
Undescribed Cyclohexene and Benzofuran Alkenyl Derivatives from Choerospondias axillaris, a Potential Hypoglycemic Fruit

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. General Procedure
2.2. Chemicals and Reagents
2.3. Materials
2.4. Extraction and Isolation
2.4.1. Choerosponol F
2.4.2. Choerosponol G
2.4.3. Choerosponol H
2.4.4. Choerosponol I
2.4.5. Choerosponol J
2.4.6. Choerosponol K
2.4.7. Choerosponol L
2.5. Quantum Chemistry ECD Calculation
2.6. α-Glucosidase Inhibitory Assay
2.7. Molecular Docking Analysis
2.8. Statistical Analysis
3. Results and Discussion
3.1. Structural Elucidation
3.2. α-Glucosidase Inhibition
3.3. Molecular Docking Studies
3.4. Structural Activity Relationship
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dangal, A.; Timsina, P.; Dahal, S. A comprehensive review on study of physical, nutritional, and phytochemical characteristics as well as therapeutic activities of Choerospondias axillaris (lapsi). Food Biosci. 2023, 53, 102713. [Google Scholar] [CrossRef]
- Surayothee, W.; Buajan, S.; Fu, P.; Pumijumnong, N.; Fan, Z.; Panthi, S.; Finnegan, P.M.; Zhang, Y.; Chen, Y.; Tor-ngern, P.; et al. Growth-climate relationships and long-Term growth trends of the tropical forest tree Choerospondias axillaris (Anacardiaceae) in east-Central Thailand. Forests 2021, 12, 1655. [Google Scholar] [CrossRef]
- Mann, S.; Chakraborty, D.; Biswas, S. An alternative perspective of an underutilized fruit tree Choerospondias axillaris in health promotion and disease prevention: A review. Food Biosci. 2022, 47, 101609. [Google Scholar] [CrossRef]
- Wang, C.; Li, J.; Cao, Y.; Huang, J.; Lin, H.; Zhao, T.; Liu, L.; Shen, P.; Julian McClements, D.; Chen, J.; et al. Extraction and characterization of pectic polysaccharides from Choerospondias axillaris peels: Comparison of hot water and ultrasound-assisted extraction methods. Food Chem. 2023, 401, 134156. [Google Scholar] [CrossRef] [PubMed]
- Rong, W.; Shi, Q.; Yang, Y.; Su, W.; Li, M.; Qin, M.; Bai, S.; Zhu, Q.; Wang, A. Fructus choerospondiatis: A comprehensive review of its traditional uses, chemical composition, pharmacological activities, and clinical studies. J. Ethnopharmacol. 2024, 323, 117696. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, J.; Li, T.; Liu, C.; Wang, X.; Dai, T.T.; McClements, D.J.; Liu, J. Impact of in vitro simulated digestion on the potential health benefits of proanthocyanidins from Choerospondias axillaris peels. Food Res. Int. 2015, 78, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Huang, K.; McClements, D.J.; Hu, X.; Luo, S.; Liu, C. Phenolics, antioxidant activity, and in vitro starch digestibility of extruded brown rice influenced by Choerospondias axillaris fruit peels addition. Starch 2019, 71, 1800346. [Google Scholar] [CrossRef]
