
A Novel PCR-Based Tool to Trace Oenological Saccharomyces cerevisiae Yeast by Monitoring Strain-Specific Nucleotide Polymorphisms
 , , ,
, , ,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Yeast Manipulation and Genomic DNA Extraction
2.2. Yeast Strain Mixtures
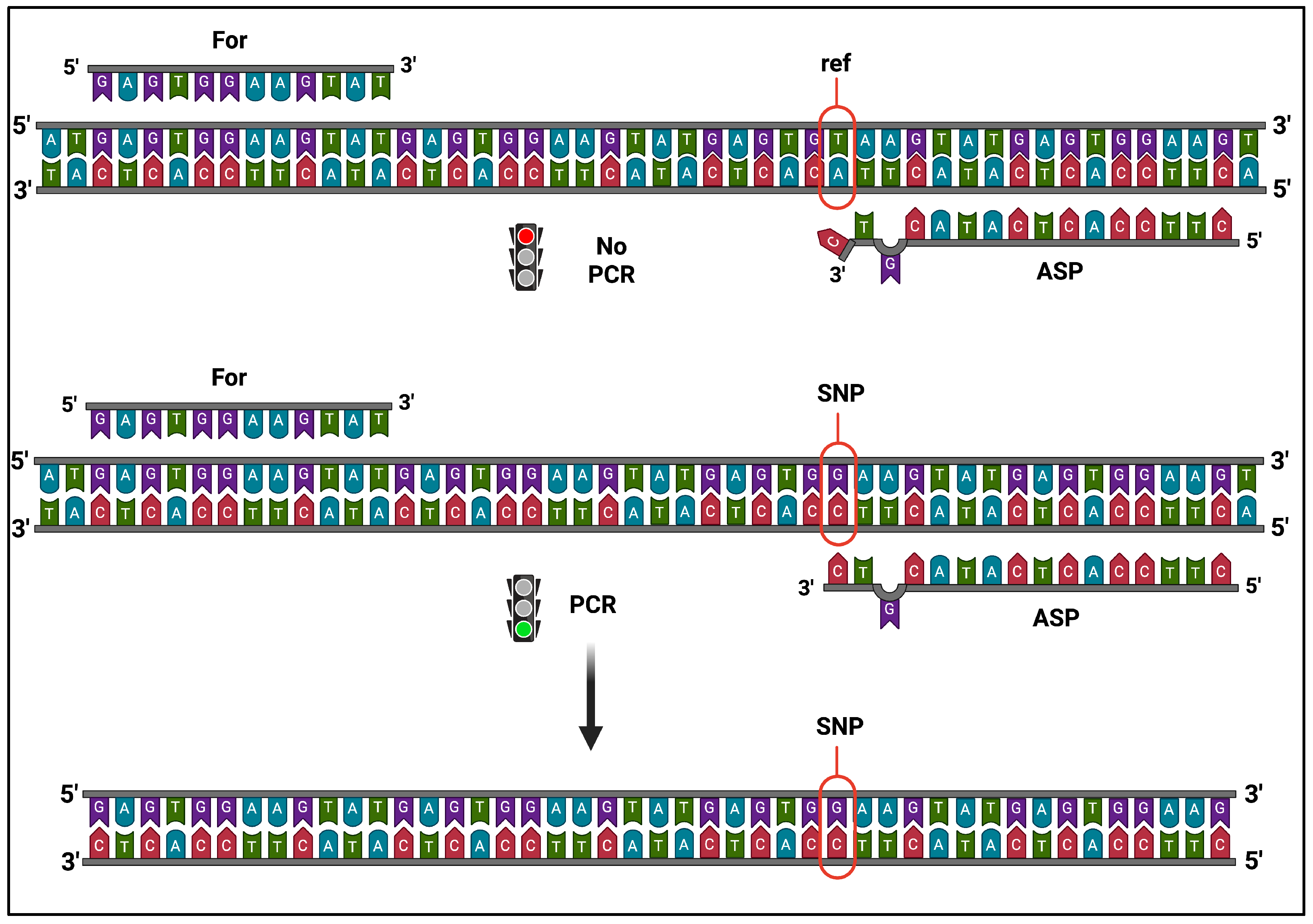
2.3. Primer Design
2.4. Multiplex Allele-Specific Primer PCR (AS-PCR)
2.5. Inter-Delta Assay
2.6. Variant Calling and Analysis
2.7. Yeast Competitive Assay in Stuck Fermentation Conditions
3. Results
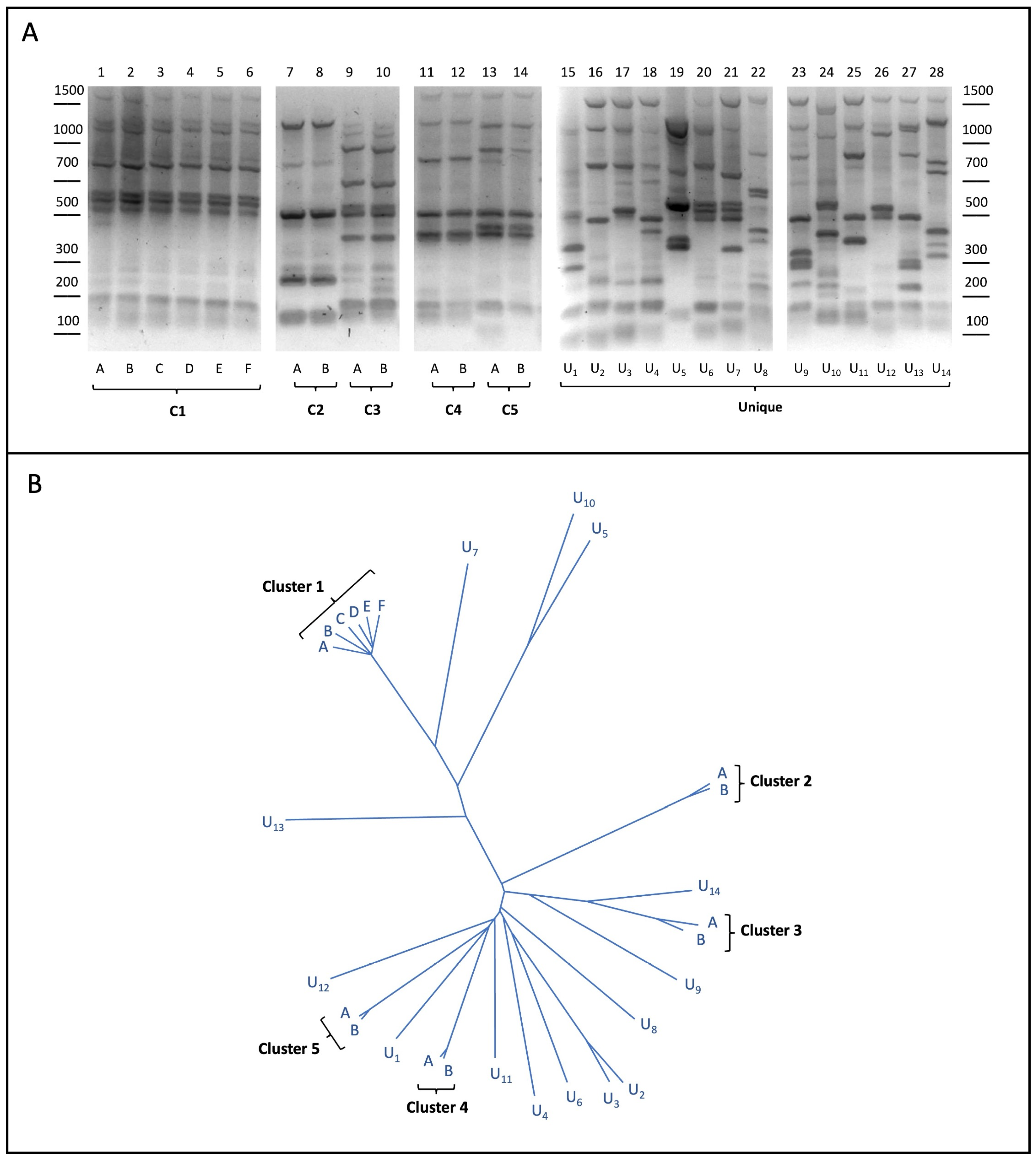
3.1. Yeast Strain Selection
3.2. Computational Identification of Strain-Specific SNPs
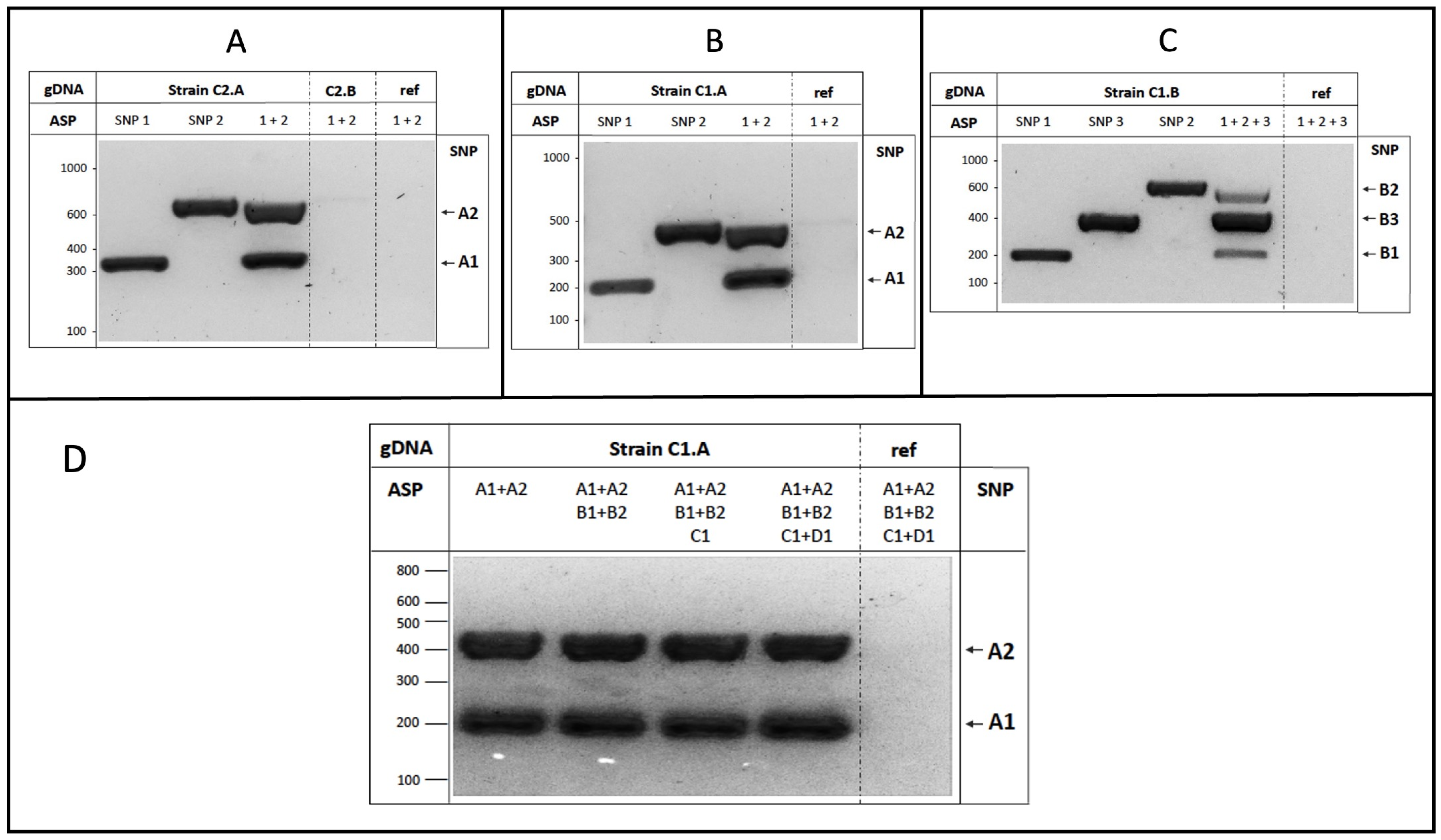
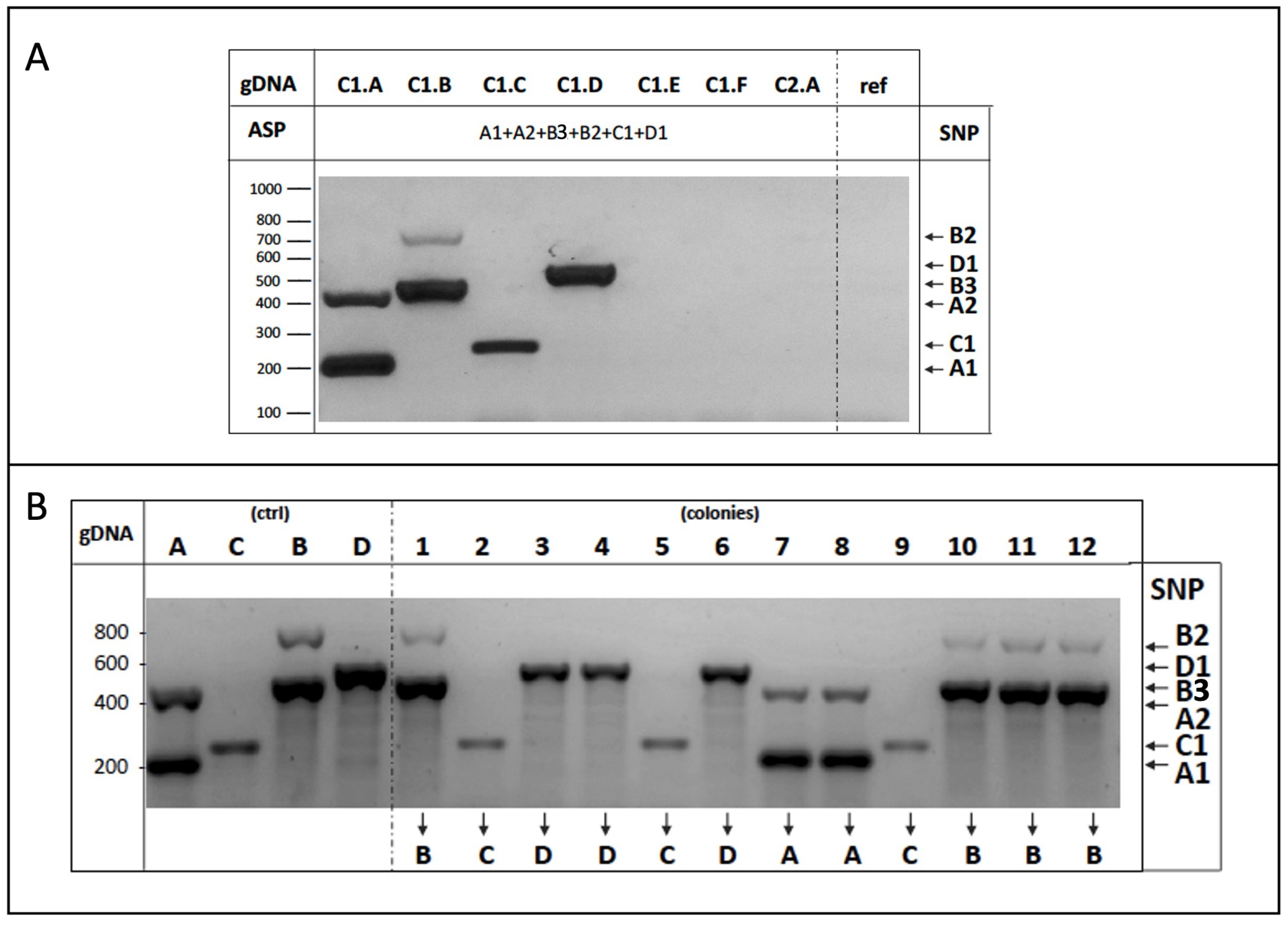
3.3. Experimental Validation of the Strain-Specific SNPs
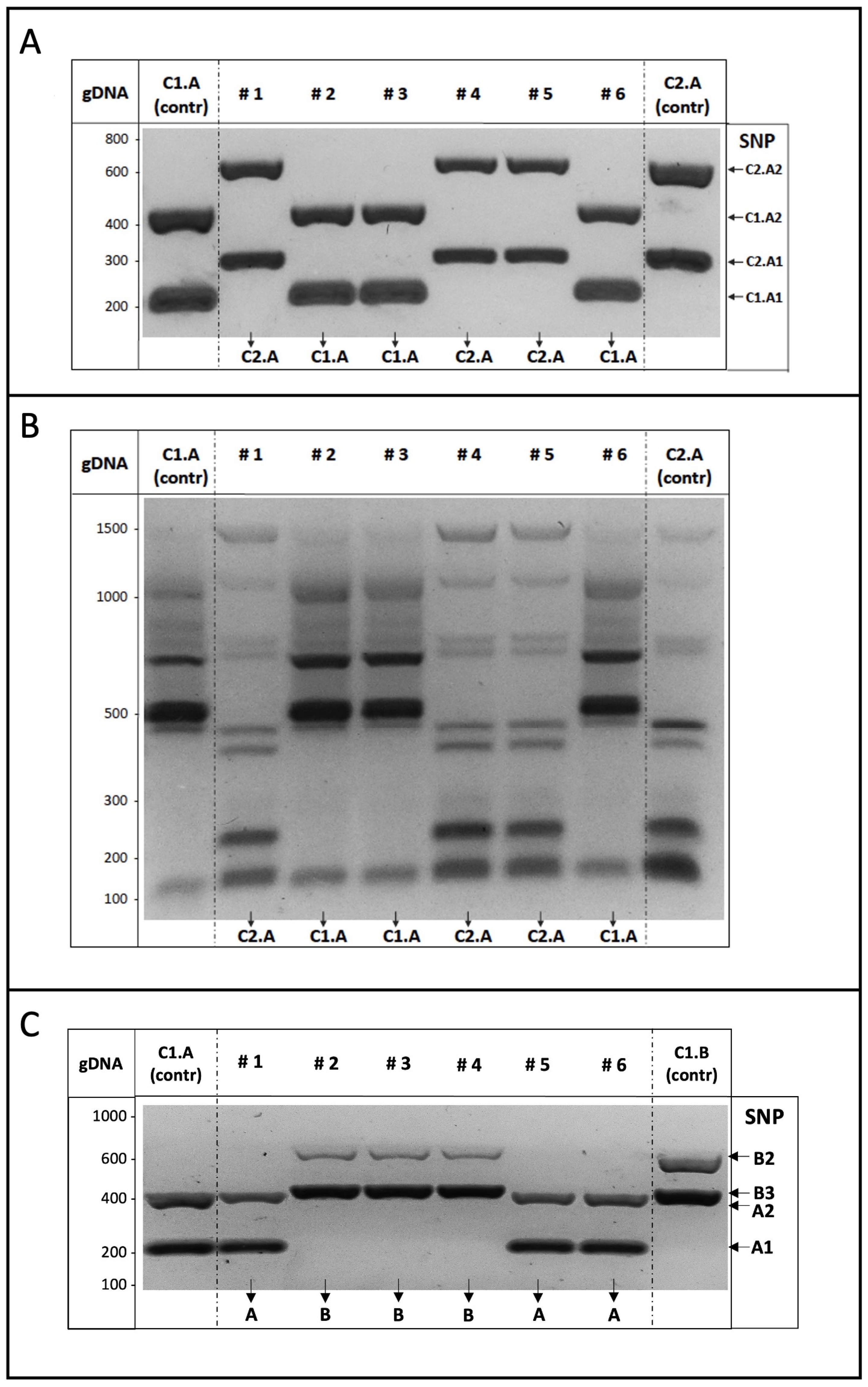
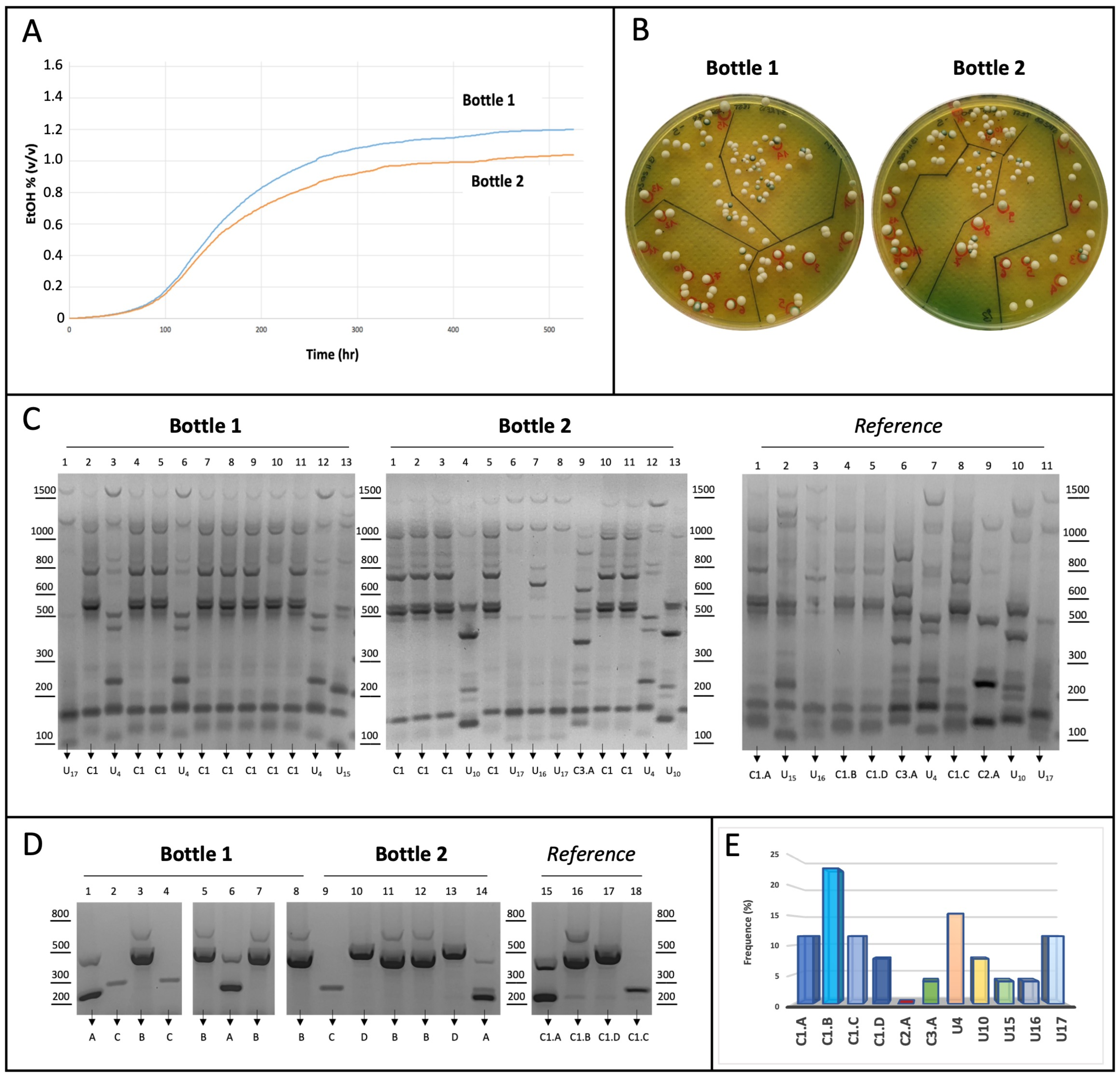
3.4. Application of the ASP-PCR Strategy in Laboratory Assays
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goddard, M.R. Quantifying the complexities of Saccharomyces cerevisiae’s ecosystem engineering via fermentation. Ecology 2008, 89, 2077–2082. [Google Scholar] [CrossRef]
