
Characterization of Feruloyl Esterase from Bacillus pumilus SK52.001 and Its Application in Ferulic Acid Production from De-Starched Wheat Bran

Abstract
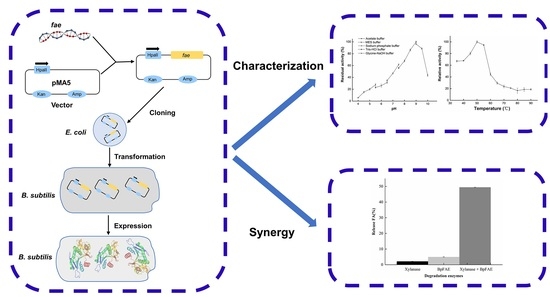
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Strain Screening
2.3. Engineered Strain, Plasmid, and Culture Media
2.4. Cloning
2.5. Heterogeneous Expression and Purification
2.6. Molecular Mass Determination
2.7. Hydrolytic Activity Assay and Protein Concentration Determination
2.8. Biochemical Characterization of BpFAE
2.9. Kinetic Parameters
2.10. Production of FA from DSWB
2.11. Statistical Analysis
3. Results and Discussion
3.1. Strain Screening and Identification
3.2. Sequence Analysis
3.3. Expression, Purification, and Molecular Mass Determination of Recombinant FAE
3.4. Effect of pH on Enzyme Activity and Stability
3.5. Effect of Temperature on Enzyme Activity and Stability
3.6. Kinetic Parameters
3.7. Release FA from DSWB
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Ethical Approval
References
- Long, L.; Zhao, H.; Ding, D.; Xu, M.; Ding, S. Heterologous expression of two Aspergillus niger feruloyl esterases in Trichoderma reesei for the production of ferulic acid from wheat bran. Bioprocess Biosyst. Eng. 2018, 41, 593–601. [Google Scholar] [CrossRef]
- Yue, X.; Suopajarvi, T.; Mankinen, O.; Mikola, M.; Mikkelson, A.; Ahola, J.; Hiltunen, S.; Komulainen, S.; Kantola, A.M.; Telkki, V.V.; et al. Comparison of lignin fractions isolated from wheat straw using alkaline and acidic deep eutectic solvents. J. Agric. Food Chem. 2020, 68, 15074–15084. [Google Scholar] [CrossRef] [PubMed]
- Swamy, P.S.K.; Govindaswamy, V. Therapeutical properties of ferulic acid and bioavailability enhancement through feruloyl esterase. J. Funct. Foods 2015, 17, 657–666. [Google Scholar] [CrossRef]
- Leonard, W.; Zhang, P.; Ying, D.; Fang, Z. Hydroxycinnamic acids on gut microbiota and health. Compr. Rev. Food Sci. Food Saf. 2021, 20, 710–737. [Google Scholar] [CrossRef]
- Song, Y.; Wu, M.S.; Tao, G.; Lu, M.W.; Lin, J.; Huang, J.Q. Feruloylated oligosaccharides and ferulic acid alter gut microbiome to alleviate diabetic syndrome. Food Res. Int. 2020, 137, 109410. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, T.; Zhang, S. Extracellular secretion of feruloyl esterase derived from Lactobacillus crispatus in Escherichia coli and its application for ferulic acid production. Bioresour. Technol. 2019, 288, 121526. [Google Scholar] [CrossRef] [PubMed]
- Dilokpimol, A.; Makela, M.R.; Aguilar-Pontes, M.V.; Benoit-Gelber, I.; Hilden, K.S.; de Vries, R.P. Diversity of fungal feruloyl esterases: Updated phylogenetic classification, properties, and industrial applications. Biotechnol. Biofuels 2016, 9, 231. [Google Scholar] [CrossRef]
- Su, R.; Ni, K.; Wang, T.; Yang, X.; Zhang, J.; Liu, Y.; Shi, W.; Yan, L.; Jie, C.; Zhong, J. Effects of ferulic acid esterase-producing Lactobacillus fermentum and cellulase additives on the fermentation quality and microbial community of alfalfa silage. PeerJ 2019, 7, e7712. [Google Scholar] [CrossRef]
