
Exploring the Blood Glucose-Lowering Potential of the Umami Peptides LADW and EEAEGT Derived from Tuna Skeletal Myosin: Perspectives from α-Glucosidase Inhibition and Starch Interaction


 ,
,  , and
, and
Abstract
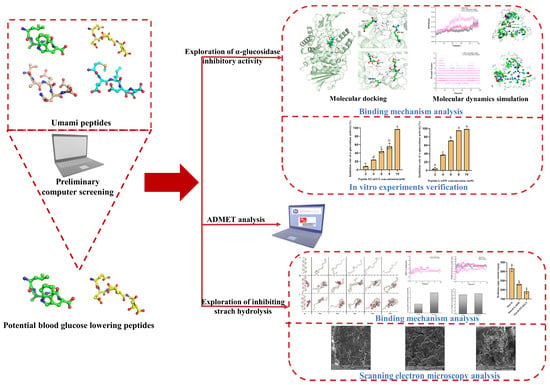
1. Introduction
2. Materials and Methods
2.1. Material
2.2. Prediction of Potential Biological Activity and Physicochemical Properties
2.3. Molecular Docking
2.4. ADMET Analysis
2.5. MD Simulation
2.5.1. Peptide and α-Glucosidase Systems
2.5.2. Peptide and Amylose Systems
2.5.3. Data Analysis of MD Simulation
2.5.4. Free Energy Landscape (FEL) and IGM Analyses
2.6. Inhibition Activity of α-Glucosidase
2.7. Starch–Peptide Interactions
2.7.1. Preparation of Sample
2.7.2. In Vitro Simulation Digestion of Starch and Starch–Peptide
2.7.3. Determination of Reducing Sugars
2.7.4. Starch Granule Morphological Observation
2.8. Statistical Analysis
3. Results and Discussion
3.1. Potential Activity Analysis and Physicochemical Properties of Peptides
3.2. Molecular Docking
3.3. ADMET Analysis
3.4. MD Simulations
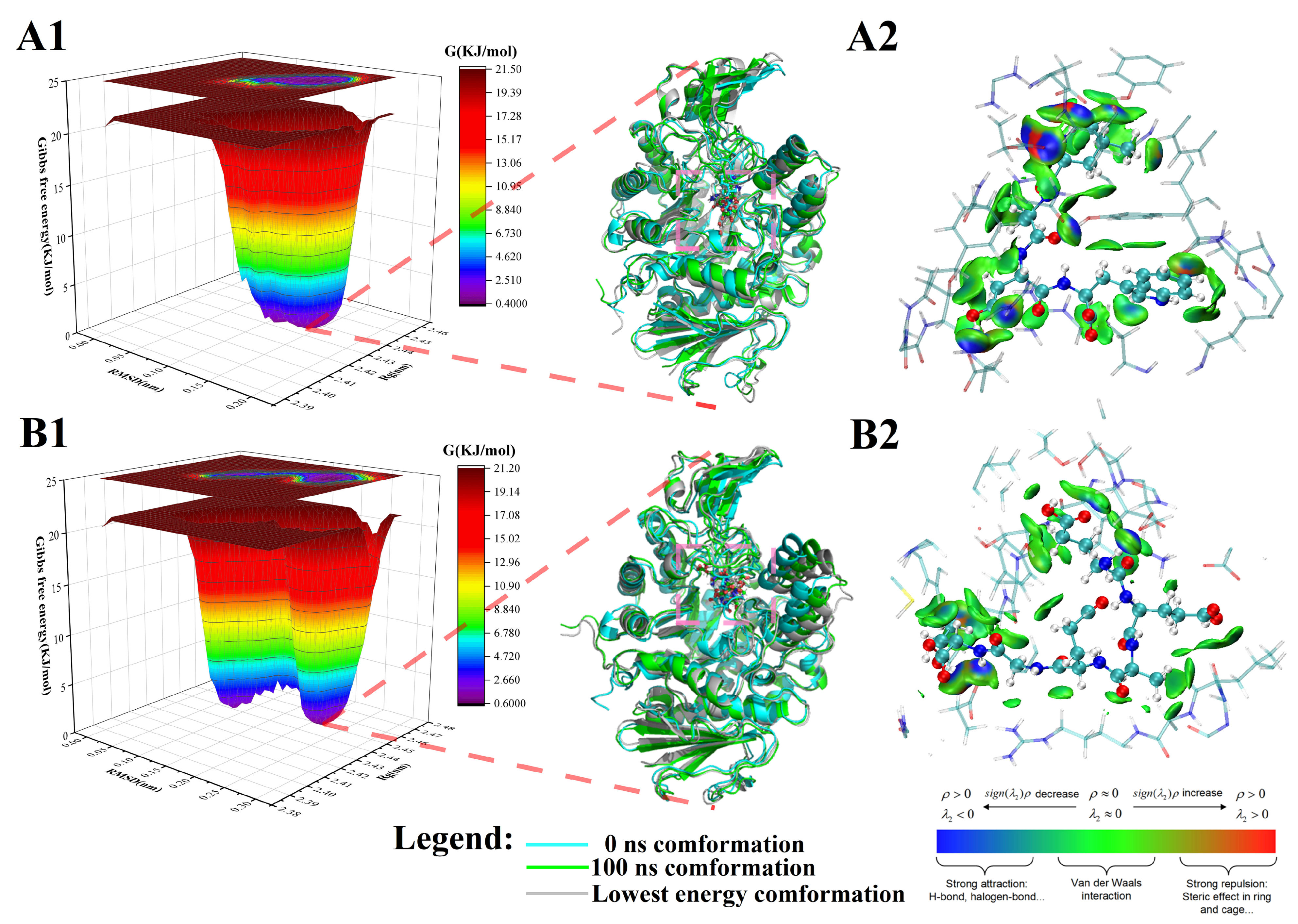
3.4.1. Peptide-α-Glucosidase Complex Systems

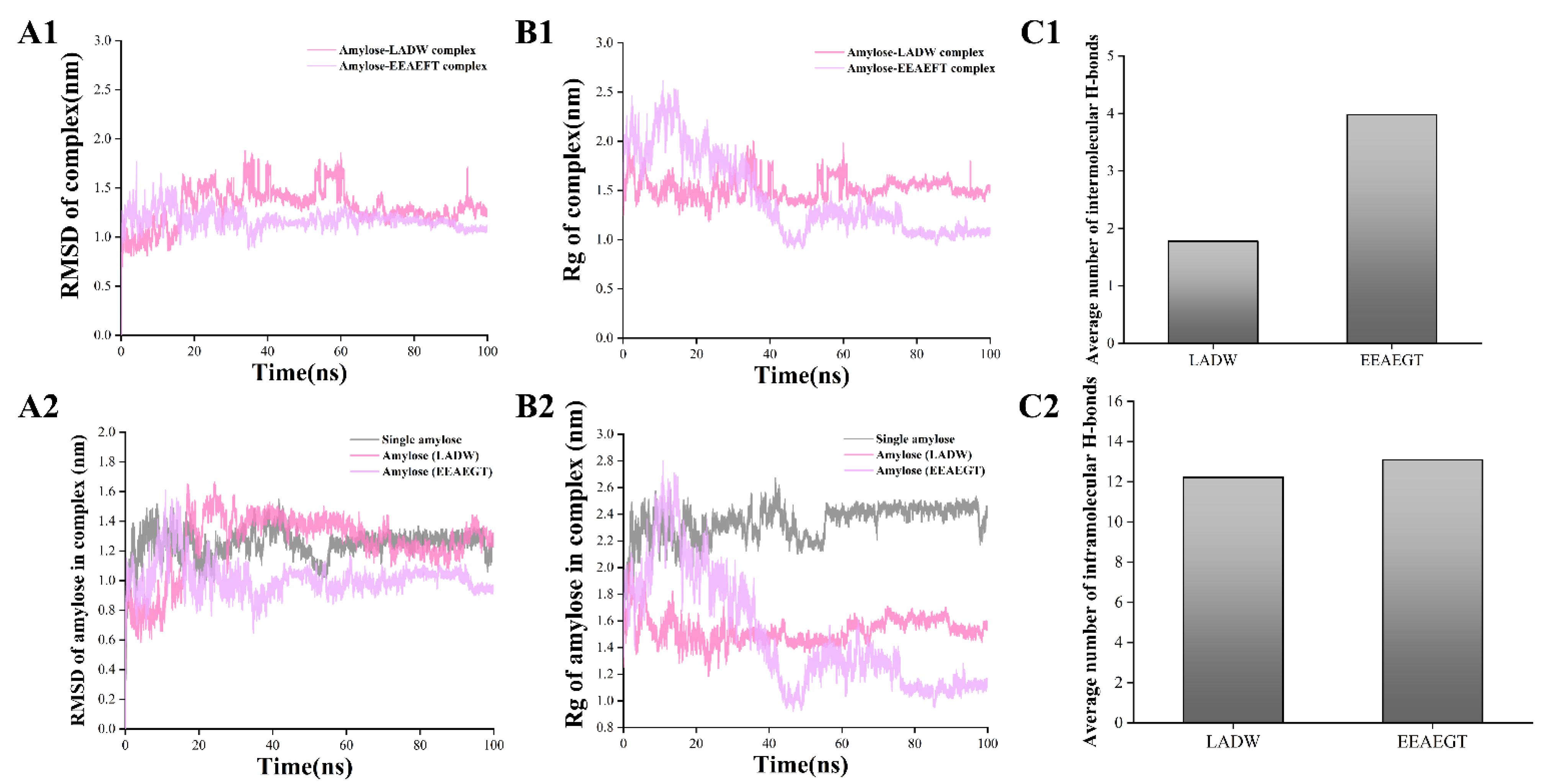
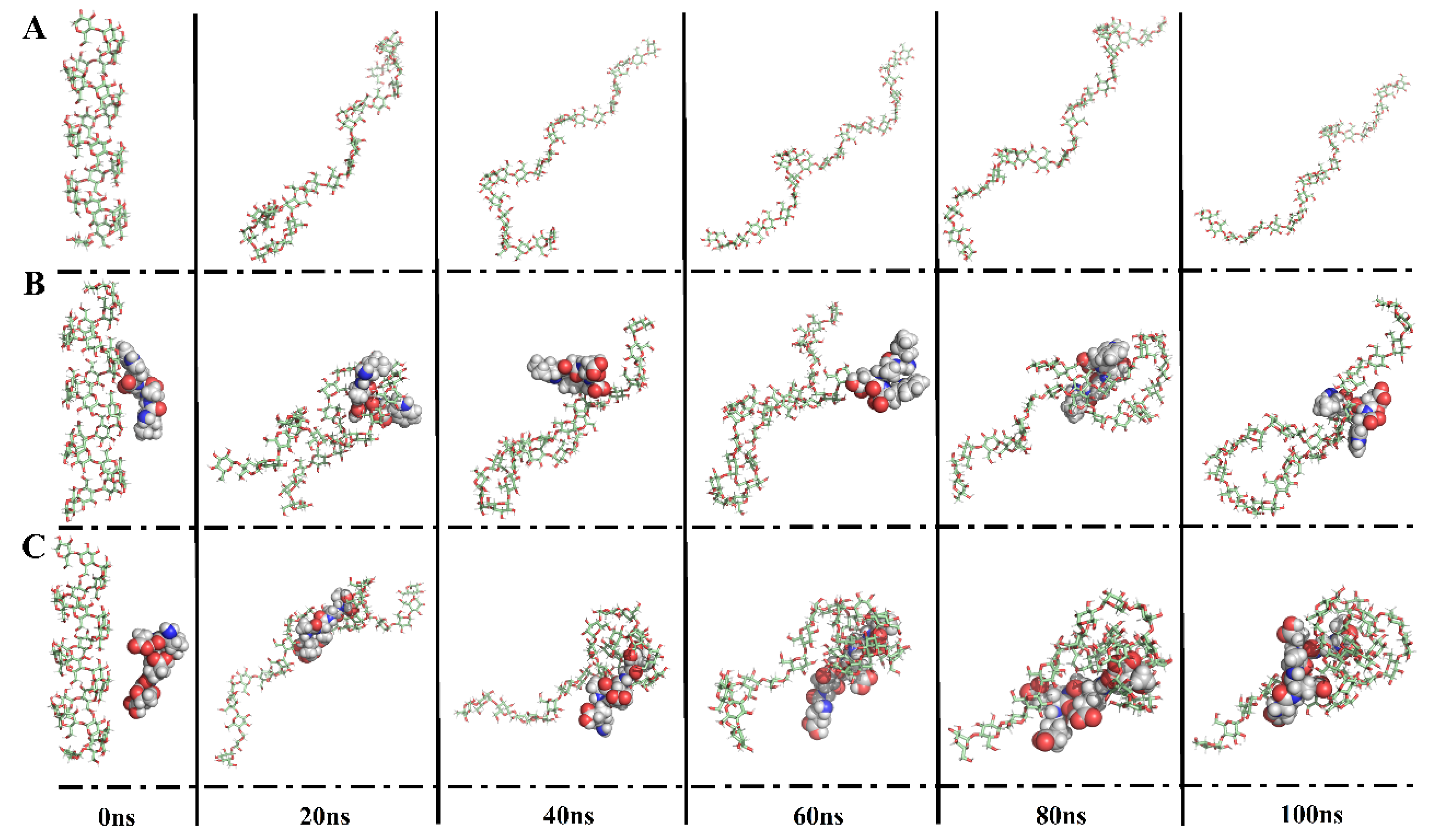
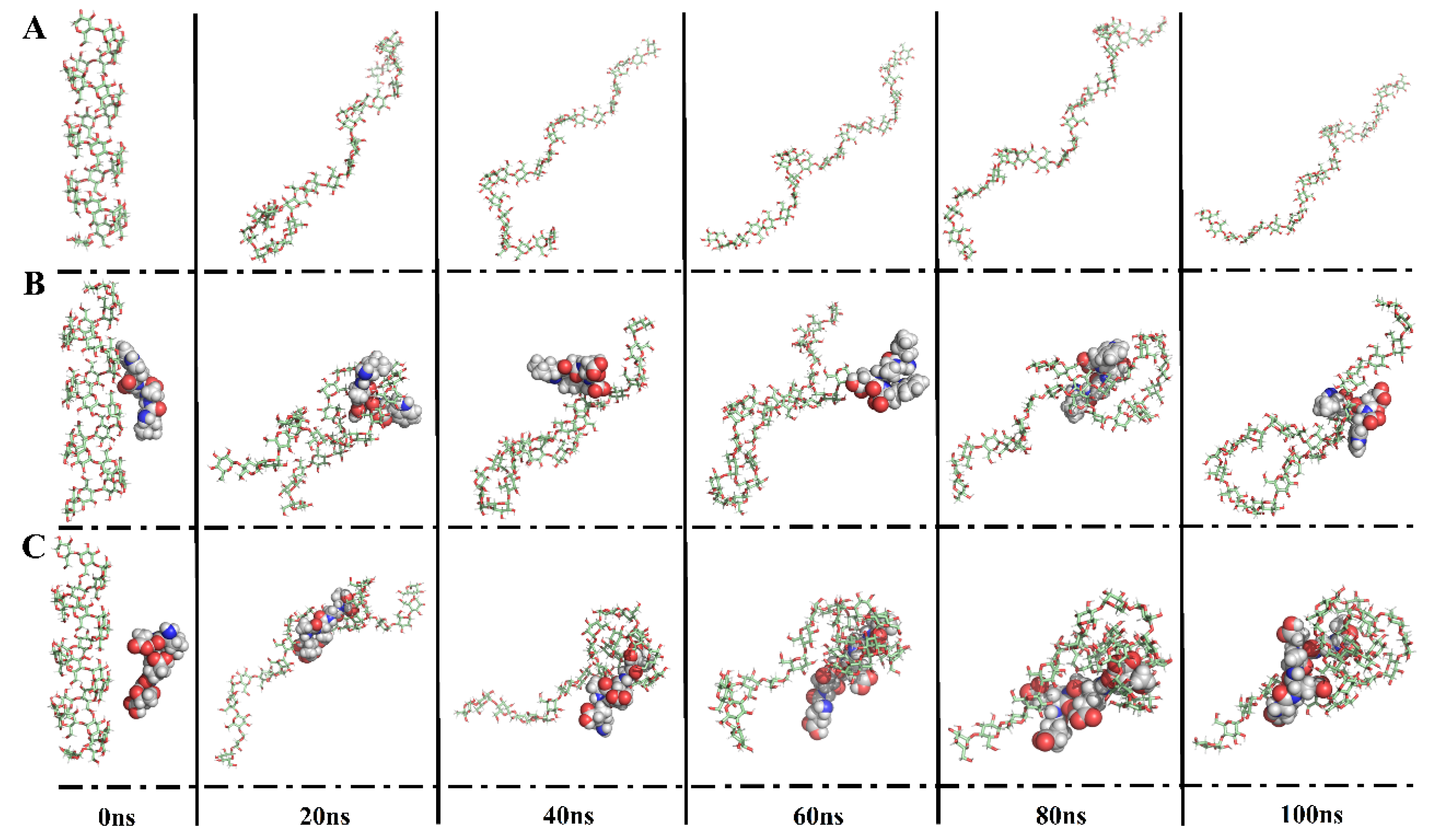
3.4.2. Peptide–Amylose Complex Systems
3.5. In Vitro Experimental Verification
3.5.1. Inhibition Activity of α-Glucosidase
3.5.2. Reducing Sugar Content and Morphological Analysis of the Sample

4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lefèbvre, P.; Pierson, A. The global challenge of diabetes. World Hosp. Health Serv. 2004, 40, 37–39. [Google Scholar]