- Salvadó, Z.; Arroyo-López, F.N.; Barrio, E.; Querol, A.; Guillamón, J.M. Quantifying the individual effects of ethanol and temperature on the fitness advantage of Saccharomyces cerevisiae. Food Microbiol. 2011, 28, 1155–1161. [Google Scholar] [CrossRef]
- Lopes, C.A.; Rodríguez, M.E.; Sangorrín, M.; Querol, A.; Caballero, A.C. Patagonian wines: The selection of an indigenous yeast starter. J. Ind. Microbiol. Biotechnol. 2007, 34, 539–546. [Google Scholar] [CrossRef]
- Marsit, S.; Dequin, S. Diversity and adaptive evolution of Saccharomyces wine yeast: A review. FEMS Yeast Res. 2015, 15, fov067. [Google Scholar] [CrossRef]
- Rainieri, S.; Pretorius, I.S. Selection and improvement of wine yeasts. Ann. Microbiol. 2000, 50, 15–31. [Google Scholar]
- Mateo, J.J.; Jimenez, M.; Pastor, A.; Huerta, T. Yeast starter cultures affecting wine fermentation and volatiles. Food Res. Int. 2001, 34, 307–314. [Google Scholar] [CrossRef]
- Nykänen, L. Formation and Occurrence of Flavor Compounds in Wine and Distilled Alcoholic Beverages. Am. J. Enol. Vitic. 1986, 37, 84–96. [Google Scholar] [CrossRef]
- Romano, P.; Capece, A.; Serafino, V.; Romaniello, R.; Poeta, C. Biodiversity of wild strains of Saccharomyces cerevisiae as tool to complement and optimize wine quality. World J. Microbiol. Biotechnol. 2008, 24, 1797–1802. [Google Scholar] [CrossRef]
- Kurtzman, C.; Fell, J.W.; Boekhout, T. The Yeasts: A Taxonomic Study, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Guillamón, J.M.; Barrio, E. Genetic Polymorphism in Wine Yeasts: Mechanisms and Methods for Its Detection. Front. Microbiol. 2017, 8, 806. [Google Scholar] [CrossRef]
- Bezerra-Bussoli, C.; Alves Baffi, M.; Gomes, E.; Da Silva, R. Yeast Diversity Isolated from Grape Musts During Spontaneous Fermentation from a Brazilian Winery. Curr. Microbiol. 2013, 67, 356–361. [Google Scholar] [CrossRef]
- Díaz, C.; Molina, A.M.; Nähring, J.; Fischer, R. Characterization and dynamic behavior of wild yeast during spontaneous wine fermentation in steel tanks and amphorae. BioMed Res. Int. 2013, 2013, 540465. [Google Scholar] [CrossRef]
