
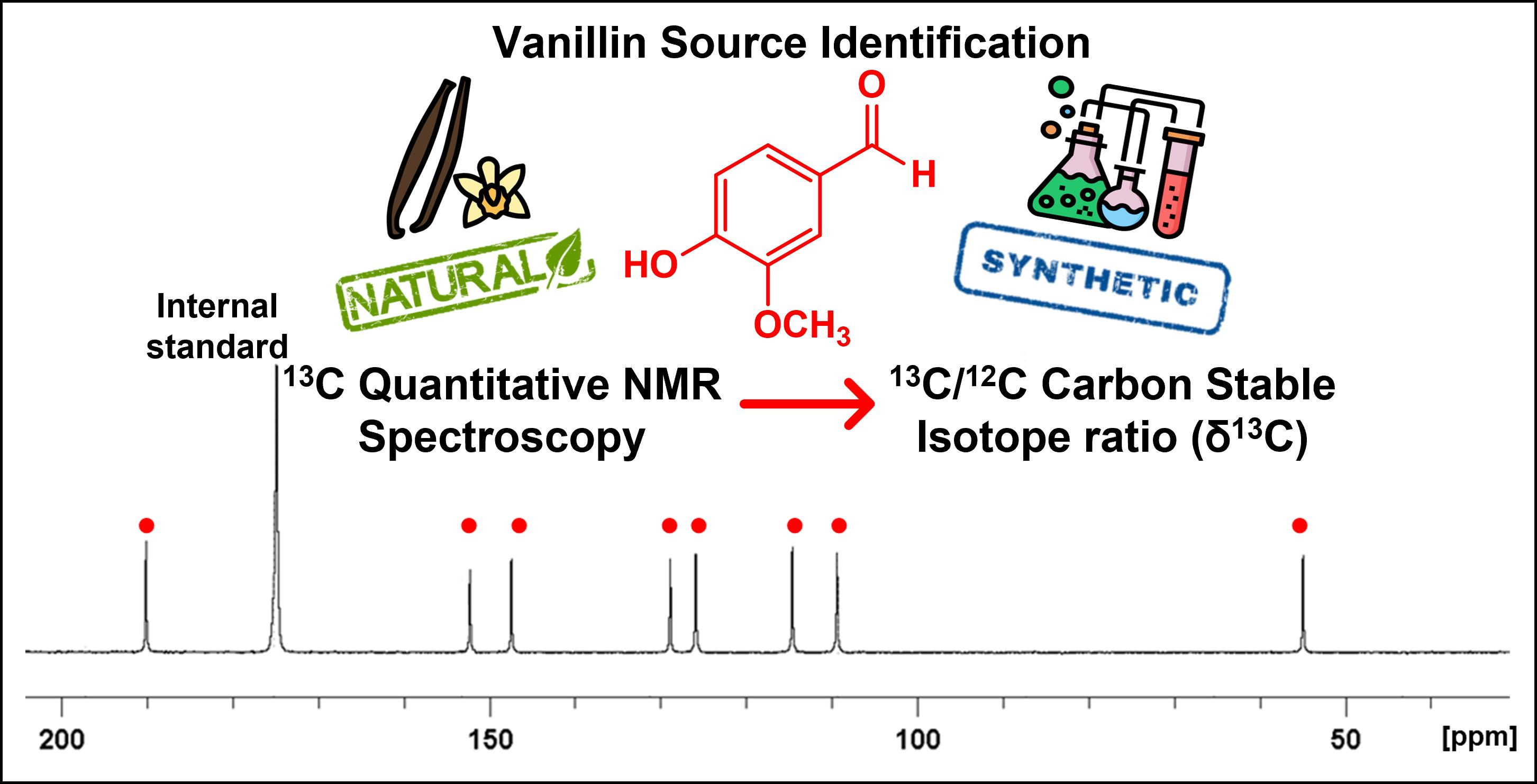
Application of 13C Quantitative NMR Spectroscopy to Isotopic Analyses for Vanillin Authentication Source †


 ,
,  ,
, 
 and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Carbon Stable Isotopic Analysis of Vanilla Samples by Elemental Analysis/Isotope Ratio Mass Spectrometry (EA/IRMS)
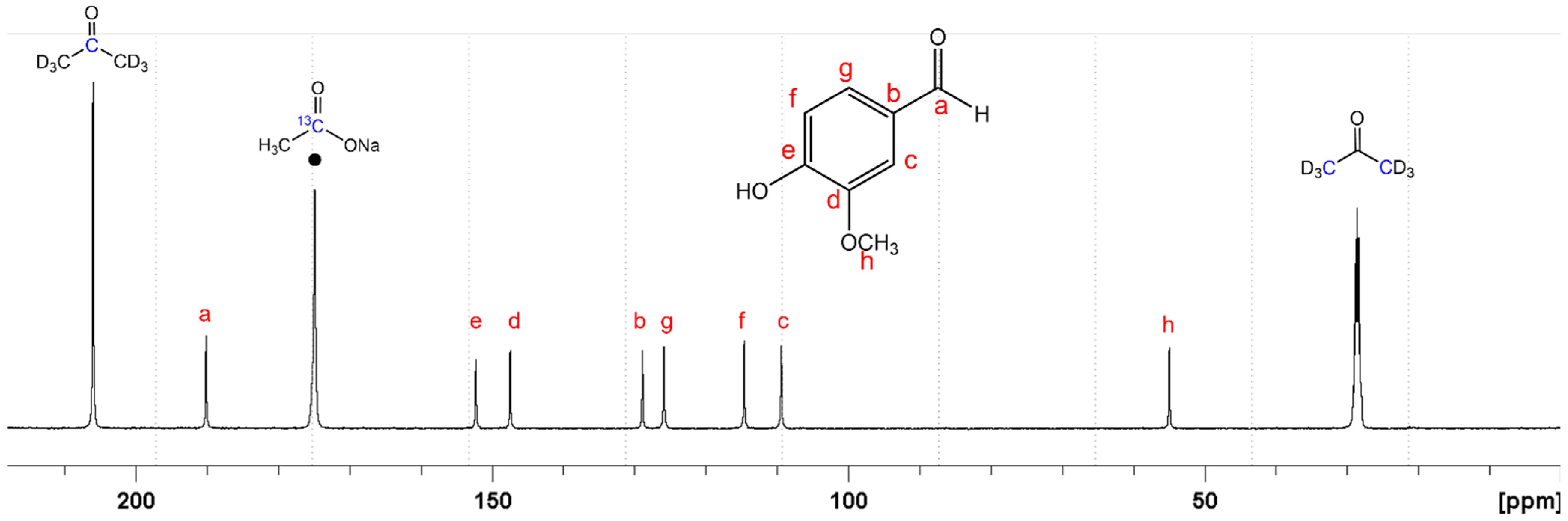
2.3. Characterization of Vanillin Samples with 13C-NMR Spectroscopy Analysis
2.4. Expression of the Results
2.5. Statistical Analysis
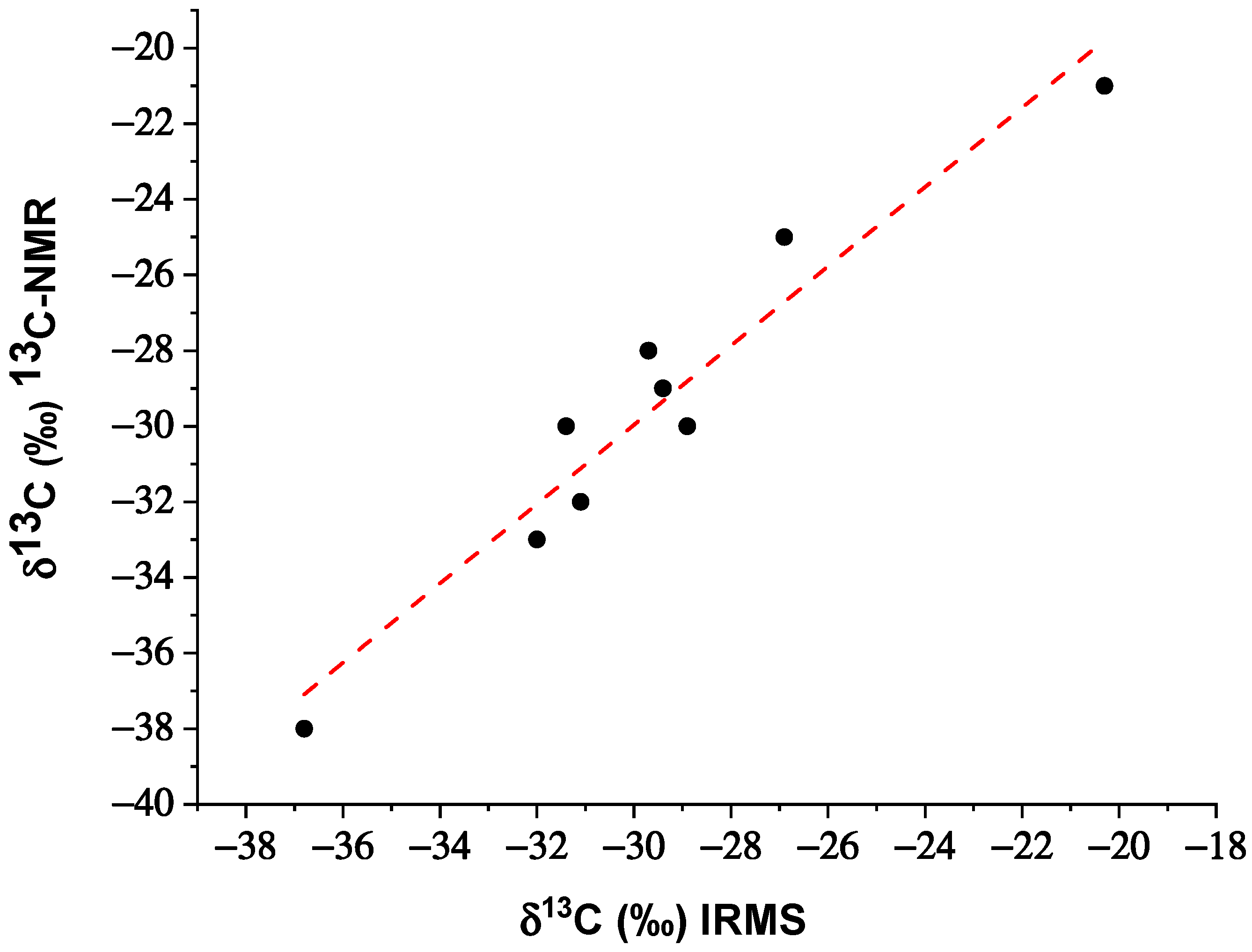
