Fatty Acid Ethyl Esters in Virgin Olive Oils: In-House Validation of a Revised Method

 , and
, and
Abstract

1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
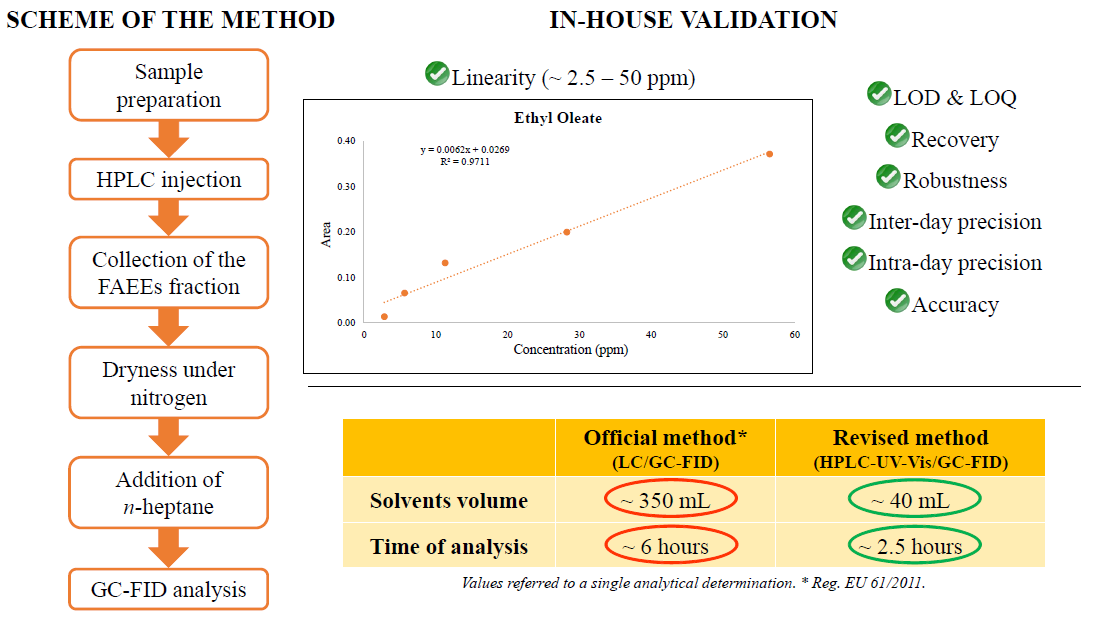
2.2. Determination of FAEEs by Off-Line HPLC-GC-FID
2.3. In-House Method Validation
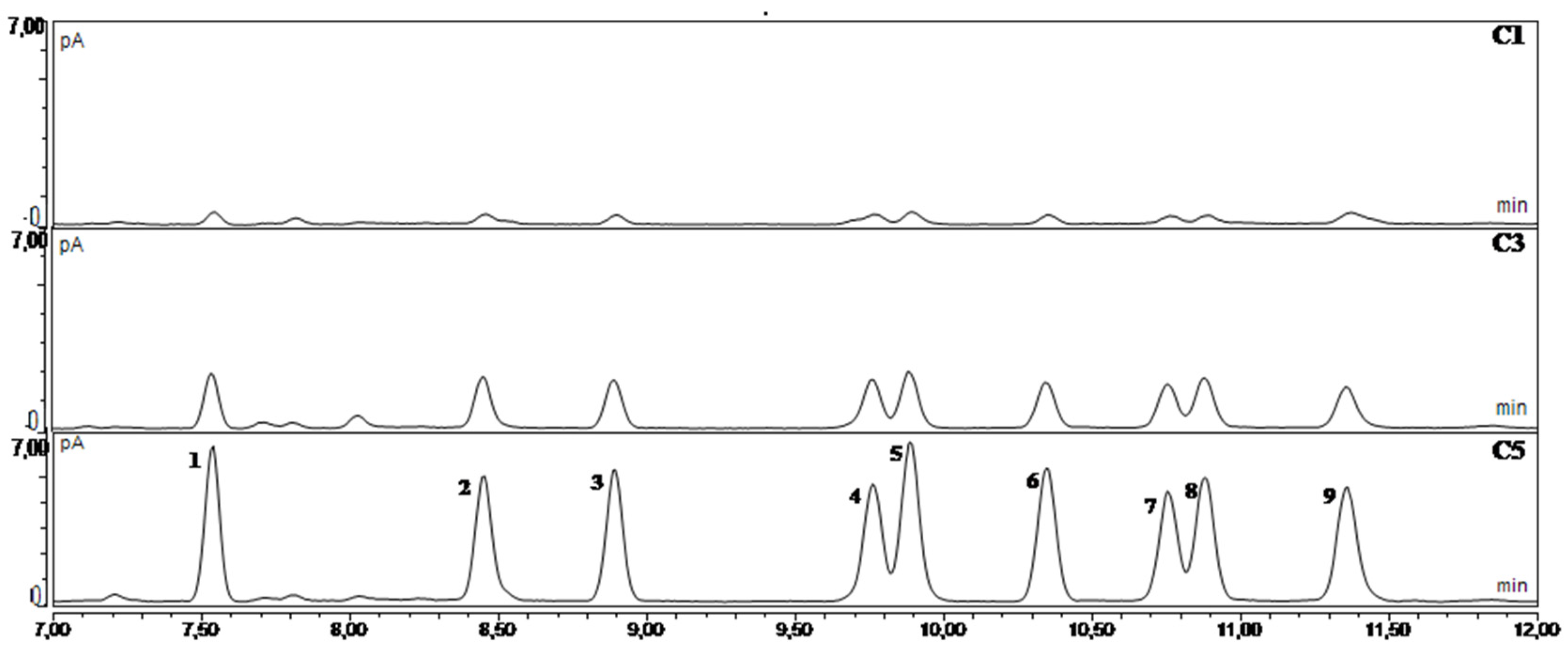
3. Results and Discussion
3.1. Development of the Method
3.2. Method Performance Determined with the In-House Validation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- United States Department of Agriculture. Oilseeds: World Markets and Trade. 2019. Available online: https://www.fas.usda.gov/data/oilseeds-world-markets-and-trade (accessed on 2 June 2020).
- Clodoveo, M.L.; Camposeo, S.; De Gennaro, B.; Pascuzzi, S.; Roselli, L. In the ancient world, virgin olive oil was called “liquid gold” by Homer and “the great healer” by Hippocrates. Why has this mythic image been forgotten? Food Res. Int. 2014, 62, 1062–1068. [Google Scholar] [CrossRef]
- Official Journal of the European Union. European Union, Commission Regulation 2568/91; Official Journal of the European Union: Brussels, Belgium, 1991; Volume L248, p. 1. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:01991R2568-20151016&from=EN (accessed on 2 June 2020).
- International Olive Council COI/T.20/Doc. No 28/Rev. 2 (2017). Determination of the Content of Waxes, Fatty Acid Methyl Esters and Fatty Acid Ethyl Esters by Capillary Gas Chromatography. Available online: https://www.internationaloliveoil.org/wp-content/uploads/2019/11/Method-COI-T.20-Doc.-No-28-Rev.2-2017.pdf (accessed on 29 May 2020).
- Official Journal of the European Union. European Union, Commission Regulation (EU) No 61/2011 amending Regulation (EEC) No 2568/91 on the Characteristics of Olive Oil and Olive-Residue Oil and on the Relevant Methods of Analysis; Official Journal of the European Union: Brussels, Belgium, 2011; Volume L23, pp. 1–14. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ%3AL%3A2011%3A023%3A0001%3A0014%3AEN%3APDF (accessed on 29 May 2020).