- Arias, C.; Burns, J.K.; Friedrich, L.M.; Goodrich, R.M.; Parish, M.E. Yeast species associated with orange juice: Evaluation of different identification methods. Appl. Environ. Microbiol. 2002, 68, 1955–1961. [Google Scholar] [CrossRef]
- Esteve-Zarzoso, B.; Belloch, C.; Uruburu, F.; Querol, A. Identification of yeasts by RFLP analysis of the 5.8S rRNA gene and the two ribosomal internal transcribed spacers. Int. J. Syst. Bacteriol. 1999, 49, 329–337. [Google Scholar] [CrossRef]
- Muyzer, G. DGGE/TGGE a method for identifying genes from natural ecosystems. Curr. Opin. Microbiol. 1999, 2, 317–322. [Google Scholar] [CrossRef]
- Ivey, M.L.; Phister, T.G. Detection and identification of microorganisms in wine: A review of molecular techniques. J. Ind. Microbiol. Biotechnol. 2011, 38, 1619–1634. [Google Scholar] [CrossRef]
- Pérez-Martín, F.; Seseña, S.; Fernández-González, M.; Arévalo, M.; Palop, M.L. Microbial communities in air and wine of a winery at two consecutive vintages. Int. J. Food Microbiol. 2014, 190, 44–53. [Google Scholar] [CrossRef]
