
Characterization of Genetically Modified Microorganisms Using Short- and Long-Read Whole-Genome Sequencing Reveals Contaminations of Related Origin in Multiple Commercial Food Enzyme Products

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. GMM Isolation from FE Products
2.2. Real-Time PCR Colony Assays
2.3. DNA Extraction, Library Preparation and Whole Genome Sequencing
2.4. Genome Assembly and Characterization
2.5. Follow-Up Analysis of Long Reads
2.6. SNP Phylogeny and SNP Typing
2.7. Whole Genome Alignment-Based Comparison
3. Results and Discussion
3.1. Isolation of Viable GM B. velezensis Producing Protease from FE Products, Long- and Short-Read WGS
3.2. Genetic Characterization of GMM
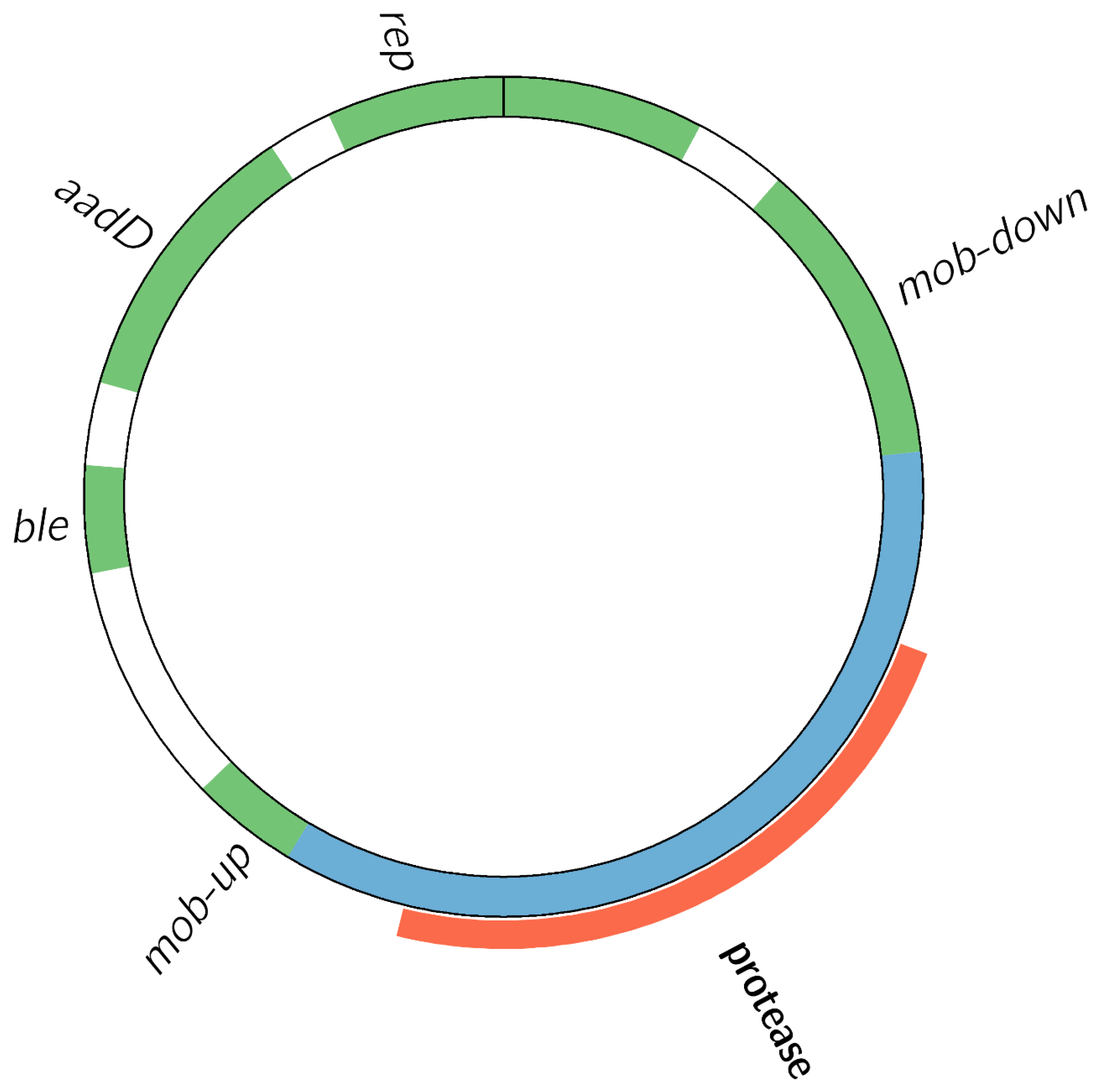
3.2.1. Characterization of GMM Host Strain
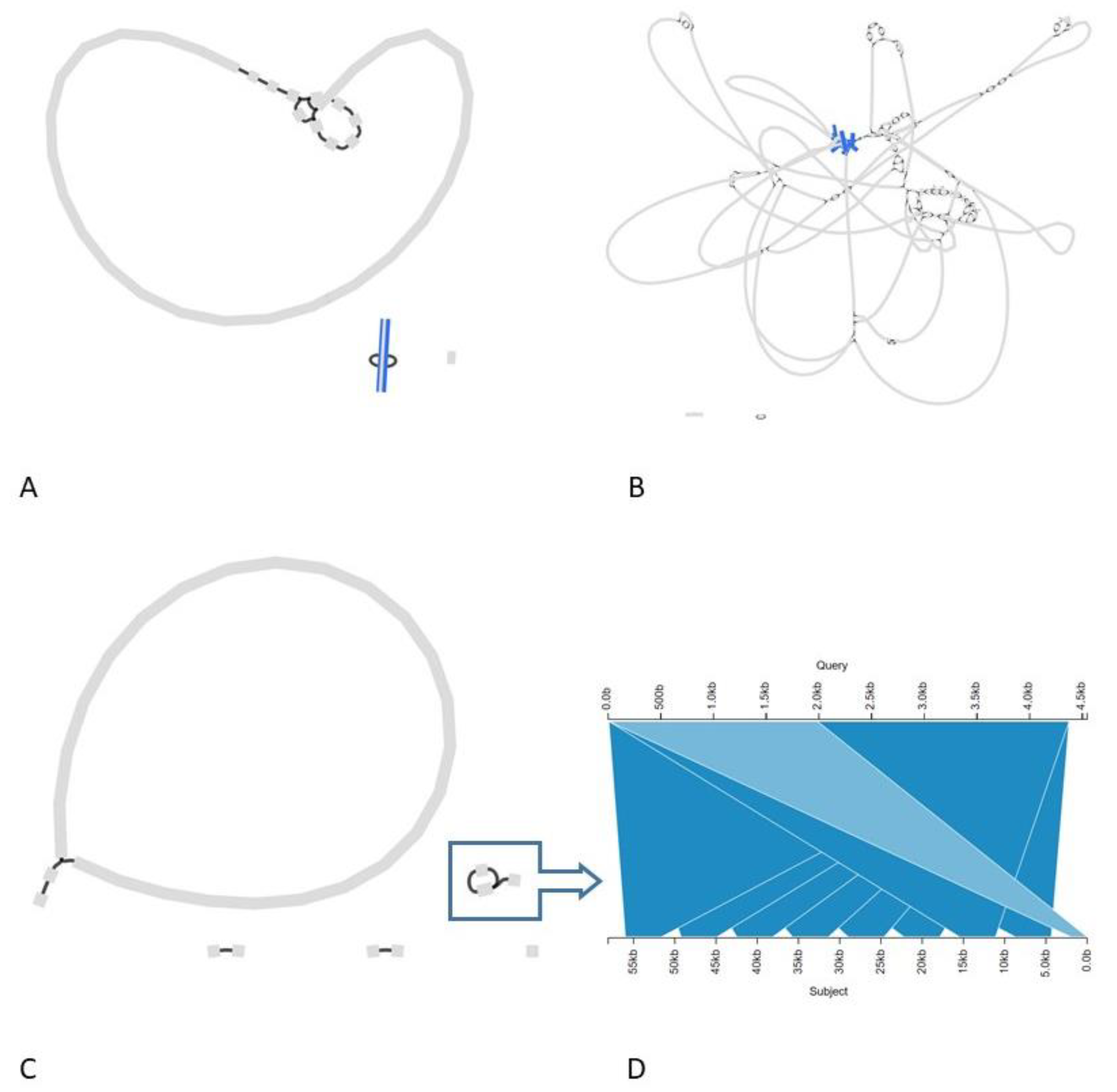
3.2.2. The Transgenic Modification Is Present as an Episomal High-Copy Plasmid
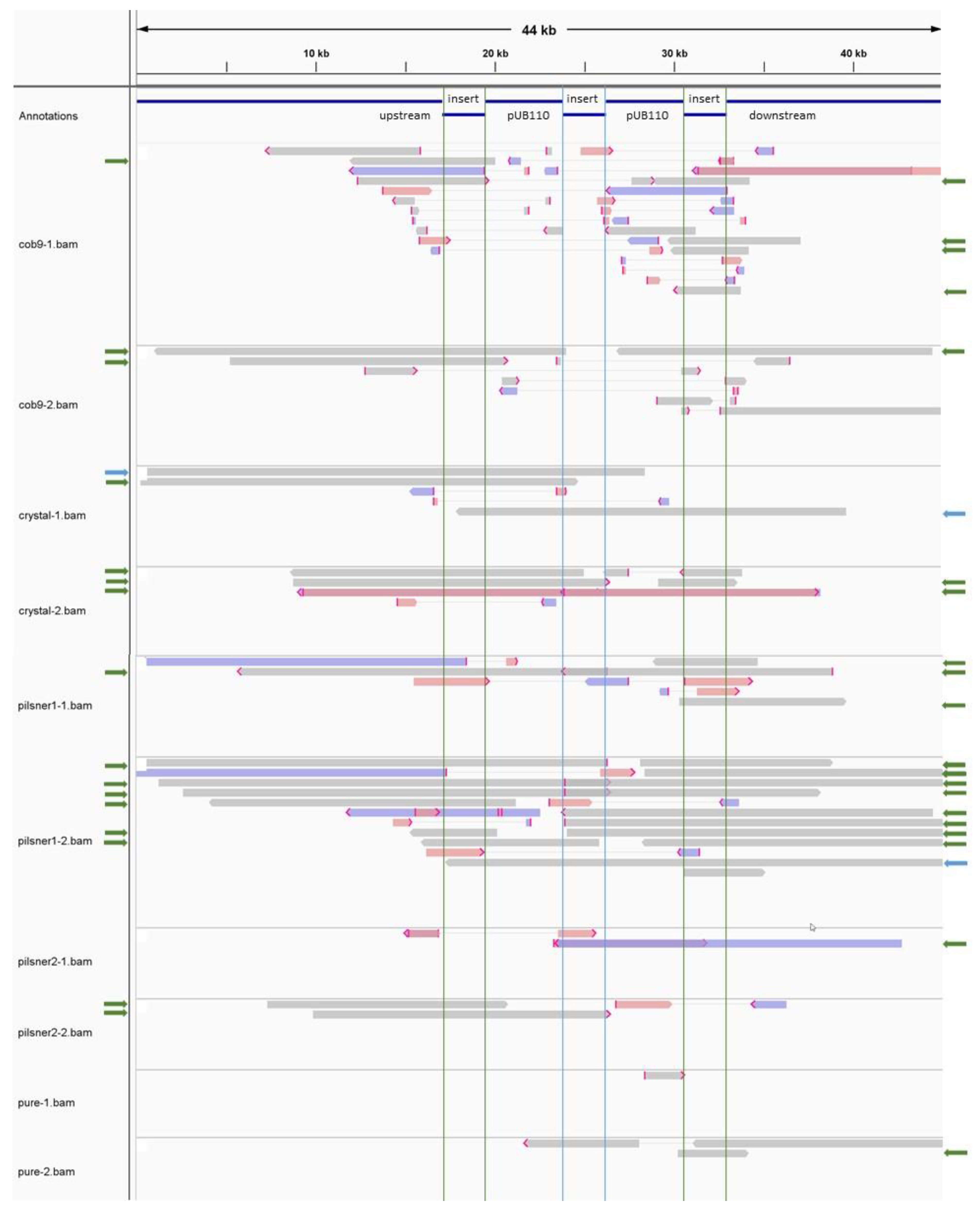
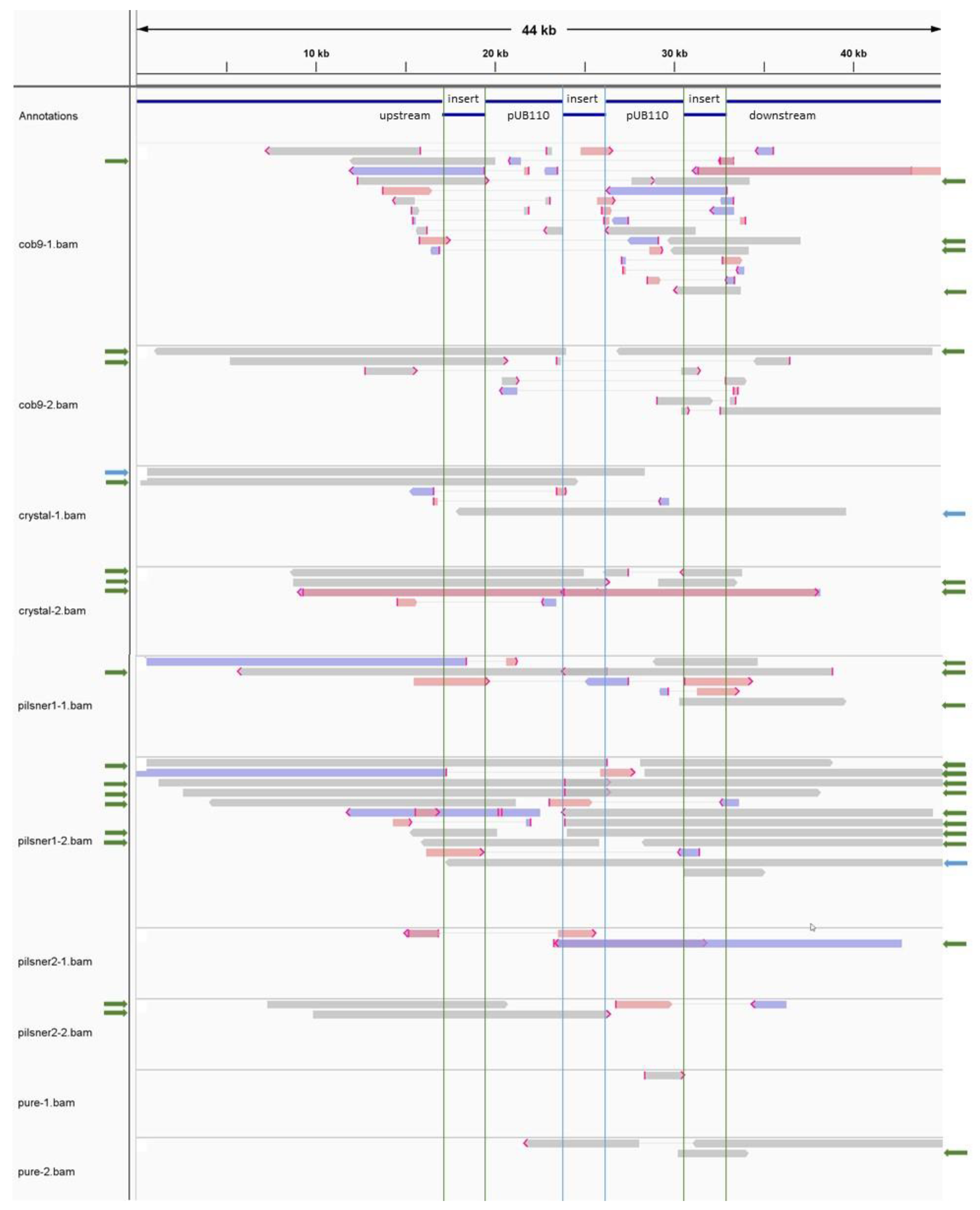
3.2.3. The Transgenic Plasmid Shows Sporadic, Unstable Integration into the Chromosome and Its Replication Is Disturbed, Leading to Accumulation of Linear Plasmid Concatemers
3.3. Comparative Analysis and Source Tracing
3.3.1. SNP Phylogenetic Analysis and Typing Indicate the Isolates Share a Common Source
3.3.2. Whole Genome Comparison Supports Results from SNP-Based Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deckers, M.; Deforce, D.; Fraiture, M.A.; Roosens, N.H.C. Genetically modified micro-organisms for industrial food enzyme production: An overview. Foods 2020, 9, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraiture, M.