- McAuley, K.E.; Svendsen, A.; Patkar, S.A.; Wilson, K.S. Structure of a feruloyl esterase from Aspergillus niger. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 878–887. [Google Scholar] [CrossRef]
- Gopalan, N.; Rodriguez-Duran, L.V.; Saucedo-Castaneda, G.; Nampoothiri, K.M. Review on technological and scientific aspects of feruloyl esterases: A versatile enzyme for biorefining of biomass. Bioresour. Technol. 2015, 193, 534–544. [Google Scholar] [CrossRef]
- Wu, H.; Li, H.; Xue, Y.; Luo, G.; Gan, L.; Liu, J.; Mao, L.; Long, M. High efficiency co-production of ferulic acid and xylooligosaccharides from wheat bran by recombinant xylanase and feruloyl esterase. Biochem. Eng. J. 2017, 120, 41–48. [Google Scholar] [CrossRef]
- Meng, Z.; Yang, Q.Z.; Wang, J.Z.; Hou, Y.H. Cloning, Characterization, and functional expression of a thermostable type B feruloyl esterase from thermophilic Thielavia terrestris. Appl. Biochem. Biotechnol. 2019, 189, 1304–1317. [Google Scholar] [CrossRef]
- Wu, S.; Nan, F.; Jiang, J.; Qiu, J.; Zhang, Y.; Qiao, B.; Li, S.; Xin, Z. Molecular cloning, expression and characterization of a novel feruloyl esterase from a soil metagenomic library with phthalate-degrading activity. Biotechnol. Lett. 2019, 41, 995–1006. [Google Scholar] [CrossRef]
- Zorn, H.; Li, Q.X. Trends in food enzymology. J. Agric. Food Chem. 2017, 65, 4–5. [Google Scholar] [CrossRef]
- He, W.; Mu, W.; Jiang, B.; Yan, X.; Zhang, T. Food-grade expression of D-psicose 3-epimerase with tandem repeat genes in Bacillus subtilis. J. Agric. Food Chem. 2016, 64, 5701–5707. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Crepin, V.F.; Faulds, C.B.; Connerton, I.F. Functional classification of the microbial feruloyl esterases. Appl. Microbiol. Biotechnol. 2004, 63, 647–652. [Google Scholar] [CrossRef]
- Gao, L.; Wang, M.; Chen, S.; Zhang, D. Biochemical characterization of a novel feruloyl esterase from Penicillium piceum and its application in biomass bioconversion. J. Mol. Catal. B Enzym. 2016, 133, S388–S394. [Google Scholar] [CrossRef]
- Liu, Y.; Xie, M.; Wan, P.; Chen, G.; Chen, C.; Chen, D.; Yu, S.; Zeng, X.; Sun, Y. Purification, characterization and molecular cloning of a dicaffeoylquinic acid-hydrolyzing esterase from human-derived Lactobacillus fermentum LF-12. Food Funct. 2020, 11, 3235–3244. [Google Scholar] [CrossRef]
- Wu, L.; Wang, M.; Zha, G.; Zhou, J.; Yu, Y.; Lu, H. Improving the expression of a heterologous protein by genome shuffling in Kluyveromyces marxianus. J. Biotechnol. 2020, 320, 11–16. [Google Scholar] [CrossRef]
- Liu, Y.; Mo, W.J.; Shi, T.F.; Wang, M.Z.; Zhou, J.G.; Yu, Y.; Yew, W.S.; Lu, H. Mutational Mtc6p attenuates autophagy and improves secretory expression of heterologous proteins in Kluyveromyces marxianus. Microb. Cell Fact. 2018, 17, 144. [Google Scholar] [CrossRef]
- Wu, D.; Cai, G.; Li, X.; Li, B.; Lu, J. Cloning and expression of ferulic acid esterase gene and its effect on wort filterability. Biotechnol. Lett. 2018, 40, 711–717. [Google Scholar] [CrossRef]