- Esteve-Zarzoso, B.; Fernández-Espinar, M.T.; Querol, A. Authentication and identification of Saccharomyces cerevisiae ‘flor’ yeast races involved in sherry ageing. Antonie Van Leeuwenhoek 2004, 85, 151–158. [Google Scholar] [CrossRef]
- Vaudano, E.; Garcia-Moruno, E. Discrimination of Saccharomyces cerevisiae wine strains using microsatellite multiplex PCR and band pattern analysis. Food Microbiol. 2008, 25, 56–64. [Google Scholar] [CrossRef]
- Legras, J.L.; Karst, F. Optimisation of interdelta analysis for Saccharomyces cerevisiae strain characterisation. FEMS Microbiol. Lett. 2003, 221, 249–255. [Google Scholar] [CrossRef]
- Siesto, G.; Capece, A.; Sipiczki, M.; Csoma, H.; Romano, P. Polymorphism detection among wild Saccharomyces cerevisiae strains of different wine origin. Ann. Microbiol. 2013, 63, 661–668. [Google Scholar] [CrossRef]
- Pfliegler, W.P.; Sipiczki, M. Does fingerprinting truly represent the diversity of wine yeasts? A case study with interdelta genotyping of Saccharomyces cerevisiae strains. Lett. Appl. Microbiol. 2016, 63, 406–411. [Google Scholar] [CrossRef]
- Cottrell, M.T. Fingerprinting Saccharomyces cerevisiae Strains Using Next Generation Sequencing of PCR Amplicons Generated from Delta Elements. J. Am. Soc. Brew. Chem. 2022, 81, 374–382. [Google Scholar] [CrossRef]
- Amberg, D.C.; Burke, D.J.; Strathern, J.N. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual, 2005th ed.; Cold Spring Harbor Laboratory Press: Long Island, NY, USA, 2005. [Google Scholar]
- Bray, M.A.; Vokes, M.S.; Carpenter, A.E. Using Cellprofiler for automatic identification and measurement of biological objects in images. Curr. Protoc. Mol. Biol. 2015, 109, 14.17.1–14.17.13. [Google Scholar] [CrossRef]
- Ye, S.; Dhillon, S.; Ke, X.; Collins, A.R.; Day, I.N. An efficient procedure for genotyping single nucleotide polymorphisms. Nucleic Acids Res. 2001, 29, E88. [Google Scholar] [CrossRef]
- Medrano, R.F.; de Oliveira, C.A. Guidelines for the tetra-primer ARMS-PCR technique development. Mol. Biotechnol. 2014, 56, 599–608. [Google Scholar] [CrossRef]
- Korbie, D.J.; Mattick, J.S. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nat. Protoc. 2008, 3, 1452–1456. [Google Scholar] [CrossRef]
- Basile, A.; De Pascale, F.; Bianca, F.; Rossi, A.; Frizzarin, M.; De Bernardini, N.; Bosaro, M.; Baldisseri, A.; Antoniali, P.; Lopreiato, R.; et al. Large-scale sequencing and comparative analysis of oenological Saccharomyces cerevisiae strains supported by nanopore refinement of key genomes. Food Microbiol. 2021, 97, 103753. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Peter, J.; De Chiara, M.; Friedrich, A.; Yue, J.X.; Pflieger, D.; Bergström, A.; Sigwalt, A.; Barre, B.; Freel, K.; Llored, A.; et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 2018, 556, 339–344. [Google Scholar] [CrossRef]
- Felsenstein, J. PHYLIP (Phylogeny Inference Package) Version 3.6. Distributed by Author, 2005. Department of Genome Sciences, University of Washington, Seattle. Available online: https://phylipweb.github.io/phylip/ (accessed on 26 June 2025).