- Li, Q.; Wang, X.; Chen, J.; Liu, C.; Li, T.; McClements, D.J.; Dai, T.; Liu, J. Antioxidant activity of proanthocyanidins-rich fractions from Choerospondias axillaris peels using a combination of chemical-based methods and cellular-based assay. Food Chem. 2016, 208, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, X.; Dai, T.; Liu, C.; Li, T.; McClements, D.J.; Chen, J.; Liu, J. Proanthocyanidins, isolated from Choerospondias axillaris fruit peels, exhibit potent antioxidant activities in vitro and a novel anti-angiogenic property in vitro and in vivo. J. Agric. Food Chem. 2016, 64, 3546–3556. [Google Scholar] [CrossRef]
- Mann, S.; Sarkar, A.; Sharma, A.; Gupta, R.K.; Biswas, S. Antitumor Activity of Choerospondias axillaris Fruit Extract by Regulating the Expression of SNCAIP and SNCA on MDA-MB-231 Cells. Asian Pac. J. Cancer Prev. 2022, 23, 1577–1586. [Google Scholar] [CrossRef]
- Labh, S.N.; Shakya, S.R. Medicinal importance of Choerospondias axillaris (Roxb.) Burtt & Hill fruits in Nepal. Trop. Plant Res. 2016, 3, 463–469. [Google Scholar]
- Tang, L.; Li, G.Y.; Yang, B.Y.; Hai, H.X. Chemical constituents from Choerospondias axillaris (Roxb.) Burtt et Hill. Chin. Tradit. Herb. 2009, 40, 541–543. (In Chinese) [Google Scholar]
- Shen, X.J.; Ge, R.L.; Wang, J.H. Chemical constituents from Choerospondias axillaris (Roxb.) Burtt et Hill. J. Hunan Univ. Med. Sci. 2009, 28, 196–199. (In Chinese) [Google Scholar]
- Lian, Z.; Zhang, C.; Li, C.; Zhou, Y. Studies on Chemical constituents of Choerospondias axillaris. Chin. Med. Mat. 2003, 26, 23–24. (In Chinese) [Google Scholar]
- Qian, H.; Hu, Q.L. Studies on the chemical constituents of Choerospondias axillaris. Chin. J. Mod. Appl. Pharm. 1992, 9, 212–213. (In Chinese) [Google Scholar]
- Wang, H.; Gao, X.D.; Zhou, G.C.; Cai, L.; Yao, W.B. In vitro and in vivo antioxidant activity of aqueous extract from Choerospondias axillaris fruit. Food Chem. 2008, 106, 888–895. [Google Scholar] [CrossRef]
- Li, Q.; Liu, C.; Li, T.; McClements, D.J.; Fu, Y.; Liu, J. Comparison of phytochemical profiles and antiproliferative activities of different proanthocyanidins fractions from Choerospondias axillaris fruit peels. Food Res. Int. 2018, 113, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Kil, Y.S.; Risinger, A.L.; Petersen, C.L.; Liang, H.; Grkovic, T.; O’Keefe, B.R.; Mooberry, S.L.; Cichewicz, R.H. Using the cancer dependency map to identify the mechanism of action of a cytotoxic alkenyl derivative from the Fruit of Choerospondias axillaris. J. Nat. Prod. 2020, 83, 584–592. [Google Scholar] [CrossRef]