- Zhou, Y.; Chi, J.; Lv, W.; Wang, Y. Obesity and diabetes as high-risk factors for severe coronavirus disease 2019 (COVID-19). Diabetes/Metab. Res. Rev. 2021, 37, e3377. [Google Scholar] [CrossRef]
- Bischoff, H. Pharmacology of α-glucosidase inhibition. Eur. J. Clin. Investig. 1994, 24 (Suppl. 3), 3–10. [Google Scholar]
- Lu, X.; Ma, R.; Qiu, H.; Sun, C.; Tian, Y. Mechanism of effect of endogenous/exogenous rice protein and its hydrolysates on rice starch digestibility. Int. J. Biol. Macromol. 2021, 193, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhang, B.; Tan, C.P.; Ding, L.; Shao, M.; Chen, C.; Fu, X.; Huang, Q. Effect of Rosa Roxburghii juice on starch digestibility: A focus on the binding of polyphenols to amylose and porcine pancreatic α-amylase by molecular modeling. Food Hydrocoll. 2022, 123, 106966. [Google Scholar] [CrossRef]
- Sang, S.; Xu, X.; Zhu, X.; Narsimhan, G. Complexation of 26-Mer Amylose with Egg Yolk Lipids with Different Numbers of Tails Using a Molecular Dynamics Simulation. Foods 2021, 10, 2355. [Google Scholar] [CrossRef]
- Zhao, S.; Ma, S.; Zhang, Y.; Gao, M.; Luo, Z.; Cai, S. Combining molecular docking and molecular dynamics simulation to discover four novel umami peptides from tuna skeletal myosin with sensory evaluation validation. Food Chem. 2023, 433, 137331. [Google Scholar] [CrossRef]
