradation experiments reported herein suggest that the compound Zn-AlPO4 is a promising photocatalyst for the degradation of fipronil.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}