- Li, Q.; Chen, J.; Li, T.; Liu, C.; Zhai, Y.; McClements, D.J.; Liu, J. Separation and characterization of polyphenolics from underutilized byproducts of fruit production (Choerospondias axillaris peels): Inhibitory activity of proanthocyanidins against glycolysis enzymes. Food Funct. 2015, 6, 3693–3701. [Google Scholar] [CrossRef]
- Ghani, U. Re-exploring promising α-glucosidase inhibitors for potential development into oral anti-diabetic drugs: Finding needle in the haystack. Eur. J. Med. Chem. 2015, 103, 133–162. [Google Scholar] [CrossRef]
- Dhameja, M.; Gupta, P. Synthetic heterocyclic candidates as promising α-glucosidase inhibitors: An overview. Eur. J. Med. Chem. 2019, 176, 343–377. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Zhang, R.; Dong, L.; Chi, J.; Huang, F.; Dong, L.; Zhang, M.; Jia, X. α-Glucosidase inhibitors from brown rice bound phenolics extracts (BRBPE): Identification and mechanism. Food Chem. 2022, 372, 131306. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Wang, L.; Su, L.; Tao, X.; Chen, S.; Wu, J.; Xia, W. Structural characterization of slow digestion dextrin synthesized by a combination of α-Glucosidase and cyclodextrin glucosyltransferase and its prebiotic potential on the gut microbiota in vitro. Food Chem. 2023, 426, 136554. [Google Scholar] [CrossRef] [PubMed]
- Sunagar, R.R.; Sreerama, Y.N. Implication of solvent polarities on browntop millet (Urochloa ramosa) phenolic antioxidants and their ability to protect oxidative DNA damage and inhibit α-amylase and α-glucosidase enzymes. Food Chem. 2023, 411, 135474. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.B.; Lamothe, L.M.; Esmeralda Nava Rodriguez, N.; Rose, D.R.; Lee, B.H. New insights suggest isomaltooligosaccharides are slowly digestible carbohydrates, rather than dietary fibers, at constitutive mammalian α-glucosidase levels. Food Chem. 2022, 383, 132456. [Google Scholar] [CrossRef]
- Kobayashi, K.; Yokoh, H.; Sato, Y.; Takemoto, M.; Uchida, D.; Kanatsuka, A.; Kuribayashi, N.; Terano, T.; Hashimoto, N.; Sakurai, K.; et al. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin compared with α-glucosidase inhibitor in Japanese patients with type 2 diabetes inadequately controlled on sulfonylurea alone (SUCCESS-2): A multicenter, randomized, open-label, non-inferiority trial. Diabetes Obes. Metab. 2014, 16, 761–765. [Google Scholar] [PubMed]
- Lim, J.; Zhang, X.; Ferruzzi, M.G.; Hamaker, B.R. Starch digested product analysis by HPAEC reveals structural specificity of flavonoids in the inhibition of mammalian α-amylase and α-glucosidases. Food Chem. 2019, 288, 413–421. [Google Scholar] [CrossRef]