- Chen, M.; Pan, D.; Zhou, T.; Gao, X.; Dang, Y. Novel umami peptide IPIPATKT with dual dipeptidyl peptidase-IV and angiotensin I-converting enzyme inhibitory activities. J. Agric. Food Chem. 2021, 69, 5463–5470. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Duan, W.; Zhang, J.; Huang, Y.; Zhang, Y.; Sun, B. Characterization and molecular docking study of taste peptides from chicken soup by sensory analysis combined with nano-LC-Q-TOF-MS/MS. Food Chem. 2022, 383, 132455. [Google Scholar] [CrossRef]
- Hao, L.; Gao, X.; Zhou, T.; Cao, J.; Sun, Y.; Dang, Y.; Pan, D. Angiotensin I-converting enzyme (ACE) inhibitory and antioxidant activity of umami peptides after in vitro gastrointestinal digestion. J. Agric. Food Chem. 2020, 68, 8232–8241. [Google Scholar] [CrossRef] [PubMed]
- Ruan, S.; Sun, L.; Sun, X.; He, J.; Zhuang, Y. Novel umami peptides from tilapia lower jaw and molecular docking to the taste receptor T1R1/T1R3. Food Chem. 2021, 362, 130249. [Google Scholar]
- Di Stefano, E.; Oliviero, T.; Udenigwe, C.C. Functional significance and structure–activity relationship of food-derived α-glucosidase inhibitors. Curr. Opin. Food Sci. 2018, 20, 7–12. [Google Scholar] [CrossRef]
- He, R.; Pan, Y.G.; Shang, W.T.; Zhong, G.; Huang, W.Y.; Xiang, D.; Pan, F.; Zhang, W.M. Ultrasonic-assisted binding of canistel (Lucuma nervosa A. DC) seed starch with quercetin. Ultrason. Sonochem. 2023, 96, 106417. [Google Scholar] [CrossRef]
- Mazloomi, S.N.; Mahoonak, A.S.; Mora, L.; Ghorbani, M.; Houshmand, G.; Toldrá, F. Pepsin Hydrolysis of Orange By-Products for the Production of Bioactive Peptides with Gastrointestinal Resistant Properties. Foods 2021, 10, 679. [Google Scholar] [CrossRef] [PubMed]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. BIOPEP-UWM database of bioactive peptides: Current opportunities. Int. J. Mol. Sci. 2019, 20, 5978. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Jia, Y.; Ma, Y.; Cheng, G.; Zhang, Y.; Cai, S. Comparative study of dietary flavonoids with different structures as α-glucosidase inhibitors and insulin sensitizers. J. Agric. Food Chem. 2019, 67, 10521–10533. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, S.; Ma, S.; Zhao, S.; Yi, J.; Zhou, L. Water Caltrop (Trapa quadrispinosa Roxb.) Husk Improves Oxidative Stress and Postprandial Blood Glucose in Diabetes: Phenolic Profiles, Antioxidant Activities and α-Glycosidase Inhibition of Different Fractions with In Vitro and In Silico Analyses. Antioxidants 2022, 11, 1873. [Google Scholar] [CrossRef]
- Zheng, X.; Chi, H.; Ma, S.; Zhao, L.; Cai, S. Identification of novel α-glucosidase inhibitory peptides in rice wine and their antioxidant activities using in silico and in vitro analyses. LWT 2023, 178, 114629. [Google Scholar] [CrossRef]
- Li, S.; Wang, R.; Hu, X.; Li, C.; Wang, L. Bio-affinity ultra-filtration combined with HPLC-ESI-qTOF-MS/MS for screening potential α-glucosidase inhibitors from Cerasus humilis (Bge.) Sok. leaf-tea and in silico analysis. Food Chem. 2022, 373, 131528. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Cheng, F.; Xu, Y.; Li, W.; Tang, Y. Estimation of ADME properties with substructure pattern recognition. J. Chem. Inf. Model. 2010, 50, 1034–1041. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Pan, F.; Zhao, L.; Cai, S.; Tang, X.; Mehmood, A.; Alnadari, F.; Tuersuntuoheti, T.; Zhou, N.; Ai, X. Prediction and evaluation of the 3D structure of Macadamia integrifolia antimicrobial protein 2 (MiAMP2) and its interaction with palmitoleic acid or oleic acid: An integrated computational approach. Food Chem. 2022, 367, 130677. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Struct. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Xia, H.; Lv, C.; Lu, Y.; Zeng, C.; Qin, S.; Shi, M. Natural deep eutectic ready to use extract of astilbin: Super high in vitro bioaccessibility, α-amylase and α-glucosidase enzyme inhibition kinetics. Food Res. Int. 2023, 173, 113368. [Google Scholar] [CrossRef]
- Goh, R.; Gao, J.; Ananingsih, V.K.; Ranawana, V.; Henry, C.J.; Zhou, W. Green tea catechins reduced the glycaemic potential of bread: An in vitro digestibility study. Food Chem. 2015, 180, 203–210. [Google Scholar] [CrossRef]
- Arámburo-Gálvez, J.G.; Arvizu-Flores, A.A.; Cárdenas-Torres, F.I.; Cabrera-Chávez, F.; Ramírez-Torres, G.I.; Flores-Mendoza, L.K.; Gastelum-Acosta, P.E.; Figueroa-Salcido, O.G.; Ontiveros, N. Prediction of ACE-I Inhibitory Peptides Derived from Chickpea (Cicer arietinum L.): In Silico Assessments Using Simulated Enzymatic Hydrolysis, Molecular Docking and ADMET Evaluation. Foods 2022, 11, 1576. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lei, T.; Shen, C.; Wang, Z.; Cao, D.; Hou, T. ADMET evaluation in drug discovery. 19. Reliable prediction of human cytochrome P450 inhibition using artificial intelligence approaches. J. Chem. Inf. Model. 2019, 59, 4587–4601. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Ortega-Castro, J.; Frau, J.; Glossman-Mitnik, D. Conceptual DFT-Based Computational Peptidology, Pharmacokinetics Study and ADMET Report of the Veraguamides A–G Family of Marine Natural Drugs. Mar. Drugs 2022, 20, 97. [Google Scholar] [CrossRef] [PubMed]
