
Chitooligosaccharide Conjugates Prepared Using Several Phenolic Compounds via Ascorbic Acid/H2O2 Free Radical Grafting: Characteristics, Antioxidant, Antidiabetic, and Antimicrobial Activities




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Preparation of COS
2.3. Preparation of COS-Polyphenol (COS-PPN) Conjugates
2.4. Determination of Total Phenolic Content (TPC) and Conjugation Efficiency of Different COS-PPN Conjugates
2.5. Characterization of Selected COS-PPN Conjugates and COS
2.5.1. UV-Visible, Fourier Transform Infrared (FTIR), and Proton (1H) Nuclear Magnetic Resonance (NMR) Spectra
2.5.2. Antioxidant (AO) Activities
2.6. Antidiabetic Activity
2.6.1. α-Amylase Inhibitory Activity
2.6.2. α-Glucosidase Inhibitory Activity
2.6.3. Lipase Inhibitory Activity
2.7. Antimicrobial Activity
2.7.1. Zone of Inhibition (ZOI)
2.7.2. Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC)
2.8. Statistical Analysis
3. Results and Discussion
3.1. Conjugation Efficiency of Various COS-PPN Conjugates
3.2. Molecular Characteristics of the Selected COS-PPN Conjugates
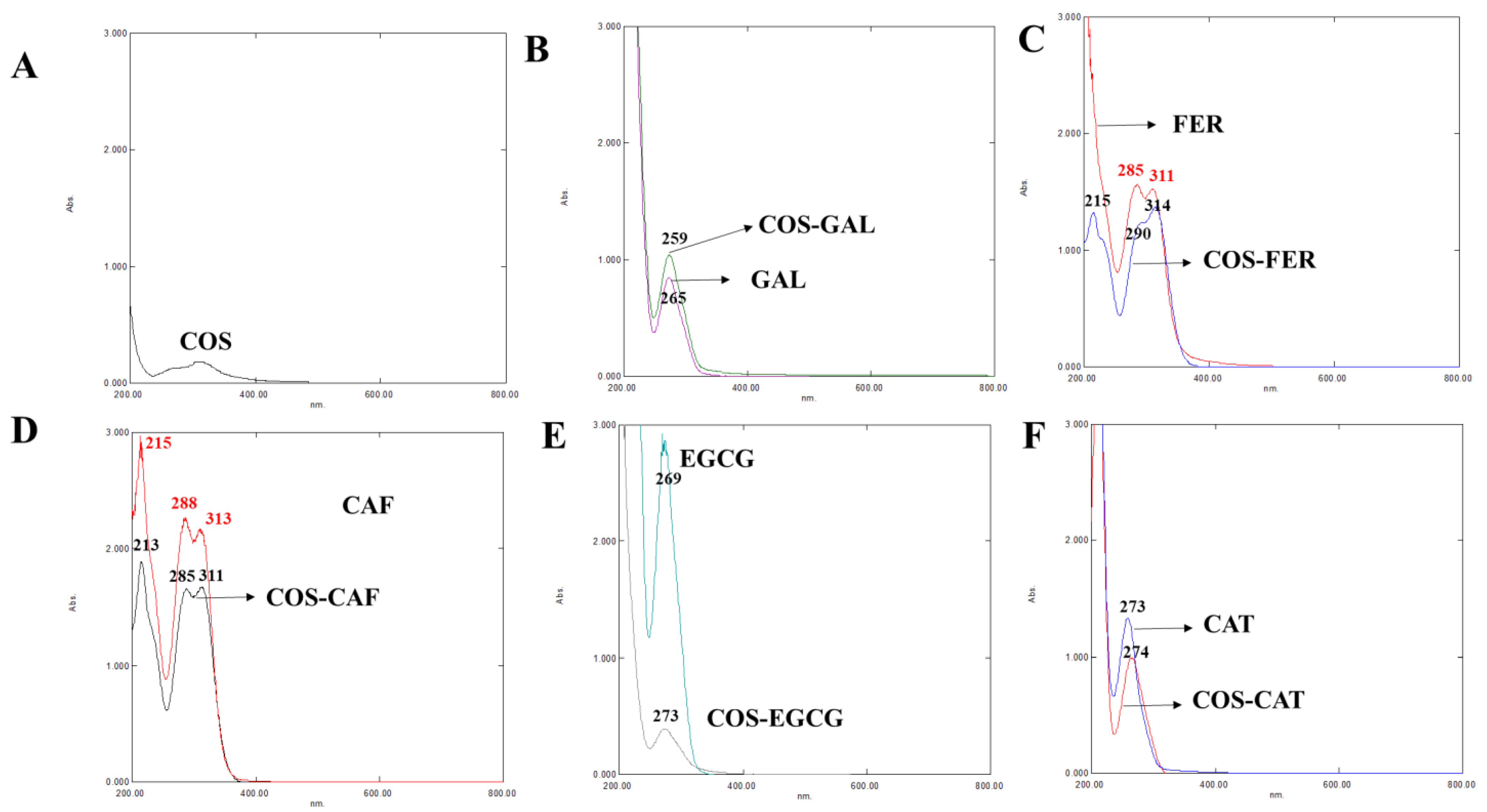
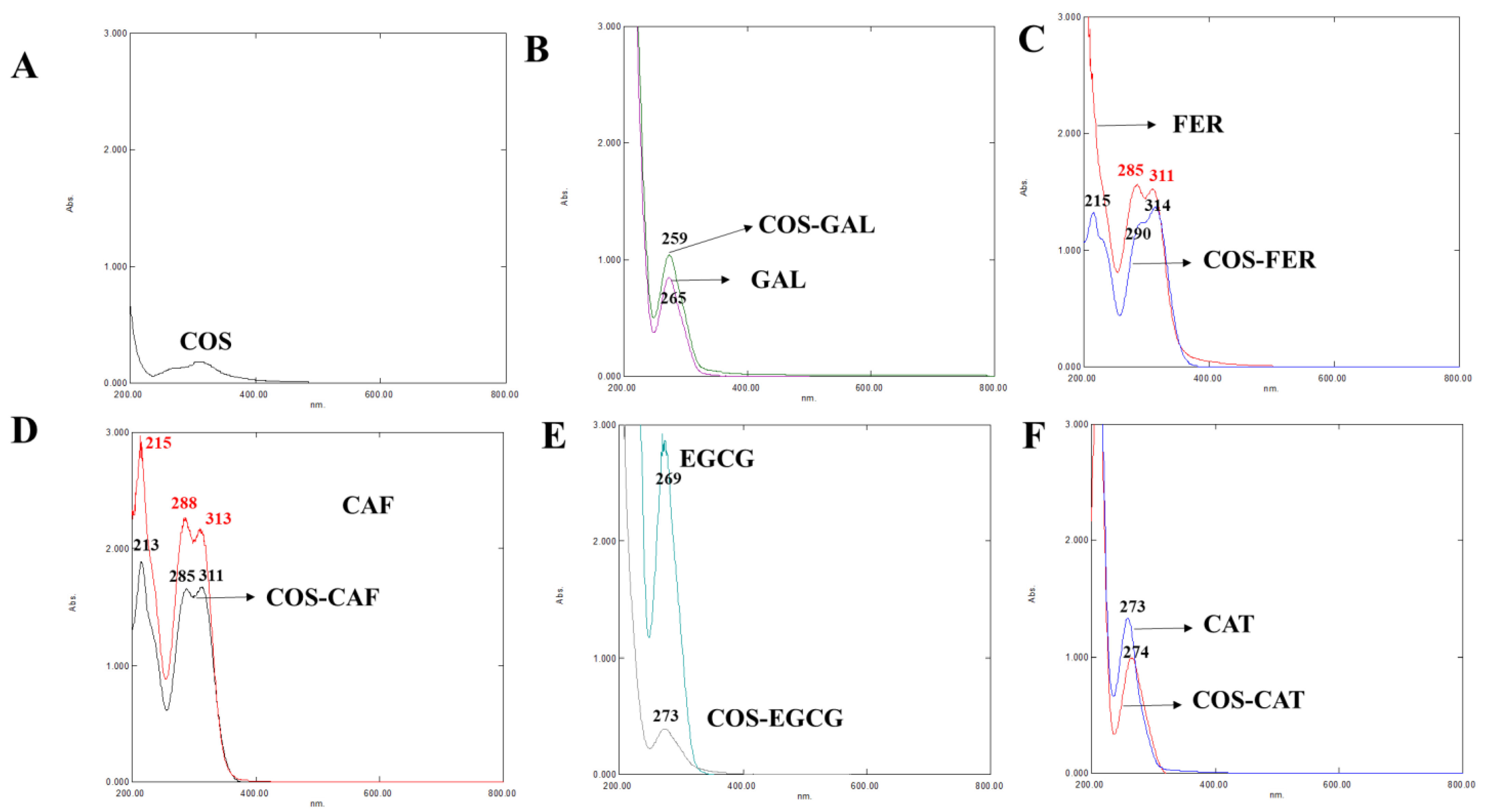
3.2.1. UV-vis Spectra
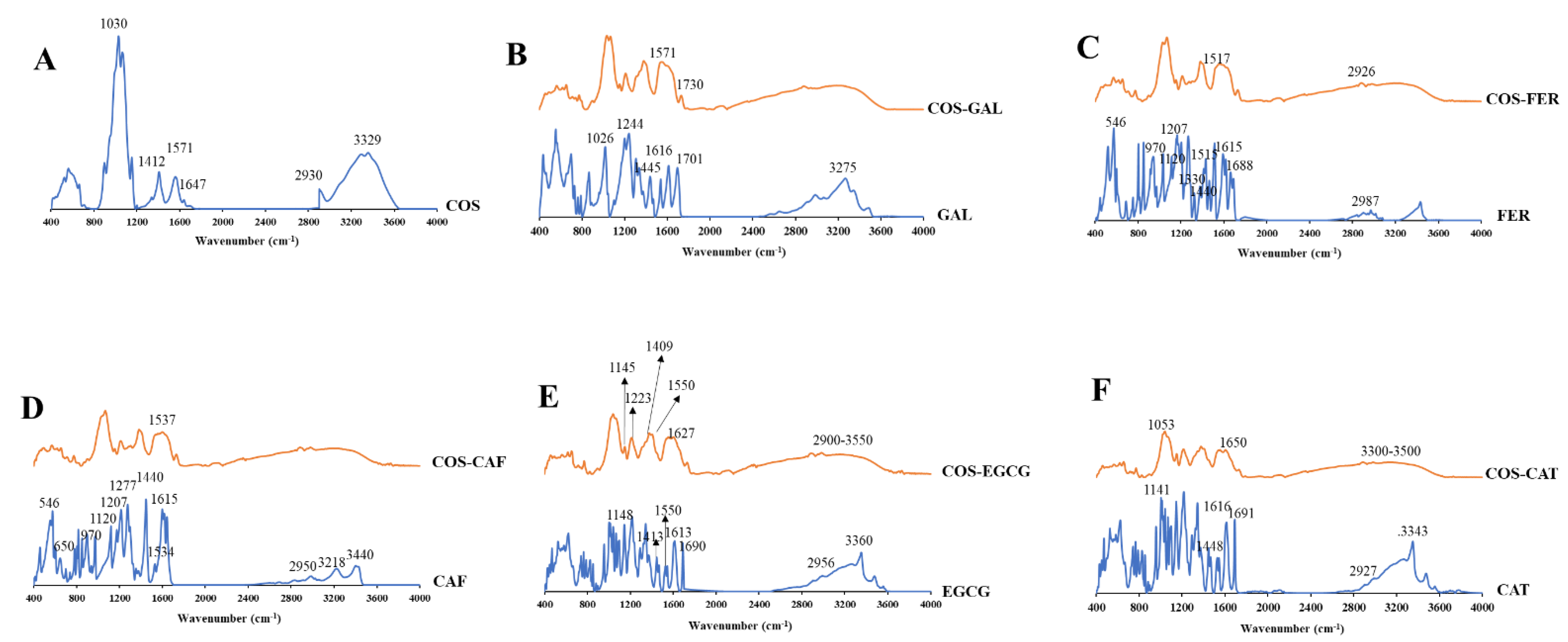
3.2.2. FTIR Spectra of COS, PPNs, and COS-PPN Conjugates
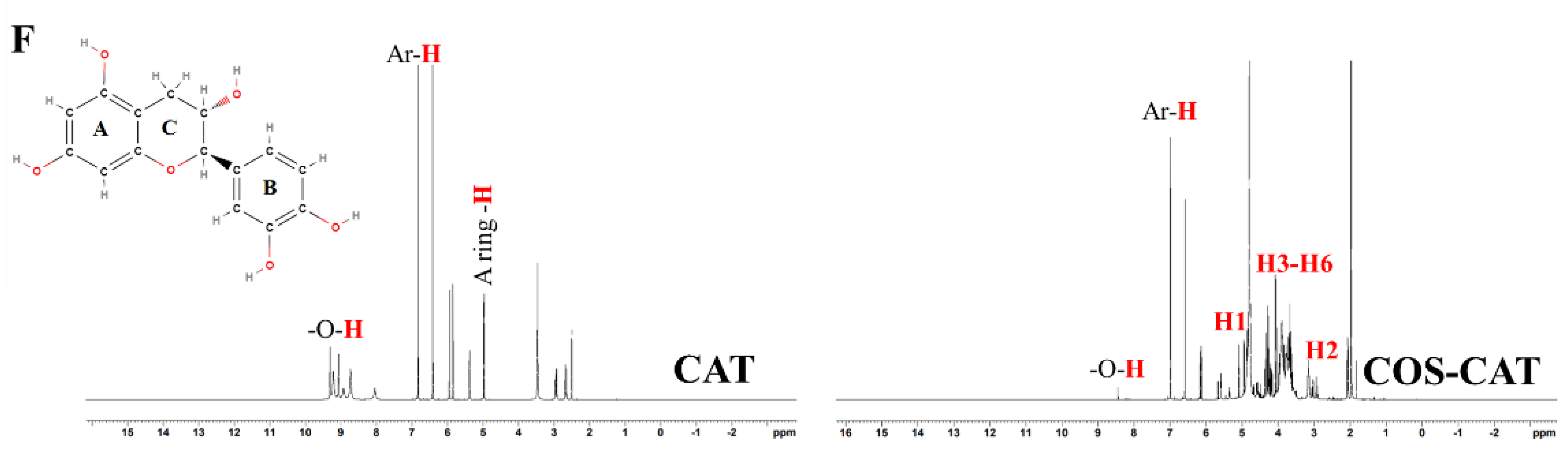
3.2.3. 1H-NMR of COS, PPN, and COS-PPN Conjugates
3.3. Total Phenolic Content (TPC) and Antioxidant Activities of COS and COS-PPN Conjugates
3.4. In Vitro Inhibitory Activities of COS and Its PPN Conjugates against α-Amylase, α-Glucosidase, and Pancreatic Lipase
3.5. Antimicrobial Activity of COS and COS-PPN Conjugates
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, A.; Mittal, A.; Benjakul, S. Chitosan, chitooligosaccharides and their polyphenol conjugates: Preparation, bioactivities, functionalities and applications in food systems. Foods Rev. Int. 2021. [Google Scholar] [CrossRef]
- Wu, T.; Wu, C.; Xiang, Y.; Huang, J.; Luan, L.; Chen, S.; Hu, Y. Kinetics and mechanism of degradation of chitosan by combining sonolysis with H2O2/ascorbic acid. RSC Adv. 2016, 6, 76280–76287. [Google Scholar] [CrossRef]
- Singh, A.; Mittal, A.; Benjakul, S. Chitosan nanoparticles: Preparation, food applications and health benefits. Sci. Asia 2021, 47, 1–10. [Google Scholar] [CrossRef]
- Mittal, A.; Singh, A.; Aluko, R.E.; Benjakul, S. Pacific white shrimp (Litopenaeus vannamei) shell chitosan and the conjugate with epigallocatechin gallate: Antioxidative and antimicrobial activities. J. Food Biochem. 2021, 45, e13569. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Rajapakse, N. Enzymatic production and biological activities of chitosan oligosaccharides (COS): A review. Carbohyd. Polym. 2005, 62, 357–368. [Google Scholar] [CrossRef]
