
Effect of the Post-Harvest Processing on Protein Modification in Green Coffee Beans by Phenolic Compounds

 ,
,  ,
,  ,
,  and
and 
Abstract
:
1. Introduction
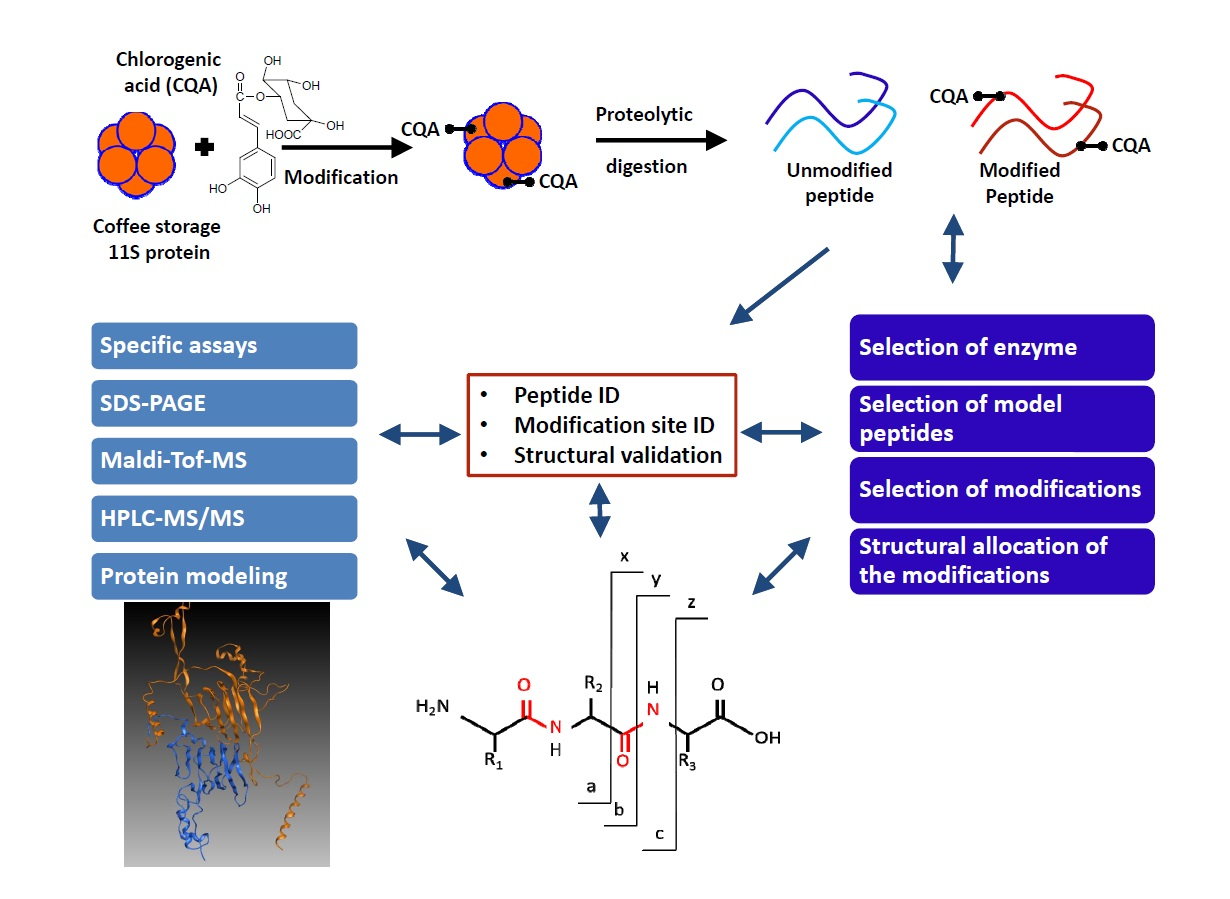
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Sample Preparation
2.2.2. Free Amino Nitrogen
2.2.3. Free Thiol Groups
2.2.4. Determination of the Composition of Protein-Bound Phenolic Compounds
2.2.5. Determination of Protein Content
2.2.6. Fluorescence Spectroscopy
2.2.7. Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis
2.2.8. In-Gel Digestion
2.2.9. MALDI-TOF-MS
2.2.10. In-Solution Digestion
2.2.11. Solid-Phase Extraction
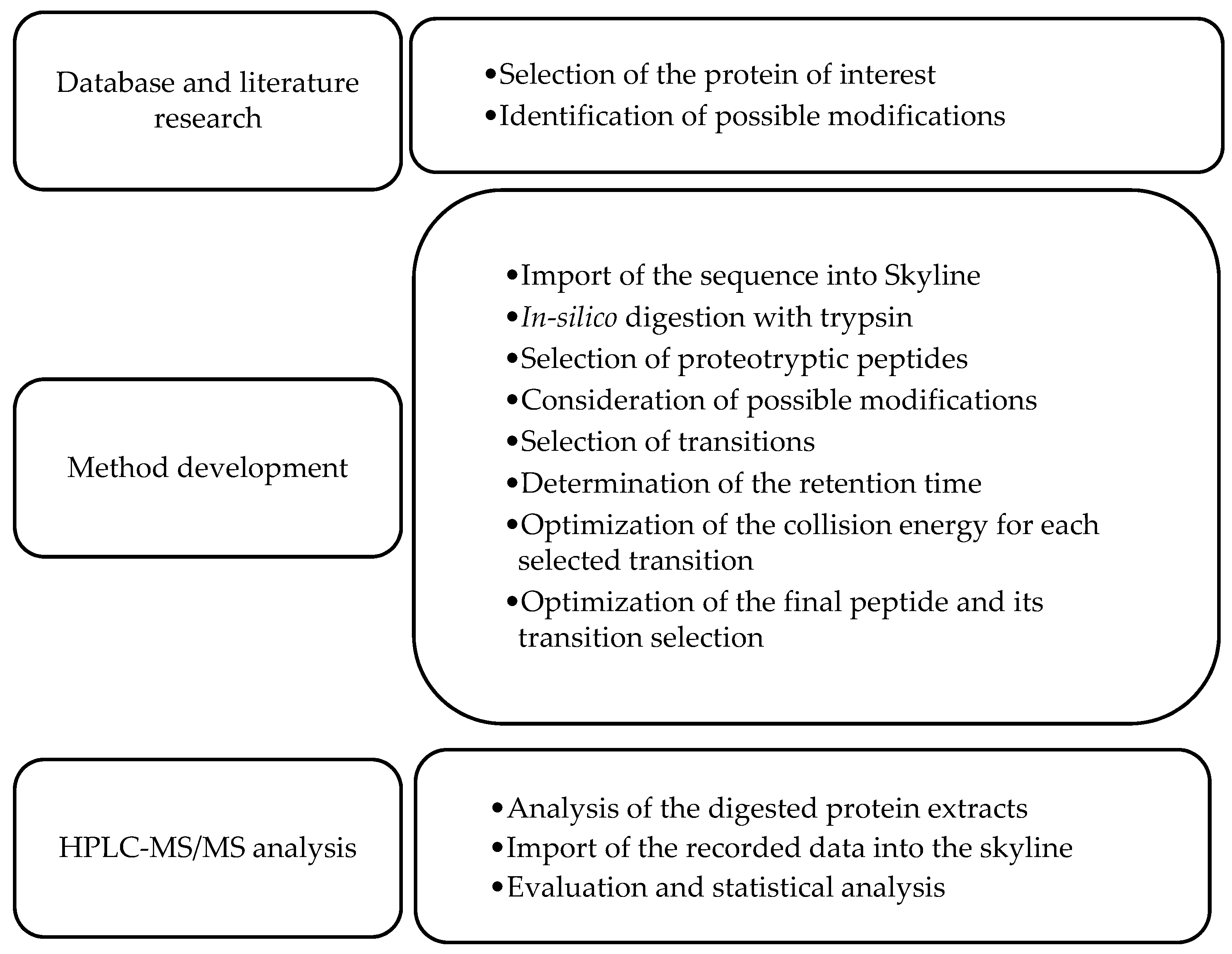
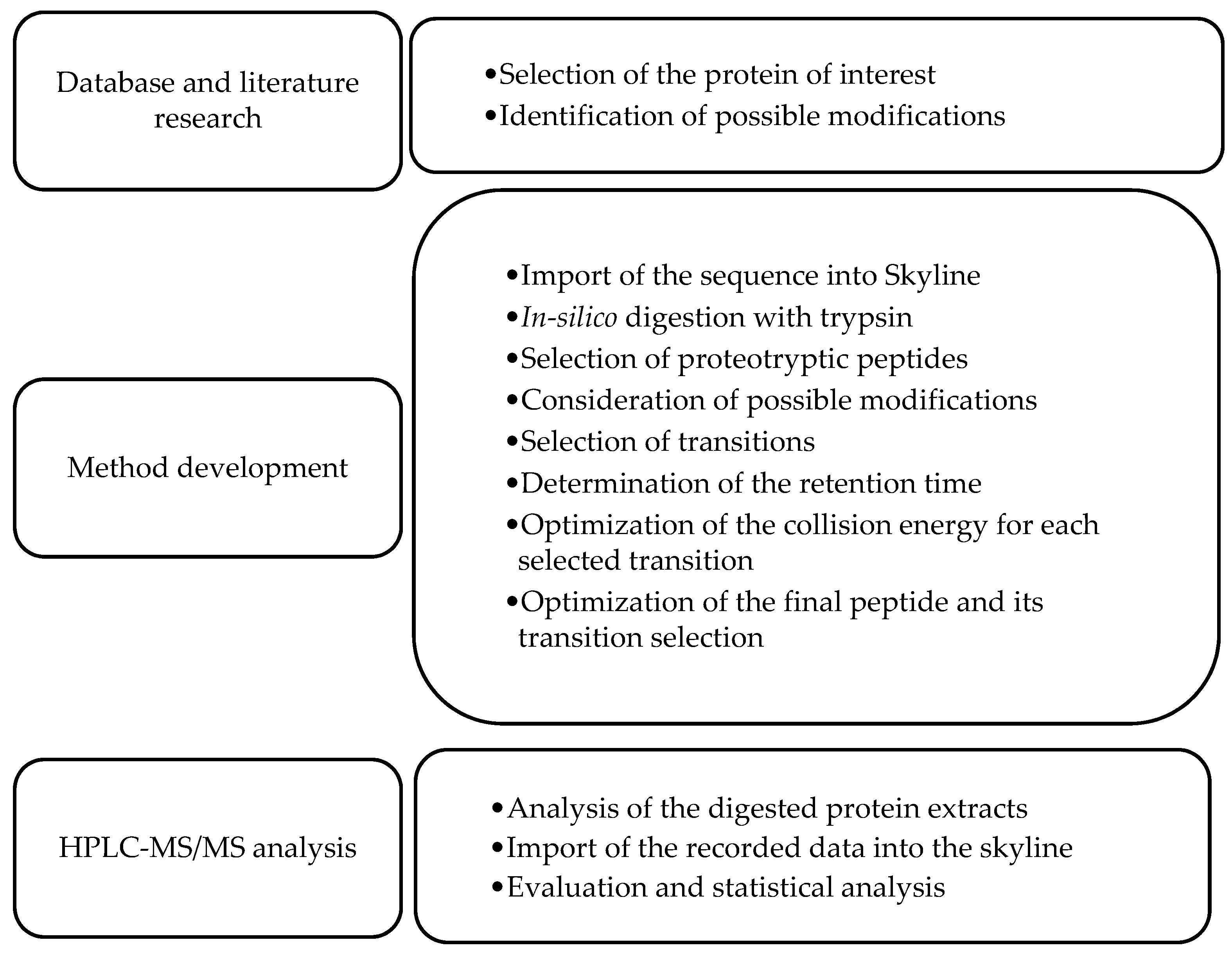
2.2.12. Development of a Multiple Reaction Monitoring (MRM) Assay Using HPLC-MS/MS
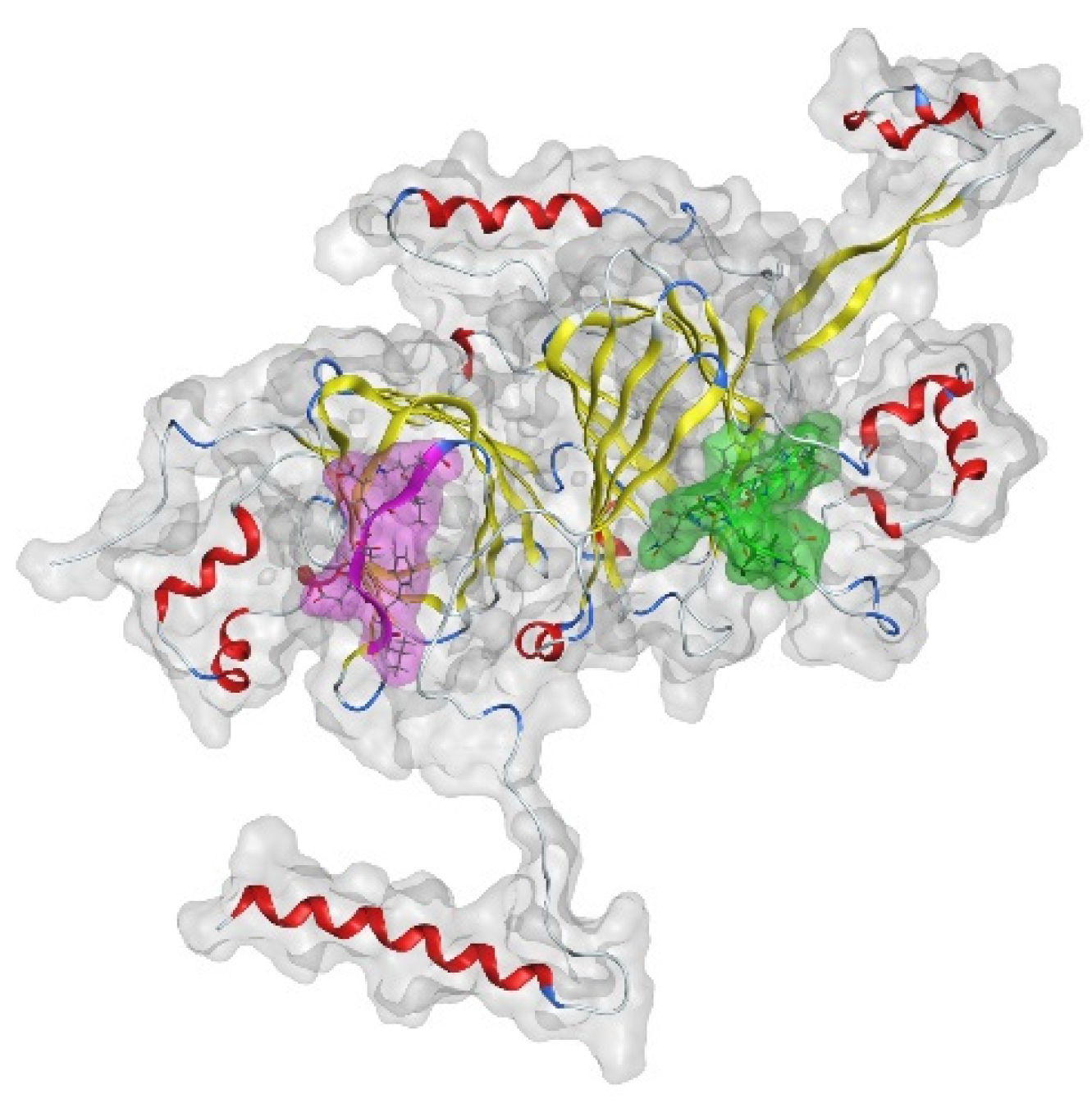
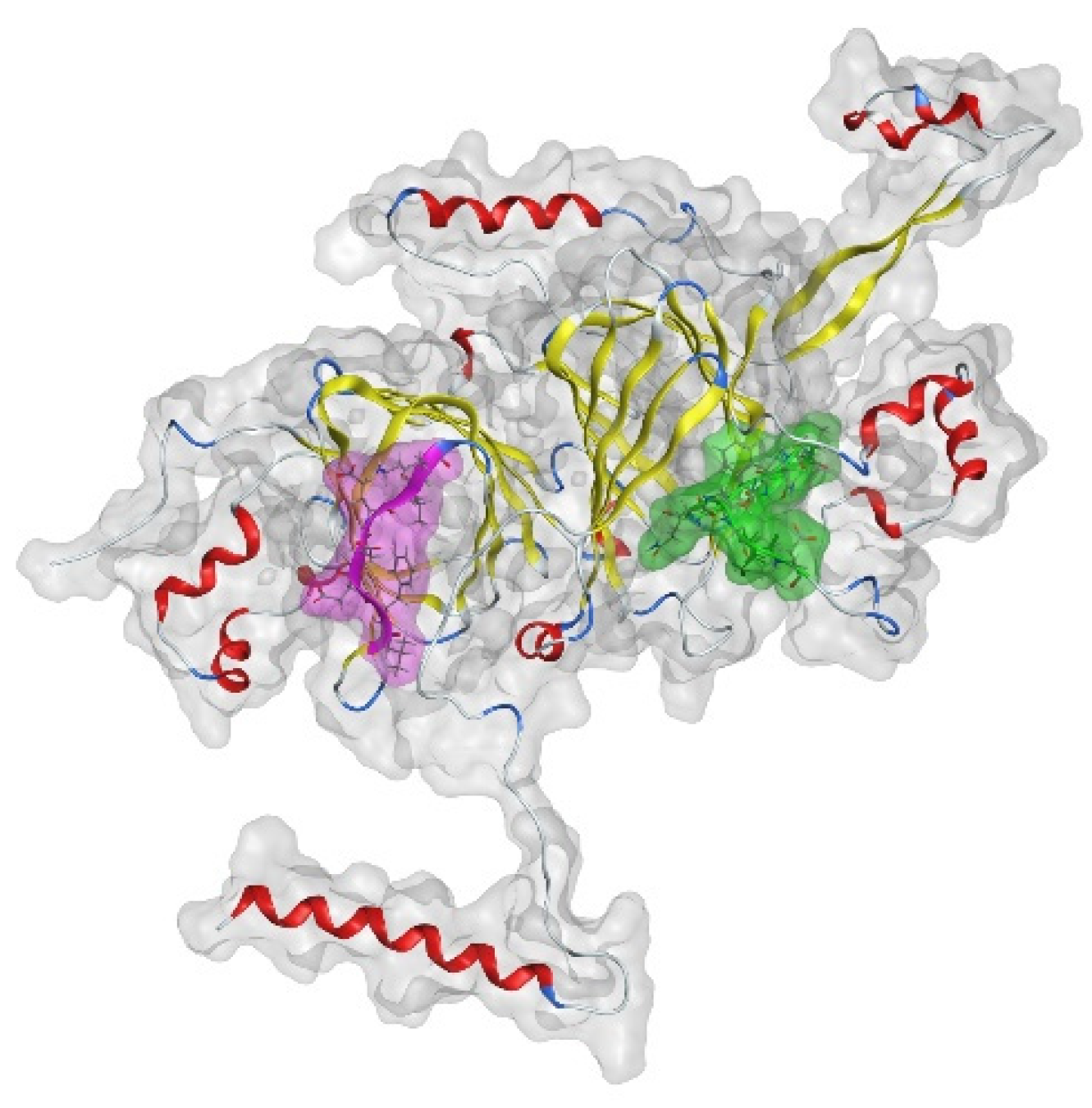
2.2.13. Molecular Modeling Experiments
2.2.14. Data Analysis
3. Results and Discussions
3.1. Free Thiol Groups and Amino Nitrogen in the Protein Fraction
3.2. Protein Content
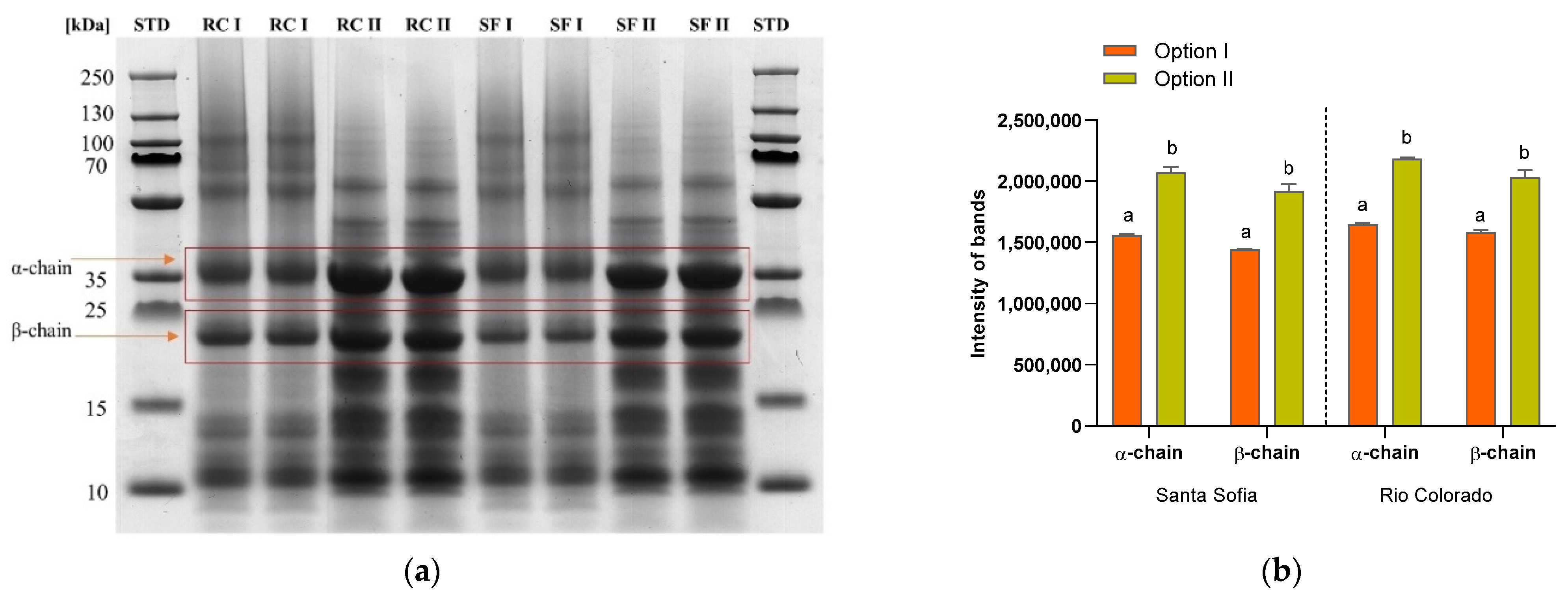
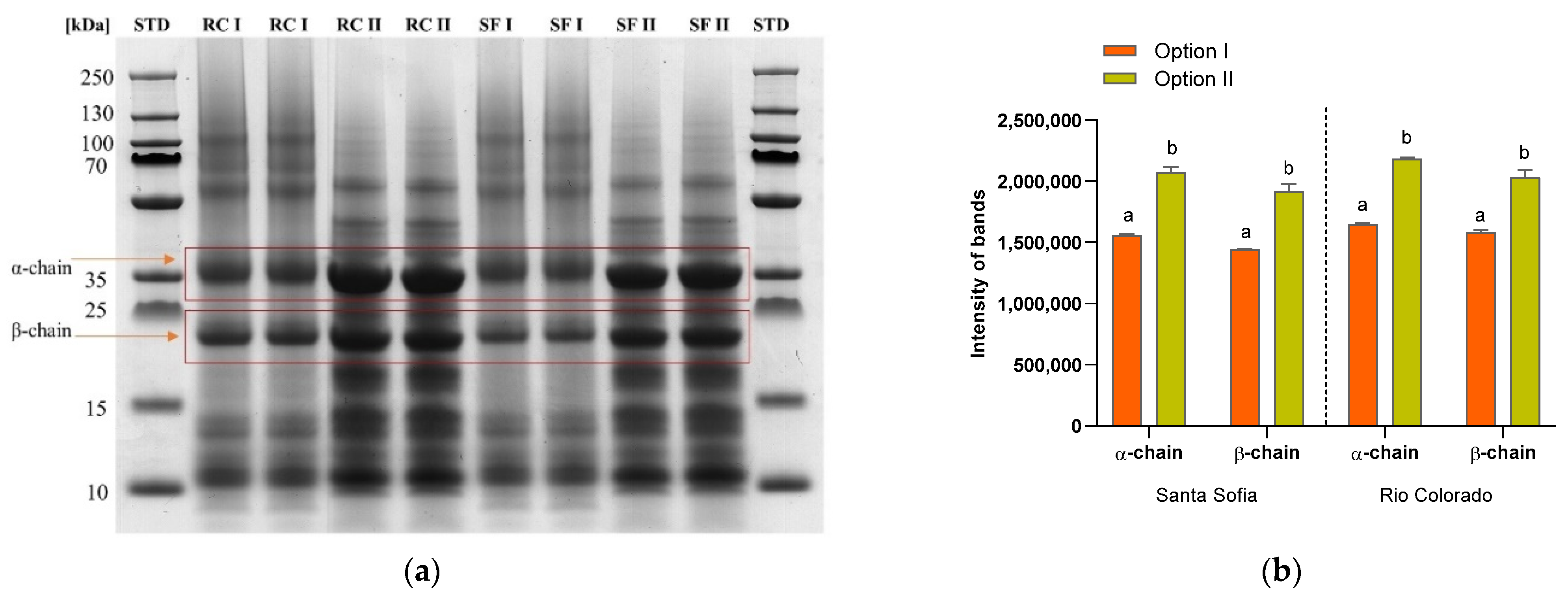
3.3. In-Gel Digestion
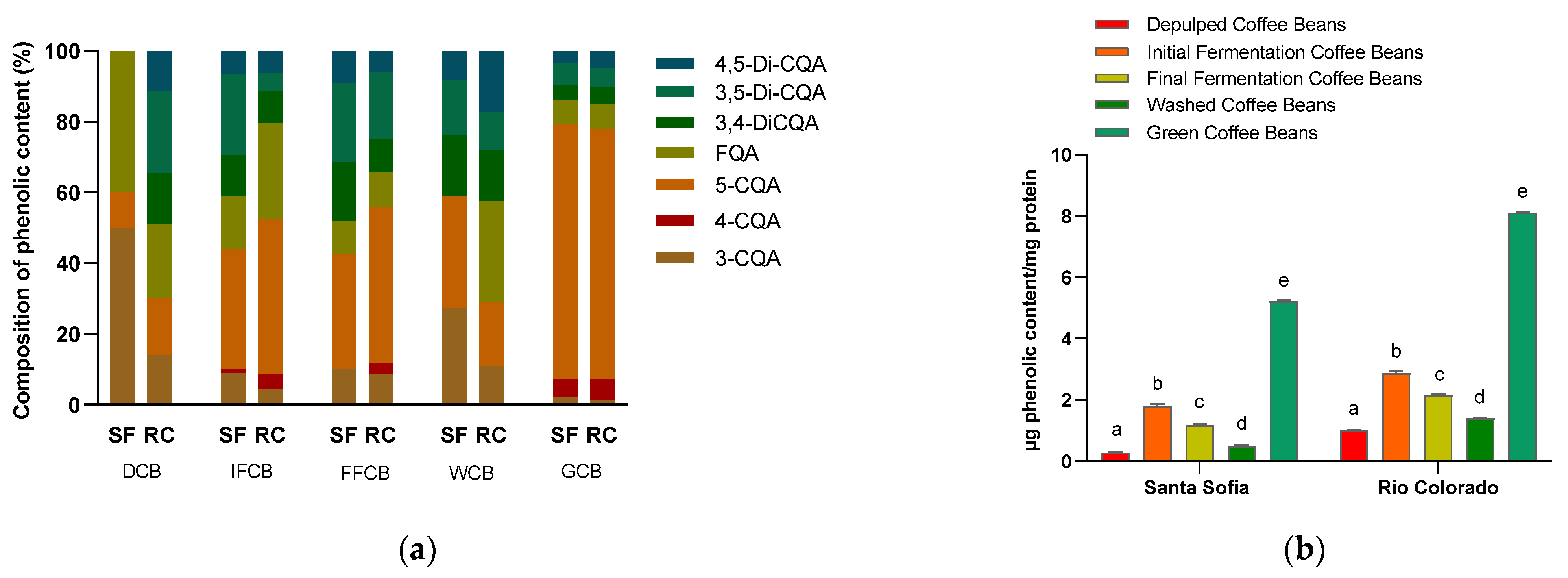
3.4. Phenolic Substances in Protein Extract by HPLC
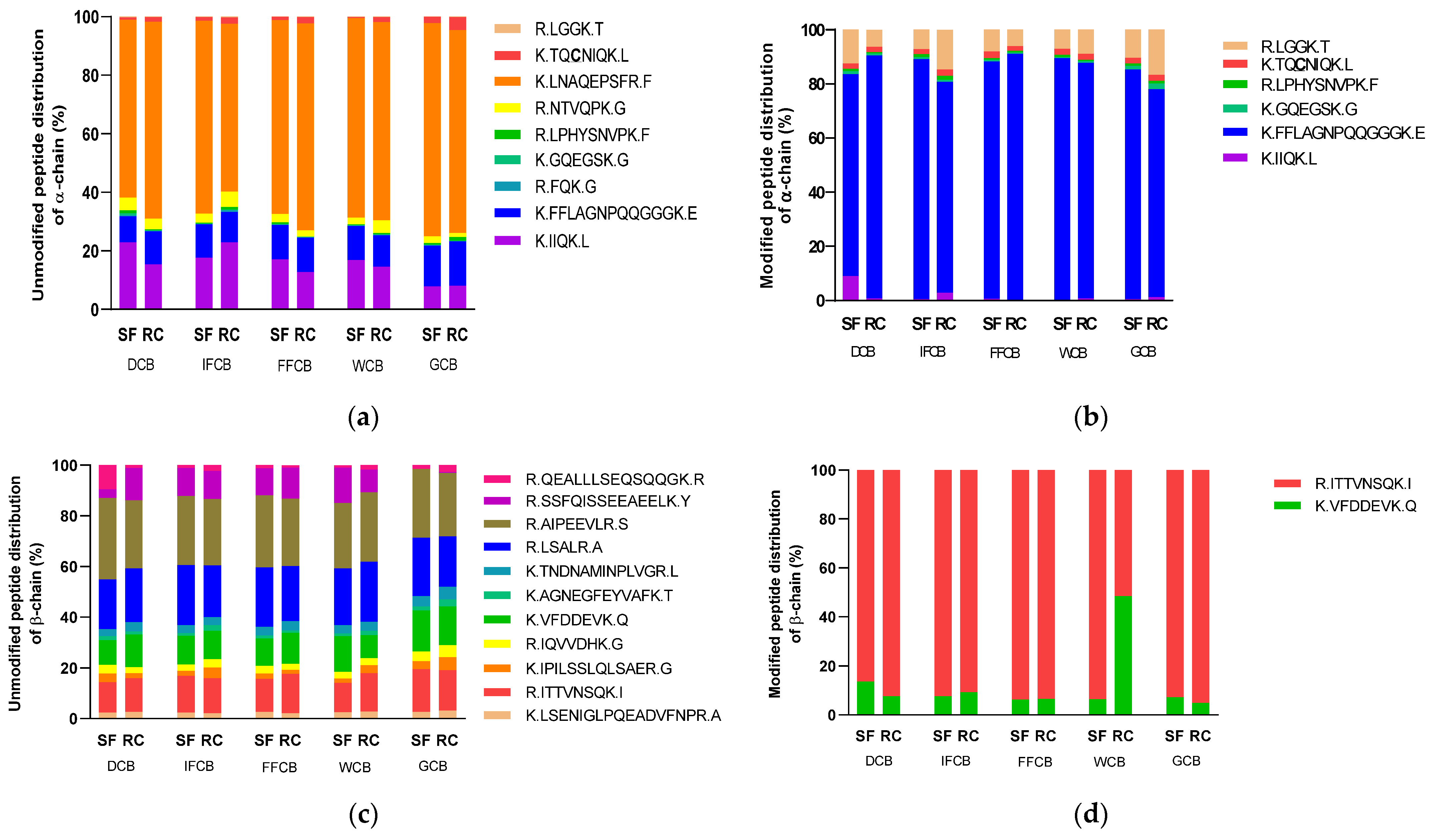
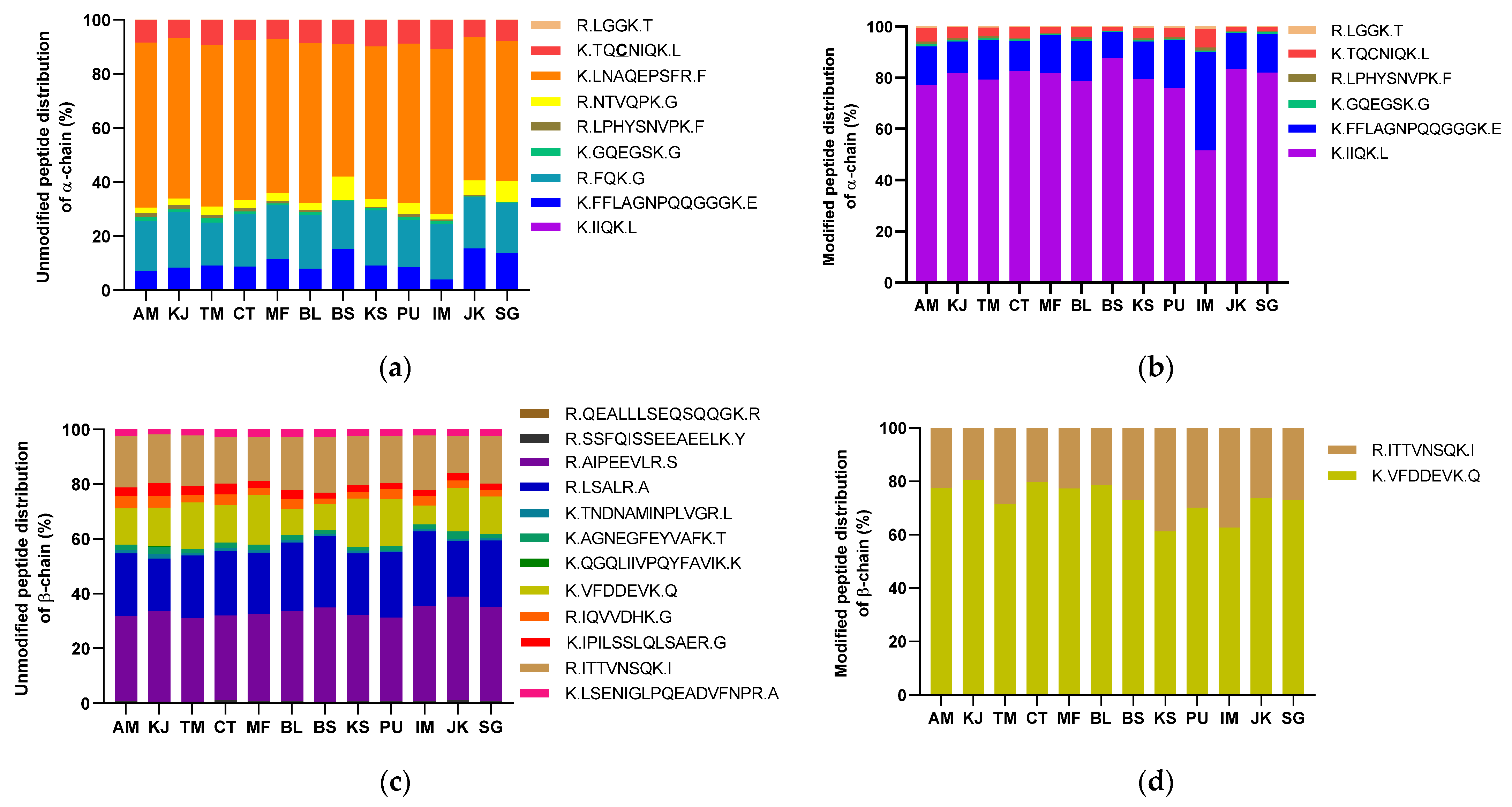
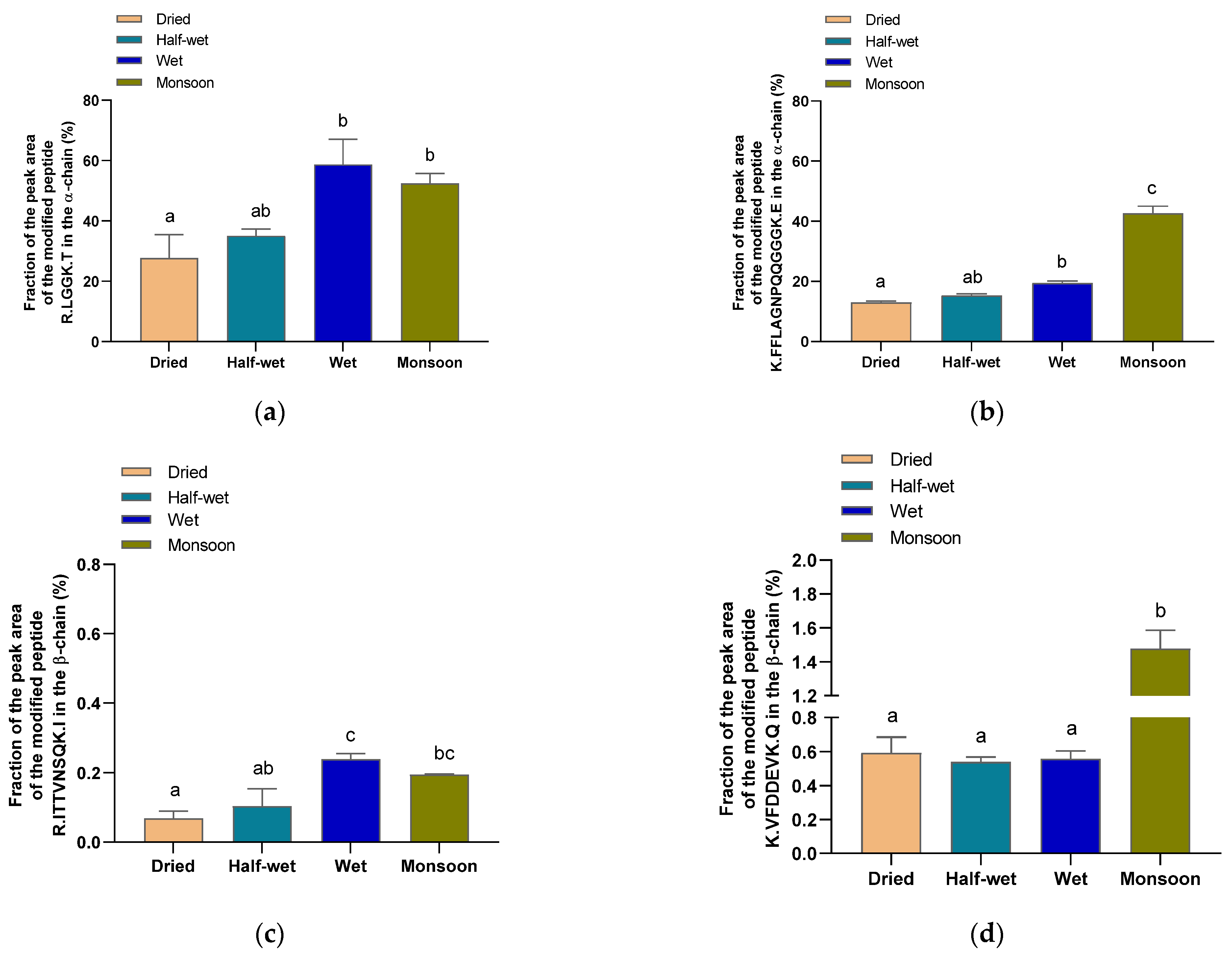
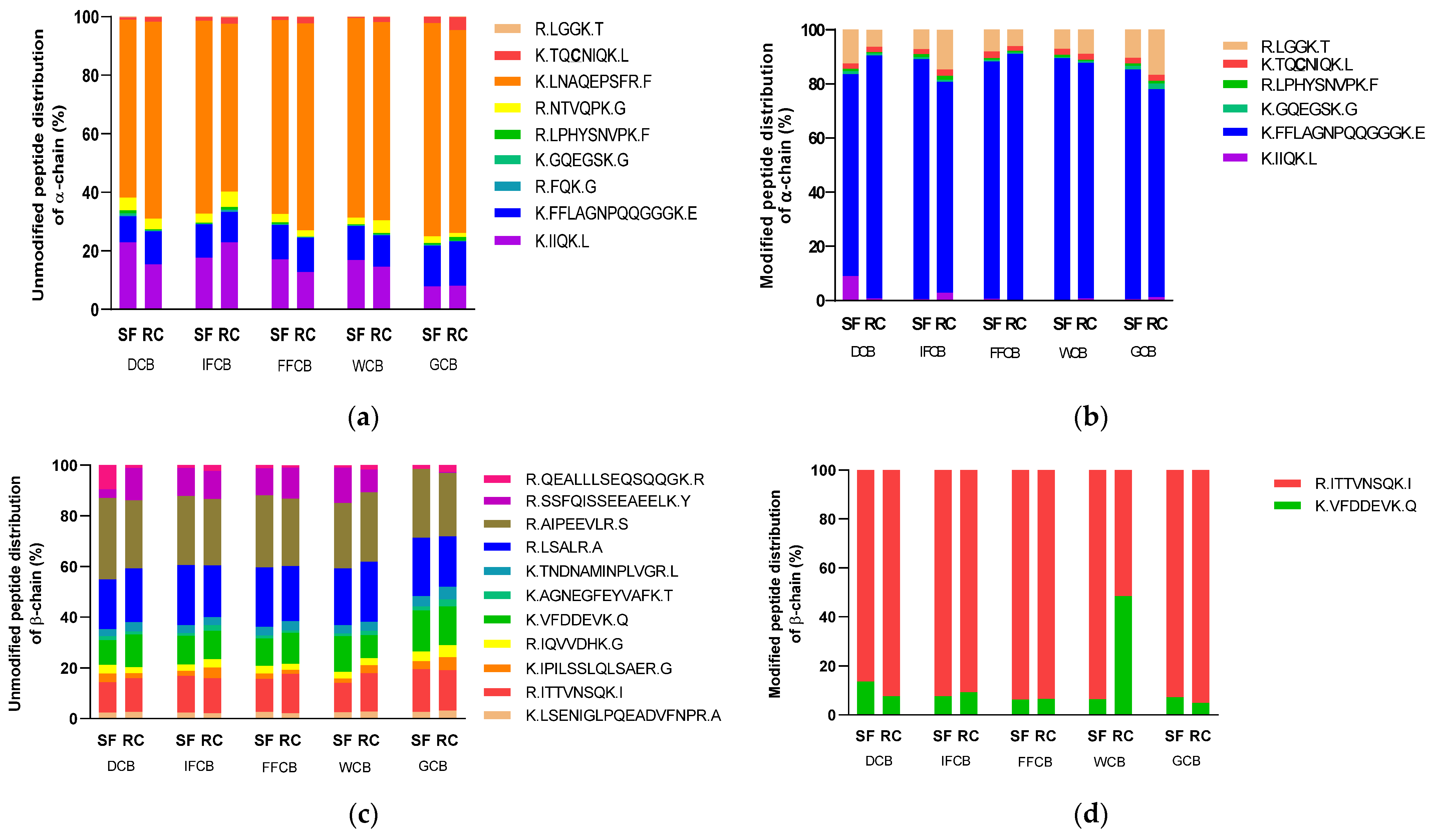
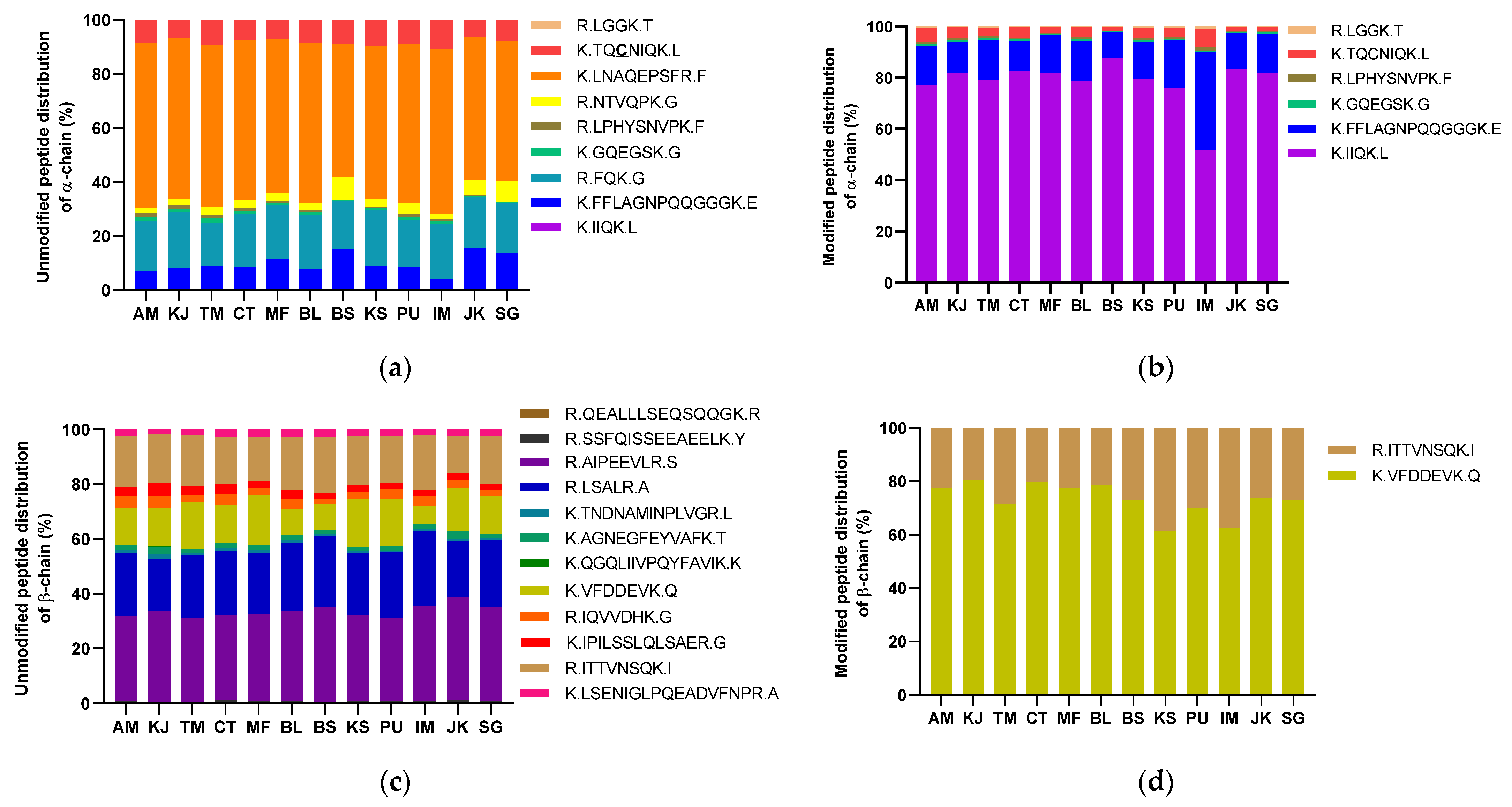
3.5. Analysis of the Protein Modification Using HPLC-MS/MS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rivera, X.C.S.; Gallego-Schmid, A.; Najdanovic-Visak, V.; Azapagic, A. Life cycle environmental sustainability of valorisation routes for spent coffee grounds: From waste to resources. Resour. Conserv. Recycl. 2020, 157, 104751. [Google Scholar] [CrossRef]