- Mariani, C.; Fedeli, E. Individuazione di oli di estrazione in quelli di pressione. Nota I. Riv. Ital. Sostanze Gr. 1986, 63, 3–17. [Google Scholar]
- Biedermann, M.; Bongartz, A.; Mariani, C.; Grob, K. Fatty acid methyl and ethyl esters as well as wax esters for evaluating the quality of olive oils. Eur. Food Res. Technol. 2008, 228, 65–74. [Google Scholar] [CrossRef]
- Conte, L.; Bendini, A.; Valli, E.; Lucci, P.; Moret, S.; Maquet, A.; Lacoste, F.; Brereton, P.; García-González, D.L.; Moreda, W.; et al. Olive oil quality and authenticity: A review of current EU legislation, standards, relevant methods of analyses, their drawbacks and recommendations for the future. Trends Food Sci. Technol. 2019, in press. [Google Scholar] [CrossRef]
- Pérez-Camino, M.C.; Cert, A.; Romero-Segura, A.; Cert-Trujillo, R.; Moreda, W. Alkyl esters of fatty acids a useful tool to detect soft deodorized olive oils. J. Agric. Food Chem. 2008, 56, 6740–6744. [Google Scholar] [CrossRef] [PubMed]
- Conte, L.; Mariani, C.; Gallina Toschi, T.; Tagliabue, S. Alkyl esters and related compounds in virgin olive oils: Their evolution over time. Riv. Ital. Sostanze Gr. 2014, 91, 21–29. [Google Scholar]
- Squeo, G.; Grassi, S.; Paradiso, V.M.; Alamprese, C.; Caponio, F. FT-IR extra virgin olive oil classification based on ethyl ester content. Food Control 2019, 102, 149–156. [Google Scholar] [CrossRef]
- Pérez-Camino, M.C.; Moreda, W.; Mateos, R.; Cert, A. Determination of esters of fatty acids with low molecular weight alcohols in olive oils. J. Agric. Food Chem. 2002, 50, 4721–4725. [Google Scholar] [CrossRef]
- Mariani, C.; Bellan, G. Detection of low quality oils in extra virgin olive oils. Riv. Ital. Sostanze Gr. 2008, 85, 3–20. [Google Scholar]
- Gómez-Coca, R.B.; Moreda, W.; Pérez-Camino, M.C. Fatty acid alkyl esters presence in olive oil vs. organoleptic assessment. Food Chem. 2012, 135, 1205–1209. [Google Scholar] [CrossRef]
- Di Serio, M.G.; Giansante, L.; Di Loreto, G.; Faberi, A.; Ricchetti, L.; Di Giacinto, L. Ethyl esters versus fermentative organoleptic defects in virgin olive oil. Food Chem. 2017, 219, 33–39. [Google Scholar] [CrossRef]
- Official Journal of the European Union. Commission Implementing Regulation (EU) No 1348/2013 of 16 December 2013 amending Regulation (EEC) No 2568/91 on the Characteristics of Olive Oil and Olive-Residue Oil and on the Relevant Methods of Analysis; Official Journal of the European Union: Brussels, Belgium, 2013; Volume L338, pp. 31–67. Available online: https://eur-lex.europa.eu/legal-content/GA/TXT/?uri=CELEX:32013R1348 (accessed on 29 May 2020).
- Official Journal of the European Union. Commission Delegated Regulation (EU) No 2019/1604 of 27 September 2019 amending Regulation (EEC) No 2568/91 on the Characteristics of Olive Oil and Olive-Residue Oil and on the Relevant Methods of Analysis; Official Journal of the European Union: Brussels, Belgium, 2019; Volume L250, pp. 14–48. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32019R1604&from=EN (accessed on 29 May 2020).
- Valli, E.; Bendini, A.; Maggio, R.M.; Cerretani, L.; Gallina Toschi, T.; Casiraghi, E.; Lercker, G. Detection of low-quality extra virgin olive oils by fatty acid alkyl esters evaluation: A preliminary and fast mid-infrared spectroscopy discrimination by a chemometric approach. Int. J. Food Sci. Technol. 2013, 48, 548–555. [Google Scholar] [CrossRef]
- Jabeur, H.; Zribi, A.; Abdelhedi, R.; Bouaziz, M. Effect of olive storage conditions on Chemlali olive oil quality and the effective role of fatty acids alkyl esters in checking olive oils authenticity. Food Chem. 2015, 169, 289–296. [Google Scholar] [CrossRef]
- International Olive Council COI/T.20/Doc. No 31 (2012). Determination of the Content of Waxes, Fatty Acid Methyl Esters and Fatty Acid Ethyl Esters by Capillary Gas Chromatography Using 3 Grams of Silica. Available online: https://www.internationaloliveoil.org/wp-content/uploads/2019/11/Method-COI-T.20-Doc.-No-31-2012.pdf (accessed on 29 May 2020).