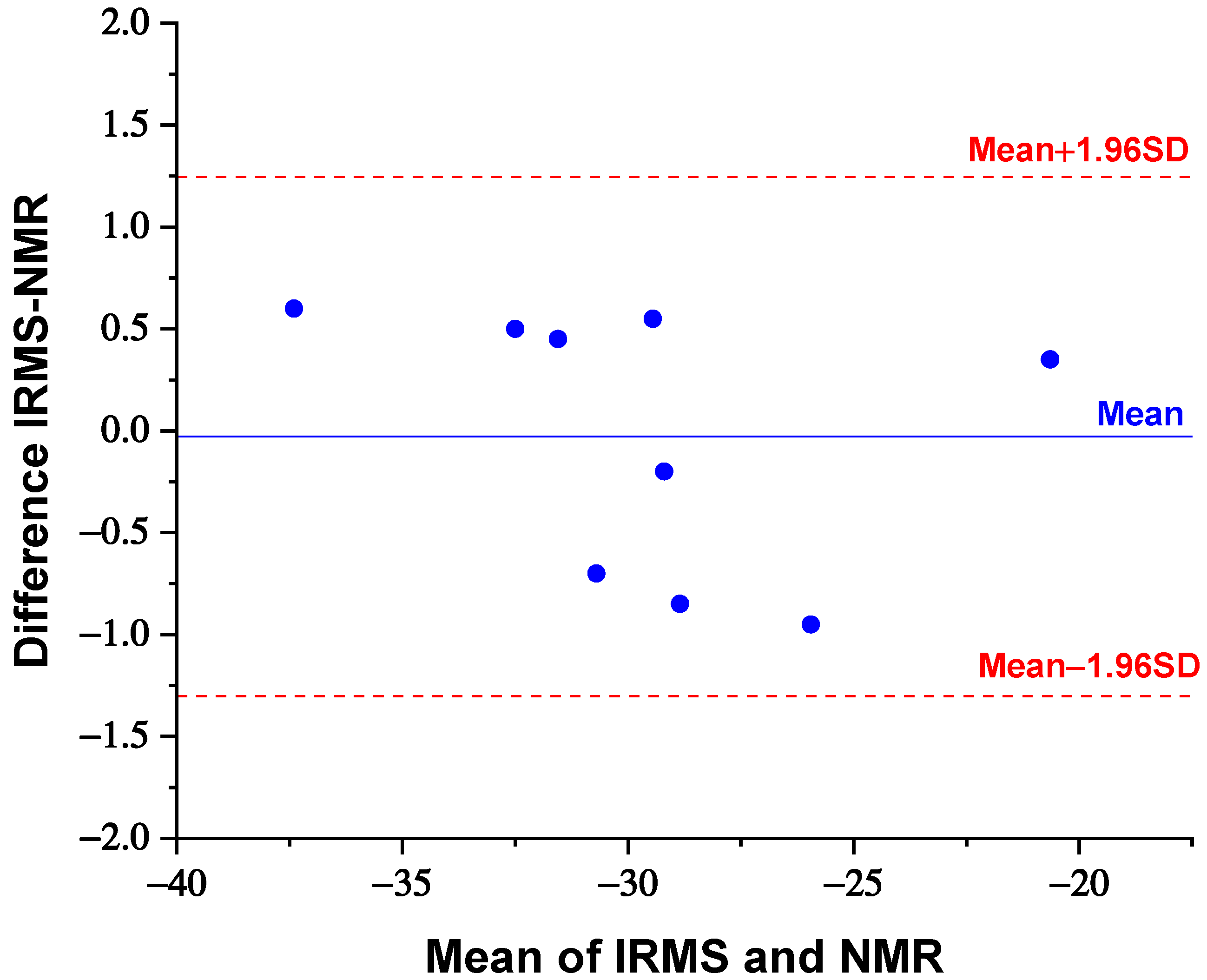
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pironti, C.; Proto, A.; Camin, F.; Cucciniello, R.; Zarrella, I.; Motta, O. FTIR and NDIR spectroscopies as valuable alternatives to IRMS spectrometry for the δ13C analysis of food. Talanta 2016, 160, 276–281. [Google Scholar] [CrossRef]
- Bontempo, L.; van Leeuwen, K.A.; Paolini, M.; Laursen, K.H.; Micheloni, C.; Prenzler, P.D.; Ryan, D.; Camin, F. Bulk and compound-specific stable isotope ratio analysis for authenticity testing of organically grown tomatoes. Food Chem. 2020, 318, 126426. [Google Scholar] [CrossRef]
- Brescia, M.A.; di Martino, G.; Guillou, C.; Reniero, F.; Sacco, A.; Serra, F. Differentiation of the geographical origin of durum wheat semolina samples on the basis of isotopic composition. Rapid Commun. Mass Spectrom. 2002, 16, 2286–2290. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.-Y.; Zhang, Y.-L.; Song, W.; Cai, H.-M.; Wang, Y.; Granato, D. Characterization of Brazilian coffee based on isotope ratio mass spectrometry (δ13C, δ18O, δ2H, and δ15N) and supervised chemometrics. Food Chem. 2019, 297, 124963. [Google Scholar] [CrossRef]
- Kawashima, H.; Suto, M.; Suto, N. Stable carbon isotope ratios for organic acids in commercial honey samples. Food Chem. 2019, 289, 49–55. [Google Scholar] [CrossRef]
- Artavia, G.; Cortés-Herrera, C.; Granados-Chinchilla, F. Selected instrumental techniques applied in food and feed: Quality, safety and adulteration analysis. Foods 2021, 10, 1081. [Google Scholar] [CrossRef] [PubMed]
- Organizzazione Internazionale della Vigna e del Vino. Available online: http://www.oiv.int/it/ (accessed on 26 September 2021).