- Singh, A.; Benjakul, S.; Huda, N.; Xu, C.; Wu, P. Preparation and characterization of squid pen chitooligosaccharide-epigallocatechin gallate conjugates and their antioxidant and antimicrobial activities. RSC Adv. 2020, 10, 33196–33204. [Google Scholar] [CrossRef]
- Papuc, C.; Goran, G.V.; Predescu, C.N.; Nicorescu, V.; Stefan, G. Plant polyphenols as antioxidant and antibacterial agents for shelf-life extension of meat and meat products: Classification, structures, sources, and action mechanisms. Compr. Rev. Food. Sci. 2017, 16, 1243–1268. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Deng, Z.; Ramdath, D.D.; Tang, Y.; Chen, P.X.; Liu, R.; Liu, Q.; Tsao, R. Phenolic profiles of 20 Canadian lentil cultivars and their contribution to antioxidant activity and inhibitory effects on α-glucosidase and pancreatic lipase. Food Chem. 2015, 172, 862–872. [Google Scholar] [CrossRef]
- Gutiérrez-Grijalva, E.P.; Antunes-Ricardo, M.; Acosta-Estrada, B.A.; Gutiérrez-Uribe, J.A.; Heredia, J.B. Cellular antioxidant activity and in vitro inhibition of α-glucosidase, α-amylase and pancreatic lipase of oregano polyphenols under simulated gastrointestinal digestion. Food Res. Int. 2019, 116, 676–686. [Google Scholar] [CrossRef]
- Eom, T.-K.; Senevirathne, M.; Kim, S.-K. Synthesis of phenolic acid conjugated chitooligosaccharides and evaluation of their antioxidant activity. Environ. Toxicol. Pharmacol. 2012, 34, 519–527. [Google Scholar] [CrossRef]
- Ryu, B.; Kim, S.Y.; Vo, T.S.; Kim, W.S.; Kim, D.G.; Kim, S.K. Characterization of the in vitro effects of gallic acid-grafted-chitooligosaccharides in the suppression of AGS human gastric cancer cell proliferation. RSC Adv. 2017, 7, 24561–24568. [Google Scholar] [CrossRef] [Green Version]
- Mittal, A.; Singh, A.; Hong, H.; Benjakul, S. Chitooligosaccharides from shrimp shell chitosan prepared using H2O2 or ascorbic acid/H2O2 redox pair hydrolysis: Characteristics, antioxidant and antimicrobial activities. Int. J. Food Sci. Technol. 2022. [Google Scholar] [CrossRef]