- Bonzom, C.; Huttner, S.; Mirgorodskaya, E.; Chong, S.L.; Uthoff, S.; Steinbuchel, A.; Verhaert, R.M.D.; Olsson, L. Glycosylation influences activity, stability and immobilization of the feruloyl esterase 1a from Myceliophthora thermophila. AMB Express 2019, 9, 126. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Wu, L.; Lin, Q.; Ding, S. Highly efficient extraction of ferulic acid from cereal brans by a new type A feruloyl esterase from Eupenicillium parvum in combination with dilute phosphoric acid pretreatment. Appl. Biochem. Biotechnol. 2020, 190, 1561–1578. [Google Scholar] [CrossRef]
- Zhang, S.B.; Wang, L.; Liu, Y.; Zhai, H.C.; Cai, J.P.; Hu, Y.S. Expression of feruloyl esterase A from Aspergillus terreus and its application in biomass degradation. Protein Expr. Purif. 2015, 115, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Wefers, D.; Cavalcante, J.J.V.; Schendel, R.R.; Deveryshetty, J.; Wang, K.; Wawrzak, Z.; Mackie, R.I.; Koropatkin, N.M.; Cann, I. Biochemical and structural analyses of two cryptic esterases in Bacteroides intestinalis and their synergistic activities with cognate xylanases. J. Mol. Biol. 2017, 429, 2509–2527. [Google Scholar] [CrossRef]
- Ichikawa, K.; Shiono, Y.; Shintani, T.; Watanabe, A.; Kanzaki, H.; Gomi, K.; Koseki, T. Efficient production of recombinant tannase in Aspergillus oryzae using an improved glucoamylase gene promoter. J. Biosci. Bioeng. 2020, 129, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bai, Y.; Cai, Y.; Zheng, X. Biochemical characteristics of three feruloyl esterases with a broad substrate spectrum from Bacillus amyloliquefaciens H47. Process Biochem. 2017, 53, 109–115. [Google Scholar] [CrossRef]
- Oleas, G.; Callegari, E.; Sepulveda, R.; Eyzaguirre, J. Properties of two novel esterases identified from culture supernatant of Penicillium purpurogenum grown on sugar beet pulp. Insights Enzym. Res. 2016, 1, 1. [Google Scholar] [CrossRef]
- Ohlhoff, C.W.; Kirby, B.M.; Van Zyl, L.; Mutepfa, D.L.R.; Casanueva, A.; Huddy, R.J.; Bauer, R.; Cowan, D.A.; Tuffin, M. An unusual feruloyl esterase belonging to family VIII esterases and displaying a broad substrate range. J. Mol. Catal. B Enzym. 2015, 118, 79–88. [Google Scholar] [CrossRef]
- Ferri, M.; Happel, A.; Zanaroli, G.; Bertolini, M.; Chiesa, S.; Commisso, M.; Guzzo, F.; Tassoni, A. Advances in combined enzymatic extraction of ferulic acid from wheat bran. New Biotech. 2020, 56, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Dordevic, T.; Milosevic, M.; Antov, M. Advance diversity of enzymatically modified arabinoxylan from wheat chaff. Food Chem. 2021, 339, 128093. [Google Scholar] [CrossRef] [PubMed]
- Lambruschini, C.; Demori, I.; El Rashed, Z.; Rovegno, L.; Canessa, E.; Cortese, K.; Grasselli, E.; Moni, L. Synthesis, photoisomerization, antioxidant activity, and lipid-lowering effect of ferulic acid and feruloyl amides. Molecules 2020, 26, 89. [Google Scholar] [CrossRef]