- Alves, L.C.; De Magalhães, D.M.; Labate, M.T.V.; Guidetti-Gonzalez, S.; Labate, C.A.; Domingues, D.S.; Sera, T.; Vieira, L.G.E.; Pereira, L.F.P. Differentially Accumulated Proteins in Coffea arabica Seeds during Perisperm Tissue Development and Their Relationship to Coffee Grain Size. J. Agric. Food Chem. 2016, 64, 1635–1647. [Google Scholar] [CrossRef]
- Rattan, S.; Parande, A.K.; Nagaraju, V.D.; Ghiwari, G.K. A comprehensive review on utilization of wastewater from coffee processing. Environ. Sci. Pollut. Res. 2015, 22, 6461–6472. [Google Scholar] [CrossRef]
- Poltronieri, P.; Rossi, F. Challenges in Specialty Coffee Processing and Quality Assurance. Challenges 2016, 7, 19. [Google Scholar] [CrossRef]
- Ferreira, G.F.P.; Novaes, Q.S.d.; Malta, M.R.; Souza, S.E.d. Quality of coffee produced in the southwest region of bahia, brazil subjected to different forms of processing and drying. Afr. J. Agric. Res. 2013, 8, 2334–2339. [Google Scholar]
- Ghosh, P.; Venkatachalapathy, N. Processing and drying of coffee–A review. Int. J. Eng. Res. Technol. 2014, 3, 784–794. [Google Scholar]
- Tsegay, G.; Redi-Abshiro, M.; Chandravanshi, B.S.; Ele, E.; Mohammed, A.M.; Mamo, H. Effect of altitude of coffee plants on the composition of fatty acids of green coffee beans. BMC Chem. 2020, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Woldesenbet, A.G.; Woldeyes, B.; Chandravanishi, B.S. Characteristics of wet coffee processing waste and its environmental impact in ethiopia. Int. J. Res. Eng. Sci. 2014, 2, 1–5. [Google Scholar]
- Campos, G.A.F.; Sagu, S.T.; Celis, P.S.; Rawel, H.M. Comparison of Batch and Continuous Wet-Processing of Coffee: Changes in the Main Compounds in Beans, By-Products and Wastewater. Foods 2020, 9, 1135. [Google Scholar] [CrossRef] [PubMed]
- Murthy, P.S.; Naidu, M.M. Sustainable management of coffee industry by-products and value addition—A review. Resour. Conserv. Recycl. 2012, 66, 45–58. [Google Scholar] [CrossRef]