- Maurya, A.K.; Mulpuru, V.; Mishra, N. Discovery of novel coumarin analogs against the α-glucosidase protein target of Diabetes mellitus: Pharmacophore-based QSAR, docking, and molecular dynamics simulation studies. ACS Omega 2020, 5, 32234–32249. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Li, J.; Zhao, L.; Tuersuntuoheti, T.; Mehmood, A.; Zhou, N.; Hao, S.; Wang, C.; Lin, W. A molecular docking and molecular dynamics simulation study on the interaction between cyanidin-3-O-glucoside and major proteins in cow’s milk. J. Food Biochem. 2021, 45, e13570. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Bester, M.J.; Neitz, A.W.; Gaspar, A.R. Rational in silico design of novel α-glucosidase inhibitory peptides and in vitro evaluation of promising candidates. Biomed. Pharmacother. 2018, 107, 234–242. [Google Scholar] [CrossRef]
- Mora, L.; González-Rogel, D.; Heres, A.; Toldrá, F. Iberian dry-cured ham as a potential source of α-glucosidase-inhibitory peptides. J. Funct. Foods 2020, 67, 103840. [Google Scholar] [CrossRef]
- Zhou, M.; Ren, G.; Zhang, B.; Ma, F.; Fan, J.; Qiu, Z. Screening and identification of a novel antidiabetic peptide from collagen hydrolysates of Chinese giant salamander skin: Network pharmacology, inhibition kinetics and protection of IR-HepG2 cells. Food Funct. 2022, 13, 3329–3342. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Molecular Weight /Da (MW) | Isoelectric Point (pI) | Net Charge | Potential Biological Activity | Umami Threshold (mg/mL) |