- Mkabayi, L.; Malgas, S.; Wilhelmi, B.S.; Pletschke, B.I. Evaluating feruloyl esterase—Xylanase synergism for hydroxycinnamic acid and xylo-oligosaccharide production from untreated, hydrothermally pre-treated and dilute-acid pre-treated corn cobs. Agronomy 2020, 10, 688. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, A.; Singh, J.; Sharma, P.; Tomar, G.S.; Singh, S.; Nain, L. A biorefinery approach for the production of ferulic acid from agroresidues through ferulic acid esterase of lactic acid bacteria. 3 Biotech 2020, 10, 367. [Google Scholar] [CrossRef]
- Wang, R.; Yang, J.; Jang, J.M.; Liu, J.; Zhang, Y.; Liu, L.; Yuan, H. Efficient ferulic acid and xylo-oligosaccharides production by a novel multi-modular bifunctional xylanase/feruloyl esterase using agricultural residues as substrates. Bioresour. Technol. 2020, 297, 122487. [Google Scholar] [CrossRef]
- Wong, D.W.S.; Chan, V.J.; Liao, H. Metagenomic discovery of feruloyl esterases from rumen microflora. Appl. Microbiol. Biotechnol. 2019, 103, 8449–8457. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | FAE Activity (U/mL) | Number | FAE Activity (U/mL) |
|---|---|---|---|
| 2 | 0.034 | 55 | 0.019 |
| 3 | 0.195 | 68 | 0.022 |
| 9 | 0.161 | 72 | 0.170 |
| 48 | 0.050 | 73 | 0.160 |
| Organisms | Subunit (kDa) | Optimal pH | Optimal Temperature (°C) | Thermal Stability | Km (mmol/L) | Reference |
|---|---|---|---|---|---|---|
| Aspergillus niger | 56 | 5 | 50 | NR | NR | [1] |
| Lactobacillus crispatus | 28 | 7 | 65 | 60% (60 °C 1 h) | NR | [5] |
| soil metagenomic library | 38.8 | 8 | 37 | 10% (55 °C, 1 h) | 0.467 | [8] |
| Penicillium piceum | 56 | 3 | 70 | 50% (70 °C, 220 min) | NR | [16] |
| Bcteroides intestinalis | 71.1 | 7.5 | 37 | NR | 0.35 | [20] |
| Bacillus amyloliquefaciens | NR | 8 | 40 | 60% (60 °C, 1 h) | 1.14 | [24] |
| Aspergillus niger | 40 | 5 | 45 | 50% (55 °C, 30 min) | 1.4 | [25] |
| Bacillus pumilus | 33 | 9 | 50 | 50% (65 °C, 63 min) | 0.95 | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, X.; Dai, Y.; Zhang, T. Characterization of Feruloyl Esterase from Bacillus pumilus SK52.001 and Its Application in Ferulic Acid Production from De-Starched Wheat Bran. Foods 2021, 10, 1229. https://doi.org/10.3390/foods10061229
Duan X, Dai Y, Zhang T. Characterization of Feruloyl Esterase from Bacillus pumilus SK52.001 and Its Application in Ferulic Acid Production from De-Starched Wheat Bran. Foods. 2021; 10(6):1229. https://doi.org/10.3390/foods10061229
Chicago/Turabian StyleDuan, Xiaoli, Yiwei Dai, and Tao Zhang. 2021. "Characterization of Feruloyl Esterase from Bacillus pumilus SK52.001 and Its Application in Ferulic Acid Production from De-Starched Wheat Bran" Foods 10, no. 6: 1229. https://doi.org/10.3390/foods10061229
APA StyleDuan, X., Dai, Y., & Zhang, T. (2021). Characterization of Feruloyl Esterase from Bacillus pumilus SK52.001 and Its Application in Ferulic Acid Production from De-Starched Wheat Bran. Foods, 10(6), 1229. https://doi.org/10.3390/foods10061229