- Variyar, P.S.; Ahmad, R.; Bhat, R.; Niyas, Z.; Sharma, A. Flavoring Components of Raw Monsooned Arabica Coffee and Their Changes during Radiation Processing. J. Agric. Food Chem. 2003, 51, 7945–7950. [Google Scholar] [CrossRef]
- Rawel, H.M.; Rohn, S.; Kroll, J. Characterisation of 11s protein fractions and phenolic compounds from green coffee beans under special consideration of their interactions–a review. Dtsch. Lebensm.-Rundsch. 2005, 4, 148–160. [Google Scholar]
- Whitfield, F.B.; Mottram, D.S. Volatiles from interactions of Maillard reactions and lipids. Crit. Rev. Food Sci. Nutr. 1992, 31, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Rogers, W.; Bézard, G.; Deshayes, A.; Meyer, I.; Pétiard, V.; Marraccini, P. Biochemical and molecular characterization and expression of the 11S-type storage protein from Coffea arabica endosperm. Plant Physiol. Biochem. 1999, 37, 261–272. [Google Scholar] [CrossRef]
- Acuña, R.; Bassüner, R.; Beilinson, V.; Cortina, H.; Cadena-Gómez, G.; Montes, V.; Nielsen, N.C. Coffee seeds contain 11S storage proteins. Physiol. Plant. 1999, 105, 122–131. [Google Scholar] [CrossRef]
- Thaler, H.; Arneth, W. Untersuchungen an kaffee und kaffee-ersatz. XII. Mitt. Polysacch. Gerösteten Arab. -Kaffees 1968, 138, 137–145. [Google Scholar]
- Bastian, F.; Hutabarat, O.S.; Dirpan, A.; Nainu, F.; Harapan, H.; Bin Emran, T.; Simal-Gandara, J. From Plantation to Cup: Changes in Bioactive Compounds during Coffee Processing. Foods 2021, 10, 2827. [Google Scholar] [CrossRef]
- Ali, M.; Homann, T.; Kreisel, J.; Khalil, M.; Puhlmann, R.; Kruse, H.-P.; Rawel, H. Characterization and Modeling of the Interactions between Coffee Storage Proteins and Phenolic Compounds. J. Agric. Food Chem. 2012, 60, 11601–11608. [Google Scholar] [CrossRef]
- Fukushima, D. Structures of plant storage proteins and their functions. Food Rev. Int. 1991, 7, 353–381. [Google Scholar] [CrossRef]