- Mittal, A.; Singh, A.; Benjakul, S. Preparation and characterisation of liposome loaded with chitosan-epigallocatechin gallate conjugate. J. Microencapsul. 2021, 38, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Benjakul, S.; Kittiphattanabawon, P.; Sumpavapol, P.; Maqsood, S. Antioxidant activities of lead (Leucaena leucocephala) seed as affected by extraction solvent, prior dechlorophyllisation and drying methods. J. Food Sci. Technol. 2014, 51, 3026–3037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chotphruethipong, L.; Benjakul, S.; Kijroongrojana, K.J. Optimization of extraction of antioxidative phenolic compounds from cashew (Anacardium occidentale L.) leaves using response surface methodology. J. Food Biochem. 2017, 41, e12379. [Google Scholar] [CrossRef]
- Awosika, T.O.; Aluko, R.E. Inhibition of the in vitro activities of α-amylase, α-glucosidase and pancreatic lipase by yellow field pea (Pisum sativum L.) protein hydrolysates. Int. J. Food Sci. Technol. 2019, 54, 2021–2034. [Google Scholar] [CrossRef] [Green Version]
- Sultana, R.; Alashi, A.M.; Islam, K. Inhibitory activities of Polyphenolic extracts of Bangladeshi vegetables against α-amylase, α-glucosidase, pancreatic lipase, renin, and angiotensin-converting enzyme. Foods 2020, 9, 844. [Google Scholar] [CrossRef] [PubMed]
- Steel, R.G.; Torrie, J.H. Principles and Procedures of Statistics: A Biometrical Approach; McGraw-Hill: New York, NY, USA, 1986. [Google Scholar]
- Hu, Q.B.; Luo, Y.C. Polyphenol-chitosan conjugates: Synthesis, characterization, and applications. Carbohydr. Polym. 2016, 151, 624–639. [Google Scholar] [CrossRef]
- Mota, F.L.; Queimada, A.J.; Pinho, S.P.; Macedo, E.A. Aqueous solubility of some natural phenolic compounds. Ind. Eng. Chem. Res. 2008, 47, 5182–5189. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Vásquez, M.J.; Plascencia-Jatomea, M.; Sánchez-Valdes, S.; Tanori-Córdova, J.C.; Castillo-Yañez, F.J.; Quintero-Reyes, I.E.; Graciano-Verdugo, A.Z. Characterization of epigallocatechin-gallate-grafted chitosan nanoparticles and evaluation of their antibacterial and antioxidant potential. Polymers 2021, 13, 1375. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, M.; Ma, G.; Fang, Y.; Yang, W.; Ma, N.; Fang, D.; Hu, Q.; Pei, F. The antioxidant and antimicrobial activities of different phenolic acids grafted onto chitosan. Carbohydr. Polym. 2019, 225, 115238. [Google Scholar] [CrossRef] [PubMed]
- Holser, R.A. Principal component analysis of phenolic acid spectra. Int. Sch. Res. Notices 2012. [Google Scholar] [CrossRef]
- İlyasoğlu, H.; Guo, Z. Water soluble chitosan-caffeic acid conjugates as a dual functional polymeric surfactant. Food Biosci. 2019, 29, 118–125. [Google Scholar] [CrossRef]