-A.; Bogaerts, B.; Winand, R.; Deckers, M.; Papazova, N.; Vanneste, K.; de Keersmaecker, S.C.J.; Roosens, N.H.C. Identification of an unauthorized genetically modified bacteria in food enzyme through whole-genome sequencing. Sci. Rep. 2020, 10, 7094. [Google Scholar] [CrossRef] [PubMed]
- Fraiture, M.-A.; Deckers, M.; Papazova, N.; Roosens, N.H.C. Detection strategy targeting a chloramphenicol resistance gene from genetically modified bacteria in food and feed products. Food Control. 2020, 108, 106873. [Google Scholar] [CrossRef]
- Fraiture, M.-A.; Deckers, M.; Papazova, N.; Roosens, N.H.C. Strategy to detect genetically modified bacteria carrying tetracycline resistance gene in fermentation products. Food Anal. Methods 2020, 13, 1929–1937. [Google Scholar] [CrossRef]
- Fraiture, M.-A.; Marchesi, U.; Verginelli, D.; Papazova, N.; Roosens, H.C. Development of a real-time PCR method targeting an unauthorized genetically modified microorganism producing alpha-amylase. Food Anal. Methods 2021, 14, 2211–2220. [Google Scholar] [CrossRef]
- Fraiture, M.-A.; Papazova, N.; Roosens, N.H.C. DNA walking strategy to identify unauthorized genetically modified bacteria in microbial fermentation products. Int. J. Food Microbiol. 2021, 337, 108913. [Google Scholar] [CrossRef]
- Paracchini, V.; Petrillo, M.; Reiting, R.; Angers-Loustau, A.; Wahler, D.; Stolz, A.; Schönig, B.; Matthies, A.; Bendiek, J.; Meinel, D.M.; et al. Molecular characterization of an unauthorized genetically modified Bacillus subtilis production strain identified in a vitamin B2 feed additive. Food Chem. 2017, 230, 681–689. [Google Scholar] [CrossRef]