- Wu, H.; Gu, J.; Bk, A.; Nawaz, M.A.; Barrow, C.J.; Dunshea, F.R.; Suleria, H.A.R. Effect of processing on bioaccessibility and bioavailability-of bioactive compounds in coffee beans. Food Biosci. 2021, 46, 101373. [Google Scholar] [CrossRef]
- Farah, A.; Monteiro, M.C.; Calado, V.; Franca, A.S.; Trugo, L.C. Correlation between cup quality and chemical attributes of Brazilian coffee. Food Chem. 2006, 98, 373–380. [Google Scholar] [CrossRef]
- Jaiswal, R.; Kuhnert, N. Hierarchical scheme for liquid chromatography/multi-stage spectrometric identification of 3,4,5-triacyl chlorogenic acids in green Robusta coffee beans. Rapid Commun. Mass Spectrom. 2010, 24, 2283–2294. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.; Kuhnert, N. Identification and characterization of five new classes of chlorogenic acids in burdock (Arctium lappa L.) roots by liquid chromatography/tandem mass spectrometry. Food Funct. 2011, 2, 63–71. [Google Scholar] [CrossRef]
- Rawel, H.M.; Meidtner, K.; Kroll, J. Binding of Selected Phenolic Compounds to Proteins. J. Agric. Food Chem. 2005, 53, 4228–4235. [Google Scholar] [CrossRef]
- Rawel, H.M.; Rohn, S. Nature of hydroxycinnamate-protein interactions. Phytochem. Rev. 2009, 9, 93–109. [Google Scholar] [CrossRef]
- Prigent, S.V.E.; Gruppen, H.; Visser, A.J.W.G.; van Koningsveld, G.A.; de Jong, G.A.H.; Voragen, A.G.J. Effects of Non-covalent Interactions with 5-O-Caffeoylquinic Acid (Chlorogenic Acid) on the Heat Denaturation and Solubility of Globular Proteins. J. Agric. Food Chem. 2003, 51, 5088–5095. [Google Scholar] [CrossRef] [PubMed]
- Kroll, J.N.G.; Rawel, H.M.; Rohn, S. Reactions of Plant Phenolics with Food Proteins and Enzymes under Special Consideration of Covalent Bonds. Food Sci. Technol. Res. 2003, 9, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Cilliers, J.J.L.; Singleton, V.L. Characterization of the products of nonenzymic autoxidative phenolic reactions in a caffeic acid model system. J. Agric. Food Chem. 1991, 39, 1298–1303. [Google Scholar] [CrossRef]