- Qin, Y.; Chen, X.; Xu, F.; Gu, C.; Zhu, K.; Zhang, Y.; Wu, G.; Wang, P.; Tan, L. Effects of hydroxylation at C3′ on the B ring and diglycosylation at C3 on the C ring on flavonols inhibition of α-glucosidase activity. Food Chem. 2023, 406, 135057. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Chen, M.; Zhu, H.T.; Zhang, M.; Wanga, D.; Yang, C.R.; Zhang, Y.J. Theaflavoids A-C, new flavan-3-ols with potent α-glucosidase inhibitory activity from Yunnan black tea ‘Jin-Ya’. LWT Food Sci. Technol. 2022, 168, 113918. [Google Scholar] [CrossRef]
- Lim, J.; Ferruzzi, M.G.; Hamaker, B.R. Structural requirements of flavonoids for the selective inhibition of α-amylase versus α-glucosidase. Food Chem. 2022, 370, 130981. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Luo, Y.; Huang, K.; Wu, Z. Quickly screening for potential α-glucosidase inhibitors from guava leaves tea by bioaffinity ultrafiltration coupled with HPLC-ESI-TOF/MS method. J. Agric. Food Chem. 2018, 66, 1576–1582. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Zhao, D.G.; Zhou, A.Y.; Zhang, Y.; Du, Z.; Zhang, K. α-Glucosidase inhibition and antihyperglycemic activity of phenolics from the flowers of Edgeworthia gardneri. J. Agric. Food Chem. 2015, 63, 8162–8169. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhu, H.T.; Wang, D.; Zhang, M.; Yang, C.R.; Zhang, Y.J. New flavoalkaloids with potent α-Glucosidase and acetylcholinesterase inhibitory activities from Yunnan black tea ‘Jin-Ya’. J. Agric. Food Chem. 2020, 68, 7955–7963. [Google Scholar] [CrossRef]
- Yang, J.; Wanga, X.; Zhang, C.; Ma, L.; Weib, T.; Zhao, Y.; Peng, X. A Comparative study of inhibition mechanisms of structurally different flavonoid compounds on α-glucosidase and synergistic effect with acarbose. Food Chem. 2021, 347, 129056. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Q.; Zhou, F.C.; Gao, F.; Bian, J.S.; Shan, F. Comparative Evaluation of Quercetin, Isoquercetin and Rutin as Inhibitors of α-Glucosidase. J. Agric. Food Chem. 2009, 57, 11463–11468. [Google Scholar] [CrossRef] [PubMed]
- Haggett, P.; Gunawardena, K.A. Determination of population thresholds for settlement functions by the reed-muench method. Prof. Geogr. 1964, 16, 6–9. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Peng, Z.; Wang, J.; Li, J.; Li, X. Synthesis, biological evaluation, and molecular docking study of N-arylbenzo[d]oxazol-2-amines as potential α-glucosidase inhibitors. Bioorg. Med. Chem. 2016, 24, 5374–5379. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Peng, X.; Dong, D.; Nian, Y.; Gao, Y.; Wang, X.; Hong, D.; Qiu, M. New ent-kaurane diterpenes from the roasted arabica coffee beans and molecular docking to α-glucosidase. Food Chem. 2021, 345, 128823. [Google Scholar] [CrossRef]
- Schrödinger LLC. The PyMOL Molecular Graphics System, Version 2.6.0; Schrödinger LLC: New York, NY, USA, 2015.