- Lei, F.; Wang, X.; Liang, C.; Yuan, F.; Gao, Y. Preparation and functional evaluation of chitosan-EGCG conjugates. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Yang, Y.; Xing, R.E.; Liu, S.; Qin, Y.; Li, K.; Yu, H.; Li, P. Immunostimulatory effects of chitooligosaccharides on raw 264.7 mouse macrophages via regulation of the mapk and pi3k/akt signaling pathways. Mar. Drugs 2019, 17, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, J.; Shi, B.; Mo, L.; Shu, D.; Guo, H. Preparation of chitooligosaccharides by α-amylase from chitosan with oxidative pretreatment. J. Chem. Technol. Biotechnol. 2021, 96, 3408–3413. [Google Scholar] [CrossRef]
- da Rosa, C.G.; Borges, C.D.; Zambiazi, R.C.; Nunes, M.R.; Benvenutti, E.V.; da Luz, S.R.; D’Avila, R.F.; Rutz, J.K. Microencapsulation of gallic acid in chitosan, β-cyclodextrin and xanthan. Ind. Crops Prod. 2013, 46, 138–146. [Google Scholar] [CrossRef]
- Liu, J.; Lu, J.F.; Kan, J.; Jin, C.H. Synthesis of chitosan-gallic acid conjugate: Structure characterization and in vitro anti-diabetic potential. Int. J. Biol. Macromol. 2013, 62, 321–329. [Google Scholar] [CrossRef]
- Woranuch, S.; Yoksan, R. Preparation, characterization and antioxidant property of water-soluble ferulic acid grafted chitosan. Carbohydr. Polym. 2013, 96, 495–502. [Google Scholar] [CrossRef]
- Wang, J.; Cao, Y.; Sun, B.; Wang, C. Characterisation of inclusion complex of trans-ferulic acid and hydroxypropyl-β-cyclodextrin. Food Chem. 2011, 124, 1069–1075. [Google Scholar] [CrossRef]
- Catauro, M.; Barrino, F.; Dal Poggetto, G.; Crescente, G.; Piccolella, S.; Pacifico, S. New SiO2/caffeic acid hybrid materials: Synthesis, spectroscopic characterization, and bioactivity. Materials 2020, 13, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Pu, H.; Chen, C.; Liu, Y.; Bai, R.; Kan, J.; Jin, C. Reaction mechanisms and structural and physicochemical properties of caffeic acid grafted chitosan synthesized in ascorbic acid and hydroxyl peroxide redox system. J. Agric. Food Chem. 2018, 66, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, J.F.; Kan, J.; Wen, X.Y.; Jin, C.H. Synthesis, characterization and in vitro anti-diabetic activity of catechin grafted inulin. Int. J. Biol. Macromol. 2014, 64, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yu, X.; Diao, Y.; Jing, Y. Free radical grafting of epigallocatechin gallate onto carboxymethyl chitosan: Preparation, characterization, and application on the preservation of grape juice. Food Bioprocess Technol. 2020, 13, 807–817. [Google Scholar] [CrossRef]