- Guo, A.; Jiang, J.; True, A.D.; Xiong, Y.L. Myofibrillar Protein Cross-Linking and Gelling Behavior Modified by Structurally Relevant Phenolic Compounds. J. Agric. Food Chem. 2021, 69, 1308–1317. [Google Scholar] [CrossRef]
- Czubinski, J.; Dwiecki, K.; Siger, A.; Neunert, G.; Lampart-Szczapa, E. Characterisation of different digestion susceptibility of lupin seed globulins. Food Chem. 2014, 143, 418–426. [Google Scholar] [CrossRef]
- Jakobek, L. Interactions of polyphenols with carbohydrates, lipids and proteins. Food Chem. 2015, 175, 556–567. [Google Scholar] [CrossRef]
- Rawel, H.M.; Rohn, S.; Kruse, H.-P.; Kroll, J. Structural changes induced in bovine serum albumin by covalent attachment of chlorogenic acid. Food Chem. 2002, 78, 443–455. [Google Scholar] [CrossRef]
- Rawel, H.M.; Rohn, S.; Kroll, J. Influence of a sugar moiety (rhamnosylglucoside) at 3-O position on the reactivity of quercetin with whey proteins. Int. J. Biol. Macromol. 2003, 32, 109–120. [Google Scholar] [CrossRef]
- Prandi, B.; Ferri, M.; Monari, S.; Zurlini, C.; Cigognini, I.; Verstringe, S.; Schaller, D.; Walter, M.; Navarini, L.; Tassoni, A.; et al. Extraction and Chemical Characterization of Functional Phenols and Proteins from Coffee (Coffea arabica) By-Products. Biomolecules 2021, 11, 1571. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, E.; Rocha, T.D.S.; Prudencio, S.H. Potential of green and roasted coffee beans and spent coffee grounds to provide bioactive peptides. Food Chem. 2021, 348, 129061. [Google Scholar] [CrossRef] [PubMed]
- Samsalee, N.; Sothornvit, R. Physicochemical, functional properties and antioxidant activity of protein extract from spent coffee grounds using ultrasonic-assisted extraction. AIMS Agric. Food 2021, 6, 864–878. [Google Scholar] [CrossRef]