- Barbau-Piednoir, E.; de Keersmaecker, S.C.J.; Wuyts, V.; Gau, C.; Pirovano, W.; Costessi, A.; Philipp, P.; Roosens, N.H. Genome sequence of EU-unauthorized genetically modified Bacillus subtilis strain 2014-3557 overproducing riboflavin, isolated from a vitamin B2 80% feed additive. Genome Announcements 2016, 3, e00214-15. [Google Scholar] [CrossRef] [Green Version]
- Berbers, B.; Saltykova, A.; Garcia-Graells, C.; Philipp, P.; Arella, F.; Marchal, K.; Winand, R.; Vanneste, K.; Roosens, N.H.C.; de Keersmaecker, S.C.J. Combining short and long read sequencing to characterize antimicrobial resistance genes on plasmids applied to an unauthorized genetically modified Bacillus. Sci. Rep. 2020, 10, 4310. [Google Scholar] [CrossRef]
- Nouws, S.; Bogaerts, B.; Verhaegen, B.; Denayer, S.; Crombé, F.; de Rauw, K.; Piérard, D.; Marchal, K.; Vanneste, K.; Roosens, N.H.C.; et al. The benefits of whole genome sequencing for foodborne outbreak investigation from the perspective of a national reference laboratory in a smaller country. Foods 2020, 9, 1030. [Google Scholar] [CrossRef]
- Sekse, C.; Holst-Jensen, A.; Dobrindt, U.; Johannessen, G.S.; Li, W.; Spilsberg, B.; Shi, J. High throughput sequencing for detection of foodborne pathogens. Front. Microbiol. 2017, 8, 2029. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.C.; Egan, R.; Frazier, P.I.; Wang, Z. ALE: A generic assembly likelihood evaluation framework for assessing the accuracy of genome and metagenome assemblies. Bioinformatics 2013, 29, 435–443. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie2. Nat. Methods 2012, 9, 358–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 49, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Bogaerts, B.; Nouws, S.; Verhaegen, B.; Denayer, S.; van Braekel, J.; Winand, R.; Fu, Q.; Crombé, F.; Piérard, D.; Marchal, K.; et al. Validation strategy of a bioinformatics whole genome sequencing workflow for Shiga toxin-producing Escherichia coli using a reference collection extensively characterized with conventional methods. Microb. Genom. 2021, 7, 000531. [Google Scholar] [CrossRef]
- Wintersinger, J.A.; Wasmuth, J.D. Kablammo: An interactive, web-based BLAST results visualizer. Bioinformatics 2015, 31, 1305–1306. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, S.; Pettengill, J.B.; Luo, Y.; Payne, J.; Shpuntoff, A.; Rand, H.; Strain, E. CFSAN SNP pipeline: An automated method for constructing snp matrices from next-generation sequence data. PeerJ Comput. Sci. 2015, 1, e20. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallman, T.; Ashton, P.; Schafer, U.; Jironkin, A.; Painset, A.; Shaaban, S.; Hartman, H.; Myers, R.; Underwood, A.; Jenkins, C.; et al. SnapperDB: A database solution for routine sequencing analysis of bacterial isolates. Bioinformatics 2018, 34, 3028–3029. [Google Scholar] [CrossRef] [Green Version]
- Nouws, S.; Bogaerts, B.; Verhaegen, B.; Denayer, S.; Piérard, D.; Marchal, K.; Roosens, N.H.C.; Vanneste, K.; de Keersmaecker, S.C.J. Impact of DNA extraction on whole genome sequencing analysis for characterization and relatedness of Shiga toxin-producing Escherichia coli isolates. Sci. Rep. 2020, 10, 14649. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. Progressivemauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Erickson, D.L.; Meng, J. Benchmarking hybrid assembly approaches for genomic analyses of bacterial pathogens using Illumina and Oxford Nanopore sequencing. BMC Genom. 2020, 21, 631. [Google Scholar] [CrossRef]