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Graef, J.; Ehrt, C.; Rarey, M. Binding site detection remastered: Enabling fast, robust, and reliable binding site detection and descriptor calculation with DoGSite3. J. Chem. Inf. Model. 2023, 63, 3128–3137. [Google Scholar] [CrossRef]
- Fortune, M.; Berhanu, A.G.; Runner, R.T.M. A new flavan from Elephantorrhiza goetzei. Fitoterapia 1999, 70, 412–416. [Google Scholar]
- Lee, E.H.; Kim, H.J.; Song, Y.S.; Jin, C.; Lee, K.T.; Cho, J.; Lee, Y.S. Constituents of the stems and fruits of Opuntia ficus-indica var. saboten. Arch. Pharm. Res. 2003, 26, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, D.; Wang, J.; Liu, Y.; Wu, J. Antibacterial phenolic compounds from the spines of Gleditsia sinensis Lam. Nat. Prod. Res. 2007, 21, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Din, S.; Hamid, S.; Yaseen, A.; Yatoo, A.M.; Ali, S.; Shamim, K.; Mahdi, W.A.; Alshehri, S.; Rehman, M.U.; Shah, W.A. Isolation and characterization of flavonoid naringenin and evaluation of cytotoxic and biological efficacy of water lilly (Nymphaea mexicana Zucc.). Plants 2022, 11, 3588. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, Y.M.; Yang, S.; Song, B.A.; Xu, G.F.; Bhadury, P.S.; Jin, L.H.; Hu, D.Y.; Liu, F.; Xue, W.; et al. Studies on the chemical constituents and anticancer activity of Saxifraga stolonifera (L.) Meeb. Bioorg. Med. Chem. 2008, 16, 1337–1344. [Google Scholar] [CrossRef]
- Zhu, J.X.; Ren, J.; Qin, J.J.; Cheng, X.R.; Zeng, Q.; Zhang, F.; Yan, S.K.; Jin, H.Z.; Zhang, W.D. Phenylpropanoids and lignanoids from Euonymus acanthocarpus. Arch. Pharm. Res. 2012, 35, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.L.; Zhu, W.; Wang, D.M.; Chen, W.L.; Hu, M.M.; Wang, M.; Xu, X.J.; Lu, C.J. Inhibitory effects of chemical compounds isolated from the rhizome of Smilax glabra on nitric oxide and tumor necrosis factor—α production in lipopolysaccharide-induced RAW264.7 cell. Evid. Based Complement. Alternat. Med. 2015, 2015, 602425. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.H.; Akao, T.; Hamasaki, K.; Deyama, T.; Hattori, M. Biotransformation of pinoresinol diglucoside to mammalian lignans by human intestinal microflora, and isolation of Enterococcus faecalis strain PDG-1 responsible for the transformation of (+)-pinoresinol to (+)-lariciresinol. Chem. Pharm. Bull. 2003, 51, 508–515. [Google Scholar] [CrossRef]
- Ma, Y.; Bao, Y.; Zhang, W.; Ying, X.; Stien, D. Four lignans from Portulaca oleracea L. and its antioxidant activities. Nat. Prod. Res. 2020, 34, 2276–2282. [Google Scholar] [CrossRef]
- Queiroz, E.F.; Kuhl, C.; Terreaux, C.; Mavi, S.; Hostettmann, K. New dihydroalkylhexenones from Lannea edulis. J. Nat. Prod. 2003, 66, 578–580. [Google Scholar] [CrossRef]