- Jamin, E.; Martin, F.; Martin, G.G.; Blanch-Cortés, A.-I.; Christoph, N.; Ciambotti, A.; Giraudon, S.; Guérin, R.; Guillou, C.; Kelly, S.; et al. Determination of the 13C/12C ratio of ethanol derived from fruit juices and maple syrup by isotope ratio mass spectrometry: Collaborative study. J. AOAC Int. 2004, 87, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Portaluri, V.; Thomas, F.; Jamin, E.; Akoka, S.; Remaud, G.S. Authentication of agave products through isotopic intramolecular 13C content of ethanol: Optimization and validation of 13C quantitative NMR methodology. ACS Food Sci. Technol. 2021, 1, 1316–1322. [Google Scholar] [CrossRef]
- Coleman, D.C.; Fry, B. (Eds.) Carbon Isotope Techniques; Isotopic Techniques in Plant, Soil, and Aquatic Biology Series; Academic Press: San Diego, CA, USA, 1991; ISBN 978-0-12-179731-7. [Google Scholar]
- Lamprecht, G.; Blochberger, K. Protocol for isolation of vanillin from ice cream and yoghurt to confirm the vanilla beans origin by 13C-EA-IRMS. Food Chem. 2009, 114, 1130–1134. [Google Scholar] [CrossRef]
- Scharrer, A.; Mosandl, A. Reinvestigation of vanillin contents and component ratios of vanilla extracts using high-performance liquid chromatography and gas chromatography. Dtsch. Lebensm.-Rundsch. 2001, 97, 449–456. [Google Scholar]
- Jäger, F.; Wagner, G.; Meijer, H.A.J.; Kerstel, E.R.T. Measuring δ13C of atmospheric air with non-dispersive infrared spectroscopy. Isot. Environ. Health Stud. 2005, 41, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Proto, A.; Cucciniello, R.; Rossi, F.; Motta, O. Stable carbon isotope ratio in atmospheric CO2 collected by new diffusive devices. Environ. Sci. Pollut. Res. 2013, 21, 3182–3186. [Google Scholar] [CrossRef]
- Zanasi, R.; Alfano, D.; Scarabino, C.; Motta, O.; Viglione, R.G.; Proto, A. Determination of 13C/12C carbon isotope ratio. Anal. Chem. 2006, 78, 3080–3083. [Google Scholar] [CrossRef]
- Motta, O.; de Caro, F.; Quarto, F.; Proto, A. New FTIR methodology for the evaluation of 13C/12C isotope ratio in Helicobacter pylori infection diagnosis. J. Infect. 2009, 59, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Pironti, C.; Ricciardi, M.; Proto, A.; Cucciniello, R.; Fiorentino, A.; Fiorillo, R.; Motta, O. New analytical approach to monitoring air quality in historical monuments through the isotopic ratio of CO2. Environ. Sci. Pollut. Res. 2021, 1–6. [Google Scholar] [CrossRef]
- Motta, O.; Pironti, C.; Ricciardi, M.; Rostagno, C.; Bolzacchini, E.; Ferrero, L.; Cucciniello, R.; Proto, A. Leonardo da Vinci’s “Last Supper”: A case study to evaluate the influence of visitors on the museum preservation systems. Environ. Sci. Pollut. Res. 2021, 1–8. [Google Scholar] [CrossRef]
- Pironti, C.; Cucciniello, R.; Camin, F.; Tonon, A.; Motta, O.; Proto, A. Determination of the 13C/12C carbon isotope ratio in carbonates and bicarbonates by 13C NMR spectroscopy. Anal. Chem. 2017, 89, 11413–11418. [Google Scholar] [CrossRef]
- Portaluri, V.; Thomas, F.; Jamin, E.; Lorandel, B.; Silvestre, V.; Akoka, S.; Remaud, G.S. Vanillin isotopic intramolecular 13C profile through polarization transfer NMR pulse sequence and statistical modelling. Food Control. 2021, 130, 108345. [Google Scholar] [CrossRef]