- Viret, J.F.; Alonso, J.C. A DNA sequence outside the pUB110 minimal replicon is required for normal replication in Bacillus subtilis. Nucleic Acids Res. 1988, 16, 4389–4406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Research 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Gruss, A.; Ehrlich, S.D. Insertion of foreign DNA into plasmids from gram-positive bacteria induces formation of high-molecular-weight plasmid multimers. J. Bacteriol. 1988, 170, 1183–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pightling, A.W.; Pettengill, J.B.; Luo, Y.; Baugher, J.D.; Rand, H.; Strain, E. Interpreting whole-genome sequence analyses of foodborne bacteria for regulatory applications and outbreak investigations. Front. Microbiol. 2018, 9, 1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Commercial FE Product (Supplier) | Associated RASFF | Labeled Enzymes | Application | Evaluated Batch | Obtained GMM Isolates |
|---|---|---|---|---|---|
| Alpha-amylase enzyme 4 g (Coobra) | RASFF2020.2577 | Alpha-amylase | Distillery | 1 | Cob9-1 |
| Cob9-2 | |||||
| Crystalmash (The Alchemist’s Pantry) | RASFF2019.3332 | Alpha-amylase, Protease, Cellulase, Xylanase, Beta-glucanase | Distillery, Brewing, Grain processing | 1 | Crystal-1 |
| Crystal-2 | |||||
| Enzyme 4 g (Pilsner) | RASFF2020.2582 | Alpha-amylase | Distillery, Brewing | 1 | Pilsner1-1 |
| Pilsner1-2 | |||||
| 2 | Pilsner2-1 | ||||
| Pilsner2-2 | |||||