- López-Martínez, L.M.; Santacruz-Ortega, H.; Navarro, R.E.; Sotelo-Mundo, R.R.; González-Aguilar, G.A. A 1H NMR investigation of the interaction between phenolic acids found in Mango (Manguifera indica cv Ataulfo) and Papaya (Carica papaya cv Maradol) and 1, 1-diphenyl-2-picrylhydrazyl (DPPH) free radicals. PLoS ONE 2015, 10, e0140242. [Google Scholar] [CrossRef]
- Zhong, Y.; Shahidi, F. Lipophilized epigallocatechin gallate (EGCG) derivatives as novel antioxidants. J. Agric. Food Chem. 2011, 59, 6526–6533. [Google Scholar] [CrossRef]
- Hong, S.; Wu, S.; Cai, H.; Wang, Y.; Liu, S. Instant structure profiling of substituted catechins by chemical shift fingerprint of hydrogens of phenolic hydroxyl groups. J. Funct. Foods 2017, 37, 58–65. [Google Scholar] [CrossRef]
- Liaqat, F.; Eltem, R. Chitooligosaccharides and their biological activities: A comprehensive review. Carbohydr. Polym. 2018, 184, 243–259. [Google Scholar] [CrossRef]
- Lodhi, G.; Kim, Y.S.; Hwang, J.W.; Kim, S.K.; Jeon, Y.J.; Je, J.Y.; Ahn, C.B.; Moon, S.H.; Jeon, B.T.; Park, P.J. Chitooligosaccharide and its derivatives: Preparation and biological applications. BioMed Res. Int. 2014, 2014, 654913. [Google Scholar] [CrossRef] [Green Version]
- Platzer, M.; Kiese, S.; Herfellner, T.; Schweiggert-Weisz, U.; Miesbauer, O.; Eisner, P. Common trends and differences in antioxidant activity analysis of phenolic substances using single electron transfer based assays. Molecules 2021, 26, 1244. [Google Scholar] [CrossRef]
- Singh, A.; Benjakul, S.; Prodpran, T. Chitooligosaccharides from squid pen prepared using different enzymes: Characteristics and the effect on quality of surimi gel during refrigerated storage. Food Prod. Process. Nutrit. 2019, 1, 5. [Google Scholar] [CrossRef]
- Zhong, Y.; Ma, C.-M.; Shahidi, F. Antioxidant and antiviral activities of lipophilic epigallocatechin gallate (EGCG) derivatives. J. Funct. Foods 2012, 4, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Gulcin, I. Antioxidant activity of food constituents: An overview. Arch. Toxicol. 2012, 86, 345–391. [Google Scholar] [CrossRef] [PubMed]
- Aleixandre, A.; Gil, J.V.; Sineiro, J.; Rosell, C.M. Understanding phenolic acids inhibition of α-amylase and α-glucosidase and influence of reaction conditions. Food Chem. 2022, 372, 131231. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Tang, S.; Zhang, L.; Xiao, J.; Luo, Q.; Chen, Y.; Zhou, M.; Feng, N.; Wang, C. The inhibitory effect of the catechin structure on advanced glycation end product formation in alcoholic media. Food Funct. 2020, 11, 5396–5408. [Google Scholar] [CrossRef]