- Roumy, V.; Fabre, N.; Portet, B.; Bourdy, G.; Acebey, L.; Vigor, C.; Valentin, A.; Moulis, C. Four anti-protozoal and anti-bacterial compounds from Tapirira guianensis. Phytochemistry 2009, 70, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Ledoux, A.; St-Gelais, A.; Cieckiewicz, E.; Jansen, O.; Bordignon, A.; Illien, B.; Di Giovanni, N.; Marvilliers, A.; Hoareau, F.; Pendeville, H.; et al. Antimalarial activities of alkyl cyclohexenone derivatives isolated from the leaves of Poupartia borbonica. J. Nat. Prod. 2017, 80, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.D.; David, J.M.; David, J.P.; Chai, H.B.; Pezzuto, J.M.; Cordell, G.A. Alkyl phenols and derivatives from Tapirira obtusa. Phytochemistry 2001, 56, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Jie, M.S.F.L.K.; Pasha, M.K.; Alam, M.S. Synthesis and nuclear magnetic resonance properties of all geometrical isomers of conjugated linoleic acids. Lipids 1997, 32, 1041–1044. [Google Scholar] [PubMed]
- Okoth, D.A.; Koorbanally, N.A. Cardanols, long chain cyclohexenones and cyclohexenols from Lannea schimperi (Anacardiaceae). Nat. Prod. Commun. 2015, 10, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, W.; Wang, R.; Li, C.; Lin, X.; Wang, L. Screening and identification of natural α-glucosidase and α-amylase inhibitors from partridge tea (Mallotus furetianus Muell-Arg) and in silico analysis. Food Chem. 2022, 388, 133004. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tang, Y.; Zhou, H.; Shao, J.; Ji, W.; Wang, Z.; Liang, D.; Zhao, C. Lignan constituents with α-amylase and α-glucosidase inhibitory activities from the fruits of Viburnum urceolatum. Phytochemistry 2023, 216, 113895. [Google Scholar] [CrossRef] [PubMed]
- Al-Romaima, A.; Hu, G.; Wang, Y.; Quan, C.; Dai, H.; Qiu, M. Identification of new diterpenoids from the pulp of Coffea arabica and their α-glucosidase inhibition activity. J. Agric. Food Chem. 2024, 72, 1683–1694. [Google Scholar] [CrossRef]
- Yin, H.; Dan, W.J.; Fan, B.Y.; Guo, C.; Wu, K.; Li, D.; Xian, K.F.; Pescitelli, G.; Gao, J.M. Anti-inflammatory and α-Glucosidase Inhibitory Activities of Labdane and Norlabdane Diterpenoids from the Rhizomes of Amomum villosum. J. Nat. Prod. 2019, 82, 2963–2971. [Google Scholar] [CrossRef]
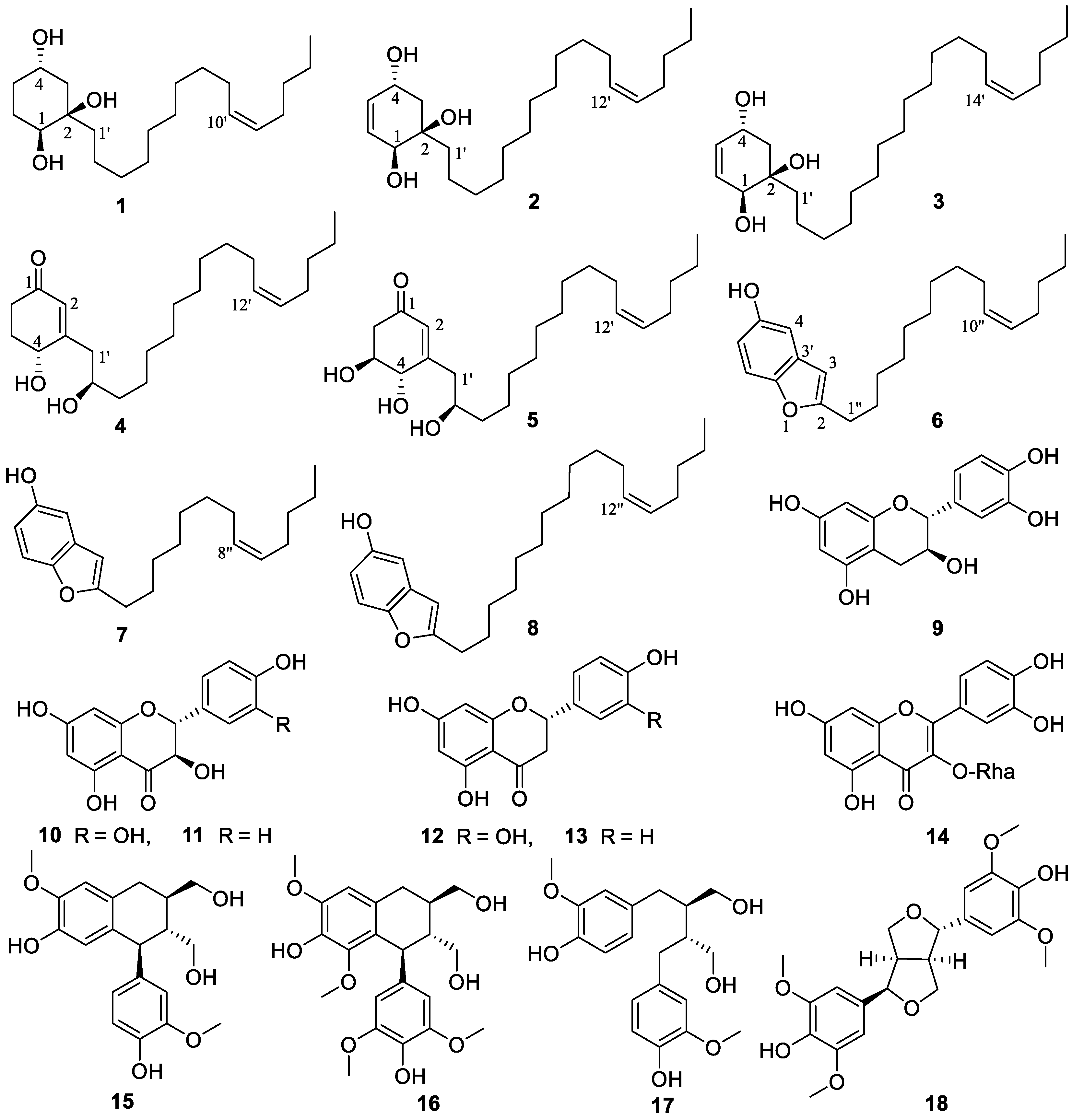
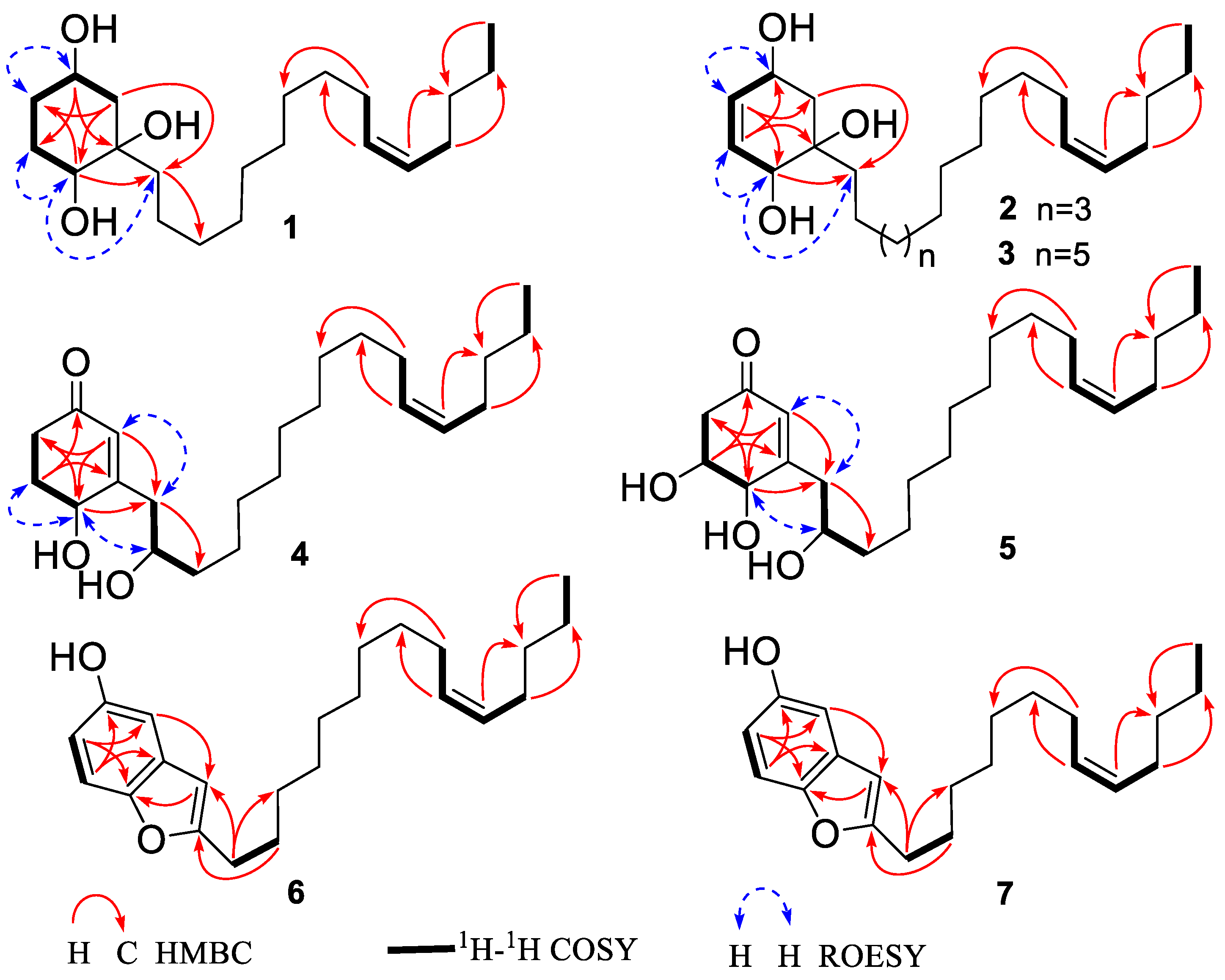
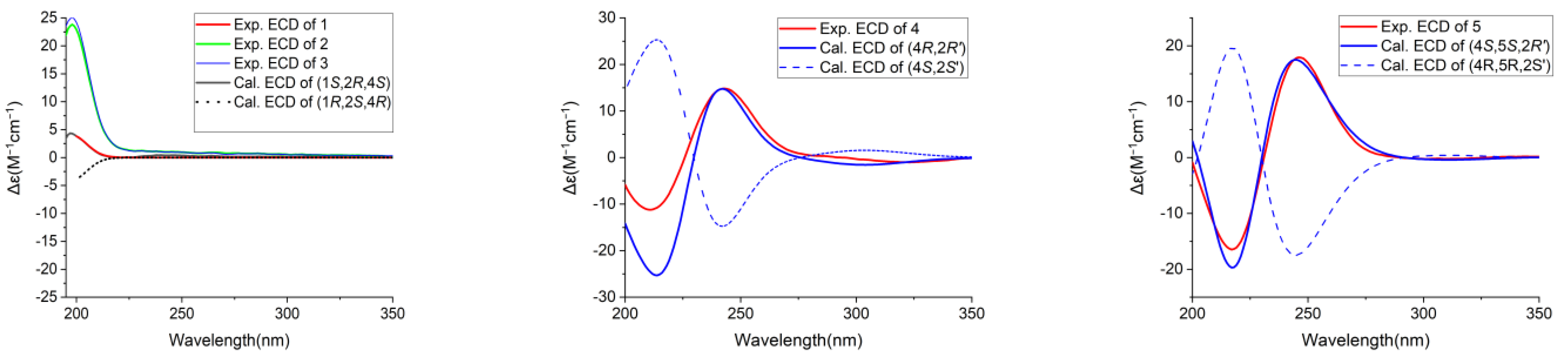
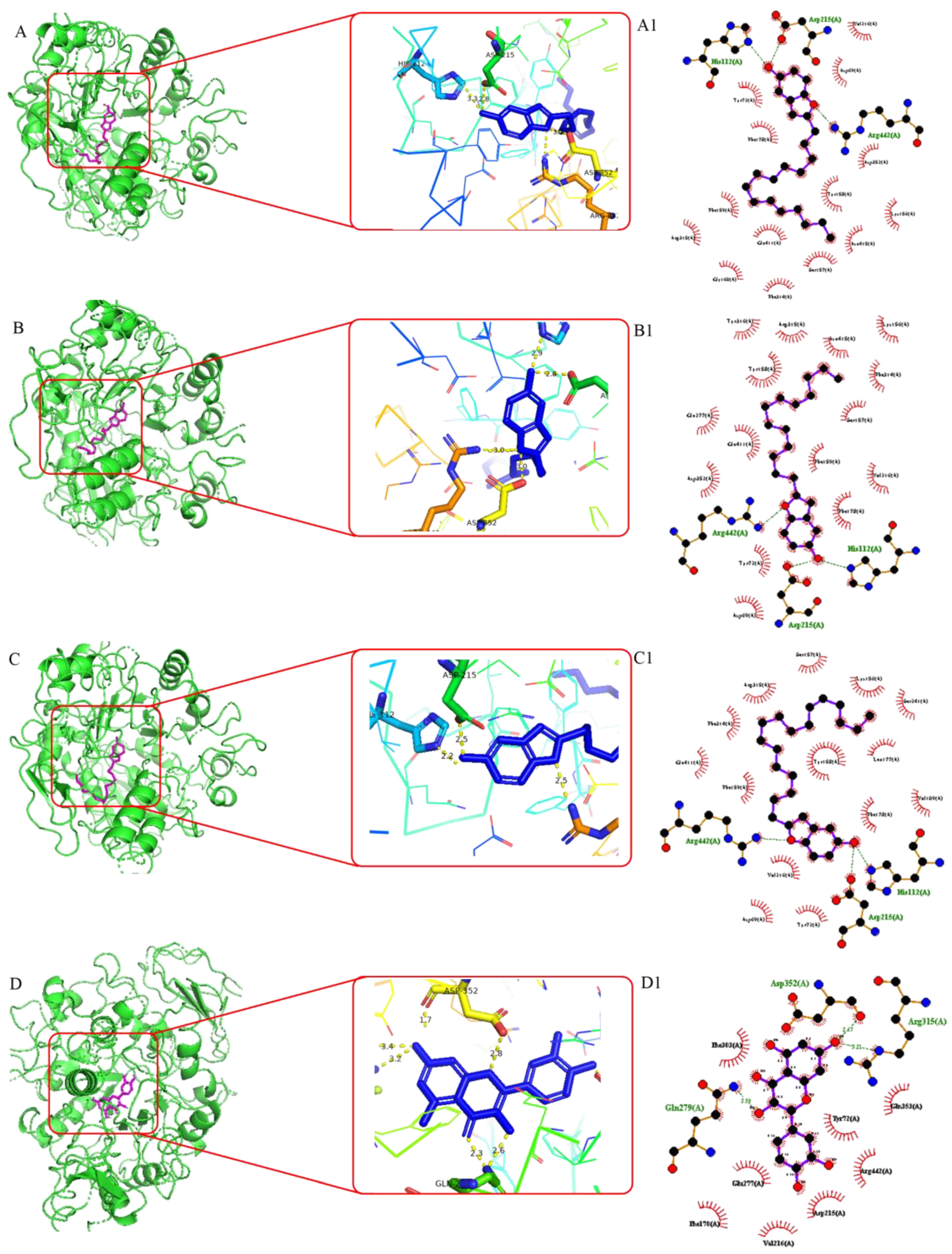

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | No. | 2 | No. | 3 | |||
|---|---|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |||
| 1 | 73.9 | 3.36 (dd, 10.7, 5.4) | 1 | 70.8 | 4.00 (d, 2.6) | 1 | 70.9 | 4.00 (d, 2.5) |
| 2 | 75.4 | 2 | 75.1 | 2 | 75.2 | |||