- Valdés, A.; Castro-Puyana, M.; Marina, M.L. Isolation of proteins from spent coffee grounds. Polyphenol removal and peptide identification in the protein hydrolysates by RP-HPLC-ESI-Q-TOF. Food Res. Int. 2020, 137, 109368. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [Green Version]
- Laing, W.; Christeller, J. Extraction of Proteins from Plant Tissues. Curr. Protoc. Protein Sci. 2004, 38, 4.7.1–4.7.7. [Google Scholar] [CrossRef]
- Wang, W.; Vignani, R.; Scali, M.; Cresti, M. A universal and rapid protocol for protein extraction from recalcitrant plant tissues for proteomic analysis. Electrophoresis 2006, 27, 2782–2786. [Google Scholar] [CrossRef]
- Abernathy, D.G.; Spedding, G.; Starcher, B. Analysis of Protein and Total Usable Nitrogen in Beer and Wine Using a Microwell Ninhydrin Assay. J. Inst. Brew. 2009, 115, 122–127. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Uhr, L.; Buchholz, T.; Homann, T.; Huschek, G.; Rawel, H.M. Targeted proteomics-based analysis of technical enzymes from fungal origin in baked products. J. Cereal Sci. 2014, 60, 440–447. [Google Scholar] [CrossRef]
- Pearson, W.R.; Lipman, D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 1988, 85, 2444–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bereman, M.S.; MacLean, B.; Tomazela, D.M.; Liebler, D.C.; MacCoss, M.J. The development of selected reaction monitoring methods for targeted proteomics via empirical refinement. Proteomics 2012, 12, 1134–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bongartz, V.; Brandt, L.; Gehrmann, M.L.; Zimmermann, B.F.; Schulze-Kaysers, N.; Schieber, A. Evidence for the Formation of Benzacridine Derivatives in Alkaline-Treated Sunflower Meal and Model Solutions. Molecules 2016, 21, 91. [Google Scholar] [CrossRef] [Green Version]