|---|---|---|---|---|---|
| LADW | 503.55 | 3.77 | −1 | ACE inhibitor a DPP-IV inhibitor a α-glucosidase inhibitor a | 0.125 |
| EEAEGT | 634.24 | 3.47 | −3 | ACE inhibitor a DPP-IV inhibitor a Immunostimulating peptide a α-glucosidase inhibitor a | 0.125 |
| VAEQE | 574.26 | 3.62 | −2 | DPP-IV inhibitor a Allergen b | 0.125 |
| MEIDD | 621.23 | 3.39 | −3 | ACE inhibitor a DPP-IV inhibitor a Allergen b | 0.250 |
| Peptide | Database | Absorption | Distribution | Metabolism | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CYP450 inhibitor | ||||||||||||
| HIA | Caco-2 | LogP | BBB | PPB | 1A2 | 2C19 | 2C9 | 2D6 | 3A4 | |||
| LADW | ADMETLab 2.0 | 0.921 | −6.816 | 0.154 | 0.132 | 0.123 | 0.003 | 0.036 | 0.081 | 0.031 | 0.017 | |
| admetSAR @ LMMD | High | Low | - | Low | - | No | No | No | No | No | ||
| Swiss-ADME | Low | - | 0.02 | No | - | No | No | No | No | No | ||
| iDrug platform | 0.985 | 0.128 × 10−6 | 0.117 | 0.297 | 0.732 | 0.22 | 0.2 | 0.09 | 0.16 | 0.14 | ||
| EEAEGT | ADMETLab 2.0 | 0.904 | −7.16 | −4.622 | 0.062 | 0.200 | 0 | 0.016 | 0.033 | 0 | 0.003 | |
| admetSAR @ LMMD | Medium | Low | - | High | - | No | No | No | No | No | ||
| Swiss-ADME | Low | - | - | No | - | No | No | No | No | No | ||
| iDrug platform | 0.842 | 1.954 × 10−6 | −3.74 | 0.195 | 0.409 | 0.197 | 0.220 | 0.156 | 0.147 | 0.119 | ||
| Excretion | Toxicity | |||||||||||
| Human Clearance | Carcinogenicity | Rat Oral Acute Toxicity | DILI | Ames Toxicity | ||||||||
| 2.074 | 0.145 | 0.173 | 0.042 | 0.005 | ||||||||