- Abdelli, I.; Benariba, N.; Adjdir, S.; Fekhikher, Z.; Daoud, I.; Terki, M.; Benramdane, H.; Ghalem, S. In silico evaluation of phenolic compounds as inhibitors of α-amylase and α-glucosidase. J. Biomol. Struct. Dyn. 2021, 39, 816–822. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Y.; Miao, M. Inhibition of α-amylase by polyphenolic compounds: Substrate digestion, binding interactions and nutritional intervention. Trends Food Sci. Technol. 2020, 104, 190–207. [Google Scholar] [CrossRef]
- Kalita, D.; Holm, D.G.; LaBarbera, D.V.; Petrash, J.M.; Jayanty, S.S. Inhibition of α-glucosidase, α-amylase, and aldose reductase by potato polyphenolic compounds. PLoS ONE 2018, 13, e0191025. [Google Scholar] [CrossRef]
- Choudhary, D.K.; Chaturvedi, N.; Singh, A.; Mishra, A. Characterization, inhibitory activity and mechanism of polyphenols from faba bean (gallic-acid and catechin) on α-glucosidase: Insights from molecular docking and simulation study. Prep. Biochem. Biotechnol. 2020, 50, 123–132. [Google Scholar] [CrossRef]
- Kohls, E.; Stein, M. The thermochemistry of long chain olefin isomers during hydroformylation. New J. Chem. 2017, 41, 7347–7355. [Google Scholar] [CrossRef] [Green Version]
- Fu, M.; Shen, W.; Gao, W.; Namujia, L.; Yang, X.; Cao, J.; Sun, L. Essential moieties of myricetins, quercetins and catechins for binding and inhibitory activity against α-Glucosidase. Bioorg. Chem. 2021, 115, 105235. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Tian, J.; Yang, W.; Chen, S.; Liu, D.; Fang, H.; Zhang, H.; Ye, X. Inhibition mechanism of ferulic acid against α-amylase and α-glucosidase. Food Chem. 2020, 317, 126346. [Google Scholar] [CrossRef]
- Adisakwattana, S.; Chantarasinlapin, P.; Thammarat, H.; Yibchok-Anun, S. A series of cinnamic acid derivatives and their inhibitory activity on intestinal α-glucosidase. J. Enzym. Inhib. Med. Chem. 2009, 24, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Limanto, A.; Simamora, A.; Santoso, A.W.; Timotius, K.H. Antioxidant, α-glucosidase inhibitory activity and molecular docking study of gallic acid, quercetin and rutin: A comparative study. Mol. Cellul. Biomed. Sci. 2019, 3, 67–74. [Google Scholar] [CrossRef]
- You, Q.; Chen, F.; Wang, X.; Jiang, Y.; Lin, S. Anti-diabetic activities of phenolic compounds in muscadine against alpha-glucosidase and pancreatic lipase. LWT-Food Sci. Technol. 2012, 46, 164–168. [Google Scholar] [CrossRef]