- Namiki, M.; Yabuta, G.; Koizumi, Y.; Yano, M. Development of Free Radical Products during the Greening Reaction of Caffeic Acid Esters (or Chlorogenic Acid) and a Primary Amino Compound. Biosci. Biotechnol. Biochem. 2001, 65, 2131–2136. [Google Scholar] [CrossRef] [Green Version]
- Yabuta, G.; Koizumi, Y.; Namiki, K.; Hida, M.; Namiki, M. Structure of Green Pigment Formed by the Reaction of Caffeic Acid Esters (or Chlorogenic acid) with a Primary Amino Compound. Biosci. Biotechnol. Biochem. 2001, 65, 2121–2130. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Sigolotto, C.-I.; Carle, R.; Schieber, A. Characterization of covalent addition products of chlorogenic acid quinone with amino acid derivatives in model systems and apple juice by high-performance liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Shutov, A.D.; Vaintraub, I.A. Degradation of storage proteins in germinating seeds. Phytochemistry 1987, 26, 1557–1566. [Google Scholar] [CrossRef]
- Czubinski, J.; Dwiecki, K. A review of methods used for investigation of protein-phenolic compound interactions. Int. J. Food Sci. Technol. 2016, 52, 573–585. [Google Scholar] [CrossRef]
- Suryaprakash, P.; Kumar, R.P.; Prakash, V. Thermodynamics of interaction of caffeic acid and quinic acid with multisubunit proteins. Int. J. Biol. Macromol. 2000, 27, 219–228. [Google Scholar] [CrossRef]
- Schwenke, K.D. Reflections about the functional potential of legume proteins A Review. Food/Nahrung 2001, 45, 377–381. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DCB | IFCB | FFCB | WCB | GCB | ||
|---|---|---|---|---|---|---|
| Santa Sofia | Exposed free thiol groups (nmol/mg) | 19.33 ± 0.78 a | 19.56 ± 0.12 a | 16.13 ± 0.66 b | 13.47 ± 0.42 b | 5.91 ± 2.62 c |
| Total free thiol groups (nmol/mg) | 23.20 ± 0.24 a | 14.52 ± 1.48 b | 15.90 ± 0.21 c | 10.13 ± 1.20 d | 7.65 ± 1.20 e | |