- Purcaro, G.; Barp, L.; Conte, L. Comparison of different injection modes in edible oil minor components analysis. J. Sep. Sci. 2015, 38, 2278–2285. [Google Scholar] [CrossRef] [PubMed]
- Küchler, T.; Nestola, M.; Boysen, H.; Tablack, P. An on-line coupled HPLC-GC-FID method for the determination of fatty acid methyl and ethyl esters in olive oil. In Book of Abstract 12th Euro Fed Lipid Congress; European Federation for the Science and Technology of lipid: Frankfurt, Germany; Montpellier, France, 14–17 September 2014; p. 504. [Google Scholar]
- Boggia, R.; Borgogni, C.; Hysenaj, V.; Leardi, R.; Zunin, P. Direct GC–(EI)MS determination of fatty acid alkyl esters in olive oils. Talanta 2014, 119, 60–67. [Google Scholar] [CrossRef]
- Uncu, O.; Ozen, B.; Tokatli, F. Use of FTIR and UV–visible spectroscopy in determination of chemical characteristics of olive oils. Talanta 2019, 201, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Berardinelli, A.; Ragni, L.; Bendini, A.; Valli, E.; Conte, L.; Gallina Toschi, T. Rapid screening of fatty acid alkyl esters in olive oils by time domain reflectometry. J. Agric. Food Chem. 2013, 61, 10919–10924. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Varo, A.; Sánchez, M.T.; De la Haba, M.J.; Torres, I.; Pérez-Marín, D. Fast, low-cost and non-destructive physico-chemical analysis of virgin olive oils using near-infrared reflectance spectroscopy. Sensors 2017, 17, 2642. [Google Scholar] [CrossRef] [PubMed]
- Cayuela, J.A. Rapid NIR determination of alkyl esters in virgin olive oil. Grasas Aceites 2017, 68, 1–8. [Google Scholar] [CrossRef]
- Sanagi, M.M.; Nasir, Z.; Ling, S.L.; Hermawan, D.; Wan Ibrahim, W.A.; Abu Naim, A. A Practical Approach for Linearity Assessment of Calibration Curves Under the International Union of Pure and Applied Chemistry (IUPAC) Guidelines for an In-House Validation of Method of Analysis. J. AOAC Int. 2010, 93, 1322–1330. [Google Scholar] [CrossRef]
- González, G.A.; Herrador, M.Á. A practical guide to analytical method validation, including measurement uncertainty and accuracy profiles. Trends Analyt. Chem. 2007, 26, 227–238. [Google Scholar] [CrossRef]
- Palagano, R.; Valli, E.; Tura, M.; Perez-Camino, M.C.; Moreda, W.; Bendini, A.; Gallina Toschi, T. OLEUM Project. Revised Method for Determination of Fatty Acid Ethyl Esters in Virgin Olive Oils. Data from in-House Validation. 2020. Available online: http://amsacta.unibo.it/6263/[Dataset] (accessed on 2 June 2020).
- Genualdi, S.; MacMahon, S.; Robbins, K.; Farris, S.; Shyong, N.; DeJager, L. Method development and survey of Sudan I–IV in palm oil and chilli spices in the Washington, DC, area. Food Addit. Contam. 2016, 33, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Engewald, W.; Teske, J.; Efer, J. Programmed temperature vaporiser-based injection in capillary gas chromatography. J. Chromatogr. A 1999, 856, 259–278. [Google Scholar] [CrossRef]
- Peters, F.T.; Drummer, O.H.; Musshoff, F. Validation of new methods. Forensic Sci. Int. 2007, 165, 216–224. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Compound | Relative Retention Time (RRT) |
|---|---|
| Ethyl palmitate | 0.95 |
| Ethyl linoleate | 1.21 |
| Ethyl oleate | 1.22 |
| Ethyl stearate | 1.28 |
| Methyl heptadecanoate | 1.00 |
| Source | SS | d.f. | MS | Fcal | Fcrit | Conclusion | |
|---|---|---|---|---|---|---|---|