| Pureferm (The Alchemist’s Pantry) | RASFF2019.3332 | Neutral protease | Beer and other cereal based beverages; Bakery products and other cereal based products | 1 | Pure-1 |
| Pure-2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’aes, J.; Fraiture, M.-A.; Bogaerts, B.; De Keersmaecker, S.C.J.; Roosens, N.H.C.; Vanneste, K. Characterization of Genetically Modified Microorganisms Using Short- and Long-Read Whole-Genome Sequencing Reveals Contaminations of Related Origin in Multiple Commercial Food Enzyme Products. Foods 2021, 10, 2637. https://doi.org/10.3390/foods10112637
D’aes J, Fraiture M-A, Bogaerts B, De Keersmaecker SCJ, Roosens NHC, Vanneste K. Characterization of Genetically Modified Microorganisms Using Short- and Long-Read Whole-Genome Sequencing Reveals Contaminations of Related Origin in Multiple Commercial Food Enzyme Products. Foods. 2021; 10(11):2637. https://doi.org/10.3390/foods10112637
Chicago/Turabian StyleD’aes, Jolien, Marie-Alice Fraiture, Bert Bogaerts, Sigrid C. J. De Keersmaecker, Nancy H. C. Roosens, and Kevin Vanneste. 2021. "Characterization of Genetically Modified Microorganisms Using Short- and Long-Read Whole-Genome Sequencing Reveals Contaminations of Related Origin in Multiple Commercial Food Enzyme Products" Foods 10, no. 11: 2637. https://doi.org/10.3390/foods10112637
APA StyleD’aes, J., Fraiture, M.-A., Bogaerts, B., De Keersmaecker, S. C. J., Roosens, N. H. C., & Vanneste, K. (2021). Characterization of Genetically Modified Microorganisms Using Short- and Long-Read Whole-Genome Sequencing Reveals Contaminations of Related Origin in Multiple Commercial Food Enzyme Products. Foods, 10(11), 2637. https://doi.org/10.3390/foods10112637