- Xufre, A.; Albergaria, H.; Gírio, F.; Spencer-Martins, I. Use of interdelta polymorphisms of Saccharomyces cerevisiae strains to monitor population evolution during wine fermentation. J. Ind. Microbiol. Biotechnol. 2011, 38, 127–132. [Google Scholar] [CrossRef]
- Castillo, M.M.; Parra, N.; Câmara, J.S.; Khadem, M. Unveiling the Regional Identity of Madeira Wine: Insights from Saccharomyces cerevisiae Strains Using Interdelta Analysis. Beverages 2025, 11, 84. [Google Scholar] [CrossRef]
- Liti, G.; Carter, D.M.; Moses, A.M.; Warringer, J.; Parts, L.; James, S.A.; Davey, R.P.; Roberts, I.N.; Burt, A.; Koufopanou, V.; et al. Population genomics of domestic and wild yeasts. Nature 2009, 458, 337–341. [Google Scholar] [CrossRef]
- Bisson, L.F. Stuck and sluggish fermentations. Am. J. Enol. Vitic. 1999, 50, 107–119. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Strain Name | SNP (Total) | SNP (Unique) |
|---|---|---|---|
| 1 | C1.A | 68,678 | 82 |
| 2 | C1.B | 69,587 | 71 |
| 3 | C1.C | 69,297 | 98 |
| 4 | C1.D | 70,513 | 90 |
| 5 | C1.E | 68,185 | 205 |
| 6 | C1.F | 69,748 | 106 |
| 7 | C2.A | 65,336 | 314 |
| 8 | C2.B | 65,563 | 377 |
| 9 | C3.A | 64,667 | 113 |
| 10 | C3.B | 60,521 | 192 |
| 11 | C4.A | 54,285 | 135 |
| 12 | C4.B | 54,552 | 117 |
| 13 | C5.A | 58,957 | 1449 |
| 14 | C5.B | 57,608 | 149 |
| 15 | U1 | 54,795 | 1981 |
| 16 | U2 | 58,235 | 784 |
| 17 | U3 | 57,917 | 475 |
| 18 | U4 | 54,567 | 2299 |
| 19 | U5 | 59,393 | 1653 |
| 20 | U6 | 56,098 | 1994 |
| 21 | U7 | 66,656 | 1085 |
| 22 | U8 | 57,025 | 2743 |
| 23 | U9 | 66,642 | 654 |
| 24 | U10 | 61,974 | 3428 |
| 25 | U11 | 54,188 | 1376 |
| 26 | U12 | 54,640 | 3123 |
| 27 | U13 | 63,597 | 2514 |
| 28 | U14 | 62,550 | 2127 |
| Strain | SNP | Locus Chr–Position | PCR Product Size (DNA SNP) (bp) | |
|---|---|---|---|---|
| C1.A | 1 | IV | 369,222 | 203 |
| C1.A | 2 | XIII | 796,963 | 430 |
| C1.B | 1 | I | 194,376 | 223 |
| C1.B | 2 | XV | 803,410 | 689 |
| C1.B | 3 | IV | 1,508,363 | 451 |
| C1.C | 1 | XV | 357,173 | 244 |
| C1.D | 1 | II | 289,545 | 505 |
| C2.A | 1 | VI | 83,415 | 289 |
| C2.A | 2 | VII | 854,569 | 599 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldisseri, A.; Santinello, D.; Granuzzo, S.; Frizzarin, M.; De Pascale, F.; Sartori, G.; Antoniali, P.; Campanaro, S.; Lopreiato, R. A Novel PCR-Based Tool to Trace Oenological Saccharomyces cerevisiae Yeast by Monitoring Strain-Specific Nucleotide Polymorphisms. Foods 2025, 14, 2379. https://doi.org/10.3390/foods14132379
Baldisseri A, Santinello D, Granuzzo S, Frizzarin M, De Pascale F, Sartori G, Antoniali P, Campanaro S, Lopreiato R. A Novel PCR-Based Tool to Trace Oenological Saccharomyces cerevisiae Yeast by Monitoring Strain-Specific Nucleotide Polymorphisms. Foods. 2025; 14(13):2379. https://doi.org/10.3390/foods14132379
Chicago/Turabian StyleBaldisseri, Anna, Davide Santinello, Sara Granuzzo, Martina Frizzarin, Fabio De Pascale, Geppo Sartori, Paolo Antoniali, Stefano Campanaro, and Raffaele Lopreiato. 2025. "A Novel PCR-Based Tool to Trace Oenological Saccharomyces cerevisiae Yeast by Monitoring Strain-Specific Nucleotide Polymorphisms" Foods 14, no. 13: 2379. https://doi.org/10.3390/foods14132379
APA StyleBaldisseri, A., Santinello, D., Granuzzo, S., Frizzarin, M., De Pascale, F., Sartori, G., Antoniali, P., Campanaro, S., & Lopreiato, R. (2025). A Novel PCR-Based Tool to Trace Oenological Saccharomyces cerevisiae Yeast by Monitoring Strain-Specific Nucleotide Polymorphisms. Foods, 14(13), 2379. https://doi.org/10.3390/foods14132379