- Cai, S.; Wang, O.; Wang, M.; He, J.; Wang, Y.; Zhang, D.; Zhou, F.; Ji, B. In vitro inhibitory effect on pancreatic lipase activity of subfractions from ethanol extracts of fermented oats (Avena sativa L.) and synergistic effect of three phenolic acids. J. Agric. Food Chem. 2012, 60, 7245–7251. [Google Scholar] [CrossRef] [PubMed]
- Grove, K.A.; Sae-Tan, S.; Kennett, M.J.; Lambert, J.D. (−)− Epigallocatechin-3-gallate inhibits pancreatic lipase and reduces body weight gain in high fat-fed obese mice. Obesity 2012, 20, 2311–2313. [Google Scholar] [CrossRef]
- Glisan, S.L.; Grove, K.A.; Yennawar, N.H.; Lambert, J.D. Inhibition of pancreatic lipase by black tea theaflavins: Comparative enzymology and in silico modeling studies. Food Chem. 2017, 216, 296–300. [Google Scholar] [CrossRef] [Green Version]
- Goy, R.C.; Morais, S.T.B.; Assis, O.B.G. Evaluation of the antimicrobial activity of chitosan and its quaternized derivative on E-coli and S. aureus growth. Rev. Bras. Farmacogn. 2016, 26, 122–127. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration of Phenolic Compound Used (mg/mL) | Conjugation Efficiency (%) | ||||
|---|---|---|---|---|---|
| Gallic Acid | Ferulic Acid | Caffeic Acid | Epigallocatechin Gallate | Catechin | |
| 1 | 29.56 ± 0.36 d | 19.29 ± 0.34 a | 19.60 ± 1.02 a | 29.32 ± 0.96 a | 14.12 ± 1.03 b |
| 2 | 35.94 ± 0.65 a | 13.10 ± 0.42 b | 8.20 ± 0.30 b | 17.59 ± 0.51 b | 14.52 ± 0.37 b |
| 4 | 35.44 ± 0.18 a | 9.30 ± 0.07 c | 8.10 ± 0.46 b | 9.56 ± 0.33 c | 17.22 ± 0.27 a |
| 6 | 33.03 ± 0.24 b | 9.83 ± 0.04 d | 7.93 ± 0.78 b | 9.05 ± 0.42 c | 10.98 ± 0.85 c |
| 8 | 32.48 ± 0.18 b | 7.05 ± 0.05 e | 8.02 ± 0.10 b | 8.73 ± 0.55 c | 9.39 ± 0.43 d |
| 10 | 31.27 ± 0.12 c | 7.93 ± 0.02 f | 8.54 ± 0.10 b | 5.16 ± 0.10 d | 8.74 ± 0.58 d |
| TPC # | DPPH-RSA * | ABTS-RSA * | FRAP * | MCA ** | ORAC * | |
|---|---|---|---|---|---|---|
| COS | - | 21.27 ± 0.24 e | 33.49 ± 0.12 f | 17.08 ± 0.57 f | 4.01 ± 0.35 d | 12.58 ± 1.25 d |
| COS-GAL | 518.89 ± 3.96 b | 116.04 ± 1.51 b | 357.70 ± 2.95 b | 170.04 ± 1.72 c | 8.27 ± 0.88 c | 78.58 ± 1.21 a |
| COS-FER | 192.86 ± 3.39 d | 102.64 ± 1.05 c | 166.43 ± 2.74 d | 115.14 ± 3.00 e | 10.11 ± 1.70 c | 30.54 ± 0.56 c |
| COS-CAF | 196.00 ± 2.16 d | 114.23 ± 1.57 b | 237.20 ± 2.23 c | 131.14 ± 3.13 d | 8.38 ± 0.90 c | 28.66 ± 1.45 c |
| COS-EGCG | 293.20 ± 5.92 c | 73.12 ± 1.71 d | 67.20 ± 2.88 e | 278.36 ± 0.11 b | 15.27 ± 1.42 b | 68.27 ± 0.99 b |
| COS-CAT | 688.79 ± 3.91 a | 123.22 ± 1.73 a | 365.83 ± 3.19 a | 409.93 ± 2.97 a | 20.13 ± 0.59 a | 80.27 ± 0.79 a |
| Parameters | Samples | Pseudomonas aeruginosa | Vibrio parahaemolyticus | Escherichia coli | Listeria monocytogenes | Staphylococcus aureus |