| 3 | 43.7 | 1.23 (dd, 13.3, 11.1) a 1.95 (ddd, 13.3, 4.4, 2.8) | 3 | 41.4 | 1.42 (dd, 13.2, 9.5) 2.17 (dd, 13.2, 5.5) | 3 | 41.5 | 1.42 (dd, 13.2, 9.5) 2.15 (dd, 13.2, 3.9) |
| 4 | 67.4 | 3.84 (tt, 11.1, 4.4) | 4 | 66.1 | 4.39 (ddd, 9.5, 5.5, 2.0) | 4 | 66.2 | 4.38 (ddd, 9.5, 3.9, 2.0) |
| 5 | 34.3 | 1.23 (m) a 1.90 (ddd, 12.5, 4.4, 2.9) | 5 | 133.3 | 5.75 (dd, 10.2, 2.0) | 5 | 133.3 | 5.74 (dd, 10.2, 2.0) |
| 6 | 29.1 | 1.70 (m) | 6 | 130.7 | 5.54 (dt, 10.2, 2.6) | 6 | 130.8 | 5.53 (dt, 10.2, 2.5) |
| 1′ | 40.7 | 1.51 (m) 1.58 (m) | 1′ | 40.4 | 1.60 (m) | 1′ | 40.4 | 1.59 (m) |
| 2′ | 24.5 | 1.29–1.36 (m) a | 2′ | 24.7 | 1.28–1.39 (m) a | 2′ | 24.7 | 1.28–1.37 (m) a |
| 3′ | 31.5 | 1.29–1.36 (m) a | 3′ | 31.5 | 1.28–1.39 (m) a | 3′ | 31.5 | 1.28–1.37 (m) a |
| 4′–8′ | 30.3–30.9 | 1.29–1.36 (m) a | 4′–10′ | 30.4–30.9 | 1.28–1.39 (m) a | 4′–12′ | 30.3–30.9 | 1.28–1.37 (m) a |
| 9′ | 28.1 | 2.04 (m) a | 11′ | 28.2 | 2.04 (m) a | 13′ | 28.1 | 2.04 (m) a |
| 10′ | 130.8 | 5.34 (t, 5.1) a | 12′ | 130.7 | 5.35 (t, 5.1) a | 14′ | 130.8 | 5.35 (t, 5.1) a |
| 11′ | 130.8 | 5.34 (t, 5.1) a | 13′ | 130.7 | 5.35 (t, 5.1) a | 15′ | 130.8 | 5.35 (t, 5.1) a |
| 12′ | 27.9 | 2.04 (m) a | 14′ | 27.9 | 2.04 (m) a | 16′ | 27.9 | 2.04 (m) a |
| 13′ | 33.1 | 1.29–1.36 (m) a | 15′ | 33.1 | 1.28–1.39 (m) a | 17′ | 33.1 | 1.28–1.37 (m) a |
| 14′ | 23.4 | 1.29–1.36 (m) a | 16′ | 23.4 | 1.28–1.39 (m) a | 18′ | 23.4 | 1.28–1.37 (m) a |
| 15′ | 14.4 | 0.92 (t, 7.1) | 17′ | 14.5 | 0.92 (t, 7.2) | 19′ | 14.4 | 0.91 (t, 7.1) |
| No. | 4 | No. | 5 | No. | 6 | No. | 7 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | ||||
| 1 | 201.7 | 1 | 199.9 | 2 | 161.4 | 2 | 161.5 | ||||
| 2 | 128.0 | 5.88 (s) | 2 | 128.4 | 5.91 (s) | 3 | 102.8 | 6.25 (s) | 3 | 102.8 | 6.27 (s) |
| 3 | 167.6 | 3 | 164.8 | 4 | 106.1 | 6.84 (d, 2.5) | 4 | 106.1 | 6.83 (d, 2.5) | ||
| 4 | 68.7 | 4.39 (dd, 8.2, 4.6) | 4 | 74.4 | 4.17 (dd, 6.8, 1.4) | 5 | 153.9 | 5 | 153.9 | ||
| 5 | 32.7 | 1.97 (dtd, 12.8, 8.2, 4.0) 2.23 (ddt, 12.8, 6.8, 4.6) | 5 | 71.0 | 3.95 (ddd, 9.5, 6.8, 4.3) | 6 | 112.5 | 6.66 (dd, 8.7, 2.5) | 6 | 112.5 | 6.65 (dd, 8.7, 2.5) |
| 6 | 35.5 | 2.37 (m) 2.54 (dt, 10.8, 6.8) | 6 | 44.4 | 2.42 (dd, 16.4, 9.5) 2.73 (dd, 16.4, 4.3) | 7 | 111.5 | 7.15 (d, 8.7) | 7 | 111.5 | 7.16 (d, 8.7) |
| 1′ | 43.0 | 2.44 (dd, 14.2, 4.6) 2.59 (dd, 14.2, 8.0) | 1′ | 42.8 | 2.46 (dd, 13.8, 4.8) 2.61 (dd, 13.9, 7.8) | 3′ | 131.1 | 3′ | 131.1 | ||
| 2′ | 71.1 | 3.86 (hept, 4.5) | 2′ | 72.7 | 3.87 (hept, 4.5) | 7′ | 150.6 | 7′ | 150.6 | ||
| 3′ | 38.7 | 1.50 (m) a | 3′ | 38.5 | 1.50 (m) a | 1″ | 29.4 | 2.67 (t, 7.6) | 1″ | 29.4 | 2.69 (t, 7.6) |
| 4′ | 26.7 | 1.50 (m) a | 4′ | 26.7 | 1.50 (m) a | 2″ | 28.8 | 1.68 (p, 7.4) | 2″ | 28.8 | 1.70 (p, 7.4) |