| Free amino nitrogen (µg/mg) | 46.97 ± 1.18 a | 48.51 ± 3.80 a | 57.52 ± 4.87 b | 58.96 ± 6.86 c | 35.94 ± 4.15 d | |
| Rio Colorado | Exposed free thiol groups (nmol/mg) | 8.00 ± 0.45 a | 14.44 ± 0.48 b | 2.61 ± 0.26 c | 13.74 ± 1.55 b | 4.93 ± 0.90 c |
| Total free thiol groups (nmol/mg) | 4.84 ± 1.65 a | 12.18 ± 2.46 b | 7.90 ± 1.41 c | 10.66 ± 1.22 d | 4.57 ± 2.23 a | |
| Free amino nitrogen (µg/mg) | 47.17 ± 2.32 ab | 51.35 ± 2.88 a | 48.62 ± 3.86 a | 45.48 ± 3.16 ab | 38.15 ± 1.14 b |
| mg Protein/100 mg DW | |||
|---|---|---|---|
| Option I | Option II | Option III | |
| Santa Sofia | 1.62 ± 0.04 a | 1.73 ± 0.19 a | 3.45 ± 0.13 b |
| Rio Colorado | 1.33 ± 0.21 a | 2.08 ± 0.08 b | 3.75 ± 0.04 c |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos, G.A.F.; Kruizenga, J.G.K.T.; Sagu, S.T.; Schwarz, S.; Homann, T.; Taubert, A.; Rawel, H.M. Effect of the Post-Harvest Processing on Protein Modification in Green Coffee Beans by Phenolic Compounds. Foods 2022, 11, 159. https://doi.org/10.3390/foods11020159
Campos GAF, Kruizenga JGKT, Sagu ST, Schwarz S, Homann T, Taubert A, Rawel HM. Effect of the Post-Harvest Processing on Protein Modification in Green Coffee Beans by Phenolic Compounds. Foods. 2022; 11(2):159. https://doi.org/10.3390/foods11020159
Chicago/Turabian StyleCampos, Gustavo A. Figueroa, Johannes G. K. T. Kruizenga, Sorel Tchewonpi Sagu, Steffen Schwarz, Thomas Homann, Andreas Taubert, and Harshadrai M. Rawel. 2022. "Effect of the Post-Harvest Processing on Protein Modification in Green Coffee Beans by Phenolic Compounds" Foods 11, no. 2: 159. https://doi.org/10.3390/foods11020159
APA StyleCampos, G. A. F., Kruizenga, J. G. K. T., Sagu, S. T., Schwarz, S., Homann, T., Taubert, A., & Rawel, H. M. (2022). Effect of the Post-Harvest Processing on Protein Modification in Green Coffee Beans by Phenolic Compounds. Foods, 11(2), 159. https://doi.org/10.3390/foods11020159