| EP | Regression | 0.047754 | 1 | 0.047754 | 463.6 | 16.3 | Regression model accepted |
| Lack-of-it | 0.002470 | 3 | 0.000823 | 7.9 | 12.1 | Linearity accepted | |
| Pure error | 0.002987 | 5 | 0.000103 | ||||
| EL | Regression | 0.038275 | 1 | 0.038275 | 329.1 | 16.3 | Regression model accepted |
| Lack-of-it | 0.002824 | 3 | 0.000941 | 8.1 | 12.1 | Linearity accepted | |
| Pure error | 0.000582 | 5 | 0.000116 | ||||
| EO | Regression | 0.048209 | 1 | 0.048209 | 319.3 | 16.3 | Regression model accepted |
| Lack-of-it | 0.005476 | 3 | 0.001825 | 12.1 | 12.1 | Linearity accepted | |
| Pure error | 0.000756 | 5 | 0.000151 | ||||
| ES | Regression | 0.050871 | 1 | 0.050871 | 860.7 | 16.3 | Regression model accepted |
| Lack-of-it | 0.001506 | 3 | 0.000502 | 8.5 | 12.1 | Linearity accepted | |
| Pure error | 0.000295 | 5 | 0.000059 |
| Compound | Recovery C3 (%) | Recovery C4 (%) | Recovery C5 (%) |
|---|---|---|---|
| Ethyl palmitate | 73.2 ± 3.6 c | 101.8 ± 1.3 a | 90.4 ± 2.0 b |
| Ethyl linoleate | 72.3 ± 6.9 b | 94.9 ± 3.2 a | 84.0 ± 0.6 a,b |
| Ethyl oleate | 71.9 ± 1.3 c | 94.6 ± 1.9 a | 86.6 ± 1.0 b |
| Ethyl stearate | 84.9 ± 2.9 c | 102.9 ± 2.9 a | 94.1 ± 1.0 b |
| Methyl heptadecanoate | 74.7 ± 3.0 c | 98.7 ± 1.6 a | 87.7 ± 1.2 b |
| Compound | Day 1 | Day 2 | Day 3 | p-Value |
|---|---|---|---|---|
| Ethyl palmitate | 1.17 ± 0.06 | 1.17 ± 0.03 | 1.11 ± 0.03 | 0.17 |
| Ethyl linoleate | 0.97 ± 0.02 | 0.95 ± 0.03 | 0.97 ± 0.02 | 0.56 |
| Ethyl oleate | 1.03 ± 0.02 | 1.05 ± 0.03 | 1.06 ± 0.02 | 0.36 |
| Ethyl stearate | 0.91 ±0.12 | 1.03 ± 0.07 | 0.99 ± 0.11 | 0.43 |
| Sample | LC | MC | HC | |||
|---|---|---|---|---|---|---|
| Compound | Official Method | Revised Method | Official Method | Revised Method | Official Method | Revised Method |
| Ethyl palmitate | 4.1 ± 0.6 | 7.9 ± 1.4 | 8.2 ± 0.0 | 10.3 ± 0.8 | 11.4 ± 0.5 | 8.4 ± 0.0 |
| Ethyl linoleate | 0.9 ± 0.1 | <LOQ | 2.0 ± 0.1 | <LOQ | 5.2 ± 0.1 | 5.2 ± 1.3 |
| Ethyl oleate | 12.0 ± 0.3 | 9.9 ± 0.5 | 29.6 ± 0.7 | 28.4 ± 1.2 | 63.4 ± 2.1 | 60.8 ± 4.0 |
| Ethyl stearate | 1.2 ± 0.1 | <LOQ | <LOQ | <LOQ | 2.2 ± 0.3 | 10.7 ± 3.4 |
| Total FAEEs | 18.4 ± 1.1 | 17.8 ± 1.0 | 39.8 ± 0.7 | 38.7 ± 0.4 | 82.1 ± 2.8 | 85.1 ± 0.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palagano, R.; Valli, E.; Tura, M.; Cevoli, C.; Pérez-Camino, M.d.C.; Moreda, W.; Bendini, A.; Gallina Toschi, T. Fatty Acid Ethyl Esters in Virgin Olive Oils: In-House Validation of a Revised Method. Foods 2020, 9, 924. https://doi.org/10.3390/foods9070924
Palagano R, Valli E, Tura M, Cevoli C, Pérez-Camino MdC, Moreda W, Bendini A, Gallina Toschi T. Fatty Acid Ethyl Esters in Virgin Olive Oils: In-House Validation of a Revised Method. Foods. 2020; 9(7):924. https://doi.org/10.3390/foods9070924
Chicago/Turabian StylePalagano, Rosa, Enrico Valli, Matilde Tura, Chiara Cevoli, María del Carmen Pérez-Camino, Wenceslao Moreda, Alessandra Bendini, and Tullia Gallina Toschi. 2020. "Fatty Acid Ethyl Esters in Virgin Olive Oils: In-House Validation of a Revised Method" Foods 9, no. 7: 924. https://doi.org/10.3390/foods9070924
APA StylePalagano, R., Valli, E., Tura, M., Cevoli, C., Pérez-Camino, M. d. C., Moreda, W., Bendini, A., & Gallina Toschi, T. (2020). Fatty Acid Ethyl Esters in Virgin Olive Oils: In-House Validation of a Revised Method. Foods, 9(7), 924. https://doi.org/10.3390/foods9070924