- Wilde, A.S.; Strucko, T.; Veje, C.R.; Mortensen, U.H.; Duedahl-Olesen, L. Authentication of vanillin ex glucose—A first study on the influence of the glucose-source on the δ13C and δ2H value. Food Control. 2022, 131, 108389. [Google Scholar] [CrossRef]
- Hansen, C.A. Vanillin biosynthesis from sucrose ex-sugarcane: Authentication of an alternative vanillin source through stable isotope data analysis. Heliyon 2021, 7, e06970. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.S.; Hansen, A.-M.S.; Fromberg, A.; Frandsen, H.L.; Smedsgaard, J. Determination of δ13C of vanillin in complex food matrices by HS-SPME-GC-C-IRMS. Flavour Fragr. J. 2020, 35, 387–393. [Google Scholar] [CrossRef]
- Perini, M.; Pianezze, S.; Strojnik, L.; Camin, F. C and H stable isotope ratio analysis using solid-phase microextraction and gas chromatography-isotope ratio mass spectrometry for vanillin authentication. J. Chromatogr. A 2019, 1595, 168–173. [Google Scholar] [CrossRef]
- Bononi, M.; Quaglia, G.; Tateo, F. Easy extraction method to evaluate δ13C vanillin by liquid chromatography–isotopic ratio mass spectrometry in chocolate bars and chocolate snack foods. J. Agric. Food Chem. 2015, 63, 4777–4781. [Google Scholar] [CrossRef]
- Embuscado, M.E. Bioactives from spices and herbs. In Encyclopedia of Food Chemistry; Melton, L., Shahidi, F., Varelis, P., Eds.; Academic Press: Oxford, UK, 2019; Volume 3, pp. 497–514. ISBN 978-0-12-814045-1. [Google Scholar]
- Van der Schaft, P. 11—Approaches to production of natural flavours. In Flavour Development, Analysis and Perception in Food and Beverages; Parker, J.K., Elmore, J.S., Methven, L., Eds.; Woodhead Publishing Series in Food Science, Technology and Nutrition; Woodhead Publishing: Sawston, UK, 2015; pp. 235–248. ISBN 978-1-78242-103-0. [Google Scholar]
- Huang, W.-B.; Du, C.-Y.; Jiang, J.-A.; Ji, Y.-F. Concurrent synthesis of vanillin and isovanillin. Res. Chem. Intermed. 2013, 39, 2849–2856. [Google Scholar] [CrossRef]
- Fache, M.; Boutevin, B.; Caillol, S. Vanillin production from lignin and its use as a renewable chemical. ACS Sustain. Chem. Eng. 2015, 4, 35–46. [Google Scholar] [CrossRef]
- Zhang, R.; Maltari, R.; Guo, M.; Kontro, J.; Eronen, A.; Repo, T. Facile synthesis of vanillin from fractionated Kraft lignin. Ind. Crop. Prod. 2020, 145, 112095. [Google Scholar] [CrossRef]
- Li, K.; Frost, J.W. Synthesis of vanillin from glucose. J. Am. Chem. Soc. 1998, 120, 10545–10546. [Google Scholar] [CrossRef]
- Converti, A.; Aliakbarian, B.; Domínguez, J.M.; Vázquez, G.B.; Perego, P. Microbial production of biovanillin. Braz. J. Microbiol. 2010, 41, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Akoka, S.; Remaud, G.S. NMR-based isotopic and isotopomic analysis. Prog. Nucl. Magn. Reson. Spectrosc. 2020, 120–121, 1–24. [Google Scholar] [CrossRef]
- Geißler, K.; Greule, M.; Schäfer, U.; Hans, J.; Geißler, T.; Meier, L.; Keppler, F.; Krammer, G. Vanilla authenticity control by DNA barcoding and isotope data aggregation. Flavour Fragr. J. 2017, 32, 228–237. [Google Scholar] [CrossRef]
- Hansen, A.-M.S.; Fromberg, A.; Frandsen, H.L. Authenticity and traceability of vanilla flavors by analysis of stable isotopes of carbon and hydrogen. J. Agric. Food Chem. 2014, 62, 10326–10331. [Google Scholar] [CrossRef]