| 5′– 10′ | 30.3– 30.9 | 1.28–1.37 (m) a | 5′– 12′ | 30.3– 30.8 | 1.27–1.35 (m) a | 3″– 8″ | 30.3– 30.8 | 1.24–1.35 (m) a | 3″– 6″ | 30.3– 30.8 | 1.26–1.36 (m) a |
| 11′ | 28.1 | 2.04 (m) a | 13′ | 28.1 | 2.03 (m) a | 9″ | 28.1 | 2.00 (m) a | 7″ | 28.1 | 2.02 (m) a |
| 12′ | 130.8 | 5.34 (t, 5.6) a | 14′ | 130.8 | 5.34 (t, 5.6) a | 10″ | 130.8 | 5.32 (t, 5.7) a | 8″ | 130.8 | 5.33 (t, 5.4) a |
| 13′ | 130.8 | 5.34 (t, 5.6) a | 15′ | 130.8 | 5.34 (t, 5.6) a | 11″ | 130.8 | 5.32 (t, 5.7) a | 9″ | 130.8 | 5.33 (t, 5.4) a |
| 14′ | 27.9 | 2.04 (m) a | 16′ | 27.9 | 2.03 (m) a | 12″ | 27.9 | 2.00 (m) a | 10″ | 27.9 | 2.02 (m) a |
| 15′ | 33.1 | 1.28–1.37 (m) a | 17′ | 33.1 | 1.27–1.35 (m) a | 13″ | 33.1 | 1.24–1.35 (m) a | 11″ | 33.1 | 1.26–1.36 (m) a |
| 16′ | 23.4 | 1.28–1.37 (m) a | 18′ | 23.4 | 1.27–1.35 (m) a | 14″ | 23.4 | 1.24–1.35 (m) a | 12″ | 23.4 | 1.26–1.36 (m) a |
| 17′ | 14.4 | 0.91 (t, 7.1) | 19′ | 14.4 | 0.90 (t, 7.1) | 15″ | 14.4 | 0.89 (t, 7.0) | 13″ | 14.4 | 0.90 (t, 7.1) |
| Compd. | Inhibition Rate b | IC50 (μM) c | Compd. | Inhibition Rate b | IC50 (μM) c |
|---|---|---|---|---|---|
| 1 | −8.1 ± 2.4 | - | 11 | 43.8 ± 1.3 | - |
| 2 | −12.4 ± 0.66 | - | 12 | 66.6 ± 0.92 | 38.3 ± 0.53 |
| 3 | −13.6 ± 1.7 | - | 13 | 65.1 ± 2.0 | 36.3 ± 1.3 |
| 4 | 11.1 ± 1.1 | - | 14 | 26.2 ± 1.3 | - |
| 5 | −11.2 ± 0.94 | - | 15 | 16.6 ± 0.57 | - |
| 6 | 97.7 ± 0.19 | 3.5 ± 0.12 | 16 | 7.5 ± 1.4 | - |
| 7 | 96.7 ± 0.51 | 2.26 ± 0.06 | 17 | 37.6 ± 0.12 | - |
| 8 | 98.7 ± 0.20 | 4.5 ± 0.10 | 18 | 4.8 ± 0.38 | - |
| 9 | 61.1 ± 1.6 | 43.9 ± 0.96 | Quercetin d | - | 5.2 ± 0.25 |
| 10 | 36.4 ± 0.72 | - | Acarbose d | - | 229.0 ± 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weldetsadik, E.T.; Li, N.; Li, J.; Shang, J.; Zhu, H.; Zhang, Y. Undescribed Cyclohexene and Benzofuran Alkenyl Derivatives from Choerospondias axillaris, a Potential Hypoglycemic Fruit. Foods 2024, 13, 1495. https://doi.org/10.3390/foods13101495
Weldetsadik ET, Li N, Li J, Shang J, Zhu H, Zhang Y. Undescribed Cyclohexene and Benzofuran Alkenyl Derivatives from Choerospondias axillaris, a Potential Hypoglycemic Fruit. Foods. 2024; 13(10):1495. https://doi.org/10.3390/foods13101495
Chicago/Turabian StyleWeldetsadik, Ermias Tamiru, Na Li, Jingjuan Li, Jiahuan Shang, Hongtao Zhu, and Yingjun Zhang. 2024. "Undescribed Cyclohexene and Benzofuran Alkenyl Derivatives from Choerospondias axillaris, a Potential Hypoglycemic Fruit" Foods 13, no. 10: 1495. https://doi.org/10.3390/foods13101495
APA StyleWeldetsadik, E. T., Li, N., Li, J., Shang, J., Zhu, H., & Zhang, Y. (2024). Undescribed Cyclohexene and Benzofuran Alkenyl Derivatives from Choerospondias axillaris, a Potential Hypoglycemic Fruit. Foods, 13(10), 1495. https://doi.org/10.3390/foods13101495