|---|---|---|---|---|---|---|
| MIC (µg/mL) | COS | 1024 | 1024 | 2048 | 2048 | 2048 |
| COS-GAL | 512 | 512 | 512 | 1024 | 1024 | |
| COS-FER | 512 | 256 | 512 | 512 | 512 | |
| COS-CAF | 256 | 256 | 512 | 512 | 512 | |
| COS-EGCG | 64 | 64 | 256 | 128 | 128 | |
| COS-CAT | 32 | 32 | 128 | 128 | 256 | |
| MBC (µg/mL) | COS | 1024 | 1024 | 2048 | 2048 | 2048 |
| COS-GAL | 512 | 512 | 512 | 1024 | 1024 | |
| COS-FER | 512 | 256 | 512 | 512 | 512 | |
| COS-CAF | 256 | 512 | 1024 | 512 | 1024 | |
| COS-EGCG | 128 | 128 | 512 | 256 | 256 | |
| COS-CAT | 64 | 64 | 256 | 128 | 256 | |
| Zone of inhibition (mm) | COS | 5.62 ± 0.07 f | 5.32 ± 0.10 f | 4.23 ± 0.02 f | 4.31 ± 0.07 f | 4.32 ± 0.02 f |
| COS-GAL | 7.54 ± 0.10 d | 6.23 ± 0.08 e | 5.32 ± 0.10 e | 5.45 ± 0.03 e | 4.72 ± 0.05 e | |
| COS-FER | 6.45 ± 0.03 e | 7.58 ± 0.10 d | 6.18 ± 0.02 d | 7.11 ± 0.05 d | 7.55 ± 0.01 d | |
| COS-CAF | 7.69 ± 0.14 c | 7.77 ± 0.07 c | 6.59 ± 0.02 c | 7.50 ± 0.14 c | 7.94 ± 0.07 c | |
| COS-EGCG | 8.54 ± 0.10 b | 8.83 ± 0.02 b | 8.58 ± 0.10 b | 8.44 ± 0.02 b | 8.79 ± 0.08 b | |
| COS-CAT | 12.38 ± 0.10 a | 13.65 ± 0.01 a | 9.45 ± 0.02 a | 8.86 ± 0.02 a | 10.47 ± 0.02 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mittal, A.; Singh, A.; Zhang, B.; Visessanguan, W.; Benjakul, S. Chitooligosaccharide Conjugates Prepared Using Several Phenolic Compounds via Ascorbic Acid/H2O2 Free Radical Grafting: Characteristics, Antioxidant, Antidiabetic, and Antimicrobial Activities. Foods 2022, 11, 920. https://doi.org/10.3390/foods11070920
Mittal A, Singh A, Zhang B, Visessanguan W, Benjakul S. Chitooligosaccharide Conjugates Prepared Using Several Phenolic Compounds via Ascorbic Acid/H2O2 Free Radical Grafting: Characteristics, Antioxidant, Antidiabetic, and Antimicrobial Activities. Foods. 2022; 11(7):920. https://doi.org/10.3390/foods11070920
Chicago/Turabian StyleMittal, Ajay, Avtar Singh, Bin Zhang, Wonnop Visessanguan, and Soottawat Benjakul. 2022. "Chitooligosaccharide Conjugates Prepared Using Several Phenolic Compounds via Ascorbic Acid/H2O2 Free Radical Grafting: Characteristics, Antioxidant, Antidiabetic, and Antimicrobial Activities" Foods 11, no. 7: 920. https://doi.org/10.3390/foods11070920
APA StyleMittal, A., Singh, A., Zhang, B., Visessanguan, W., & Benjakul, S. (2022). Chitooligosaccharide Conjugates Prepared Using Several Phenolic Compounds via Ascorbic Acid/H2O2 Free Radical Grafting: Characteristics, Antioxidant, Antidiabetic, and Antimicrobial Activities. Foods, 11(7), 920. https://doi.org/10.3390/foods11070920