| - | No | - | - | No | ||||||||
| - | - | - | - | - | ||||||||
| 0.519 | 0.197 | - | 0.371 | 0.031 | ||||||||
| 1.59 | 0.032 | 0.173 | 0.013 | 0.010 | ||||||||
| - | No | - | - | No | ||||||||
| - | - | - | - | - | ||||||||
| 0.692 | 0.197 | - | 0.344 | 0.027 | ||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, S.; Cai, S.; Ding, L.; Yi, J.; Zhou, L.; Liu, Z.; Chu, C. Exploring the Blood Glucose-Lowering Potential of the Umami Peptides LADW and EEAEGT Derived from Tuna Skeletal Myosin: Perspectives from α-Glucosidase Inhibition and Starch Interaction. Foods 2024, 13, 294. https://doi.org/10.3390/foods13020294
Zhao S, Cai S, Ding L, Yi J, Zhou L, Liu Z, Chu C. Exploring the Blood Glucose-Lowering Potential of the Umami Peptides LADW and EEAEGT Derived from Tuna Skeletal Myosin: Perspectives from α-Glucosidase Inhibition and Starch Interaction. Foods. 2024; 13(2):294. https://doi.org/10.3390/foods13020294
Chicago/Turabian StyleZhao, Shuai, Shengbao Cai, Lixin Ding, Junjie Yi, Linyan Zhou, Zhijia Liu, and Chuanqi Chu. 2024. "Exploring the Blood Glucose-Lowering Potential of the Umami Peptides LADW and EEAEGT Derived from Tuna Skeletal Myosin: Perspectives from α-Glucosidase Inhibition and Starch Interaction" Foods 13, no. 2: 294. https://doi.org/10.3390/foods13020294
APA StyleZhao, S., Cai, S., Ding, L., Yi, J., Zhou, L., Liu, Z., & Chu, C. (2024). Exploring the Blood Glucose-Lowering Potential of the Umami Peptides LADW and EEAEGT Derived from Tuna Skeletal Myosin: Perspectives from α-Glucosidase Inhibition and Starch Interaction. Foods, 13(2), 294. https://doi.org/10.3390/foods13020294