- Greule, M.; Tumino, L.D.; Kronewald, T.; Hener, U.; Schleucher, J.; Mosandl, A.; Keppler, F. Improved rapid authentication of vanillin using δ13C and δ2H values. Eur. Food Res. Technol. 2010, 231, 933–941. [Google Scholar] [CrossRef]
- Wilde, A.S.; Frandsen, H.L.; Fromberg, A.; Smedsgaard, J.; Greule, M. Isotopic characterization of vanillin ex glucose by GC-IRMS—New challenge for natural vanilla flavour authentication? Food Control. 2019, 106, 106735. [Google Scholar] [CrossRef]
- Remaud, G.S.; Martin, Y.-L.; Martin, G.G.; Martin, G.J. Detection of Sophisticated Adulterations of Natural Vanilla Flavors and Extracts: Application of the SNIF-NMR Method to Vanillin andp-Hydroxybenzaldehyde. J. Agric. Food Chem. 1997, 45, 859–866. [Google Scholar] [CrossRef]
- Guyader, S.; Thomas, F.; Jamin, E.; Grand, M.; Akoka, S.; Silvestre, V.; Remaud, G.S. Combination of13C and2HSNIF-NMRisotopic fingerprints of vanillin to control its precursors. Flavour Fragr. J. 2019, 34, 133–144. [Google Scholar] [CrossRef]
- Ricciardi, M.; Pironti, C.; Motta, O.; Fiorillo, R.; Camin, F.; Faggiano, A.; Proto, A. Investigations on historical monuments’ deterioration through chemical and isotopic analyses: An Italian case study. Environ. Sci. Pollut. Res. 2021, 1–10. [Google Scholar] [CrossRef]
- Pironti, C.; Motta, O.; Ricciardi, M.; Camin, F.; Cucciniello, R.; Proto, A. Characterization and authentication of commercial cleaning products formulated with biobased surfactants by stable carbon isotope ratio. Talanta 2020, 219, 121256. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | 13Cs/12Cs (NMR) | δ13C (‰) 13C-NMR | σ | δ13C (‰) IRMS | σ | Classification |
|---|---|---|---|---|---|---|
| 1 | 0.0115968 | −32 | ±1 | −31.1 | ±0.1 | synthetic |
| 2 | 0.0115631 | −29 | ±1 | −29.4 | ±0.1 | synthetic |
| 3 | 0.0115743 | −30 | ±1 | −31.4 | ±0.1 | synthetic |
| 4 | 0.0115743 | −30 | ±1 | −28.9 | ±0.1 | synthetic |
| 5 | 0.0116642 | −38 | ±1 | −36.8 | ±0.1 | synthetic |
| 6 | 0.0115518 | −28 | ±1 | −29.7 | ±0.1 | synthetic |
| 7 | 0.0115181 | −25 | ±1 | −26.9 | ±0.1 | synthetic |
| 8 | 0.0116080 | −33 | ±1 | −32.0 | ±0.1 | synthetic |
| 9 * | 0.0110027 | −21 | ±1 | −20.3 | ±0.1 | natural |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pironti, C.; Ricciardi, M.; Motta, O.; Camin, F.; Bontempo, L.; Proto, A. Application of 13C Quantitative NMR Spectroscopy to Isotopic Analyses for Vanillin Authentication Source. Foods 2021, 10, 2635. https://doi.org/10.3390/foods10112635
Pironti C, Ricciardi M, Motta O, Camin F, Bontempo L, Proto A. Application of 13C Quantitative NMR Spectroscopy to Isotopic Analyses for Vanillin Authentication Source. Foods. 2021; 10(11):2635. https://doi.org/10.3390/foods10112635
Chicago/Turabian StylePironti, Concetta, Maria Ricciardi, Oriana Motta, Federica Camin, Luana Bontempo, and Antonio Proto. 2021. "Application of 13C Quantitative NMR Spectroscopy to Isotopic Analyses for Vanillin Authentication Source" Foods 10, no. 11: 2635. https://doi.org/10.3390/foods10112635
APA StylePironti, C., Ricciardi, M., Motta, O., Camin, F., Bontempo, L., & Proto, A. (2021). Application of 13C Quantitative NMR Spectroscopy to Isotopic Analyses for Vanillin Authentication Source. Foods, 10(11), 2635. https://doi.org/10.3390/foods10112635