1. Introduction
Since their discovery over two decades ago, metal–organic frameworks (MOFs) have been considered promising materials for application in gas separation and storage, owing to their large porosity and ease of functionalisation [
1,
2,
3]. Deployment of MOFs as solid sorbents for application in industrial-scale gas separation or storage processes requires powders to be compacted into industrially acceptable forms, such as tablets, pellets or extrudates. The use of free-flowing fine powders in industrial scale gas separation processes is indeed not desirable, mainly due to the large pressure drop across the sorbent bed [
4,
5]. In storage applications, high volumetric adsorption capacity is a key requirement to minimize the size of the gas container, calling for densification of the sorbent [
6]. In addition to these technological aspects, there are several safety concerns arising from handling of free-flowing powders [
5]. However, compaction of MOF powders under pressure often leads to partial collapse of their porous framework, thus reducing surface area and pore volume, with an impact on the gas sorption performance [
7,
8,
9]. The challenge is therefore to identify the correct conditions to ensure mechanical stability of the shaped sorbents while preserving the performance of the original powder. Given its practical importance, there is a growing interest in the shaping of powders in the MOF community.
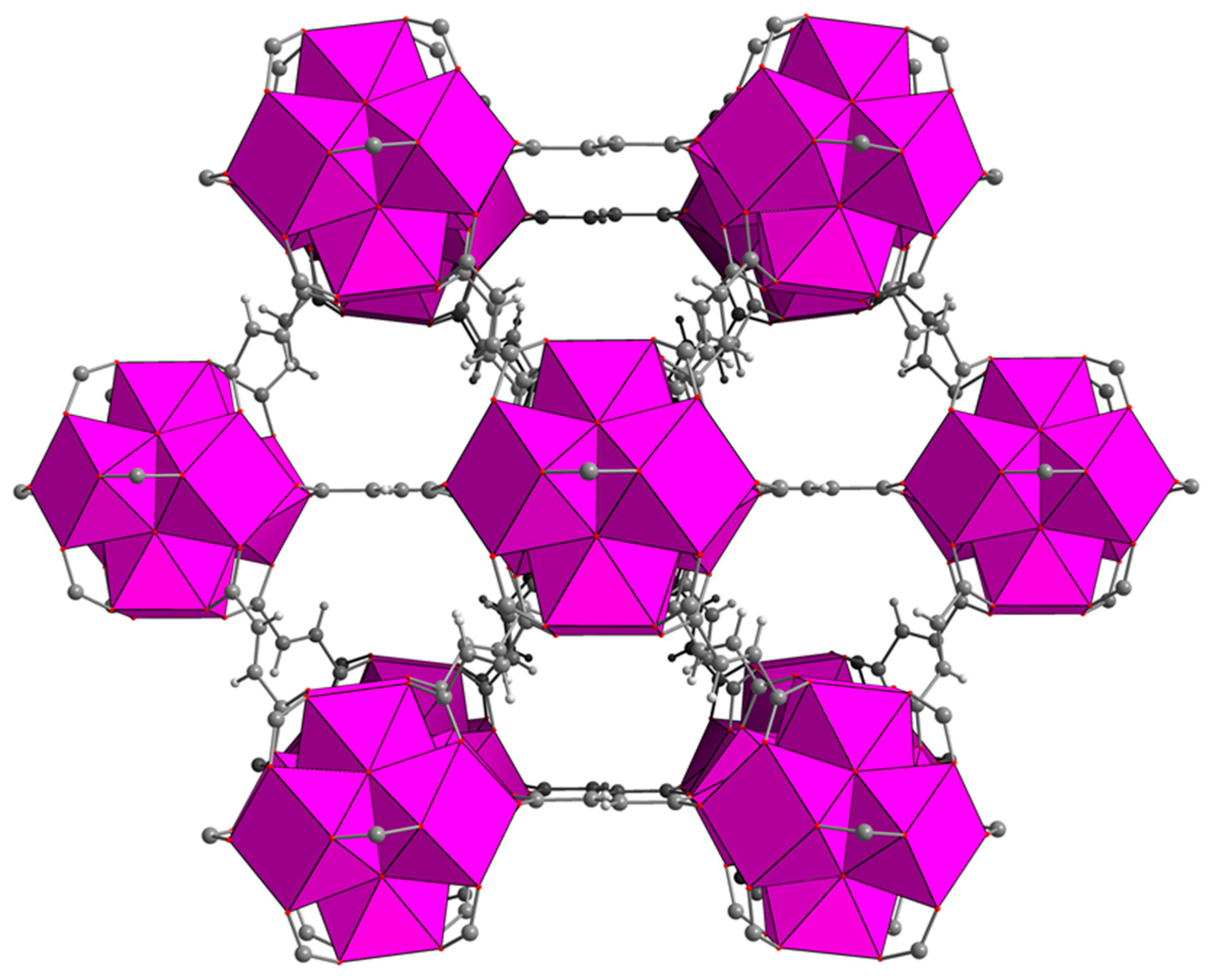
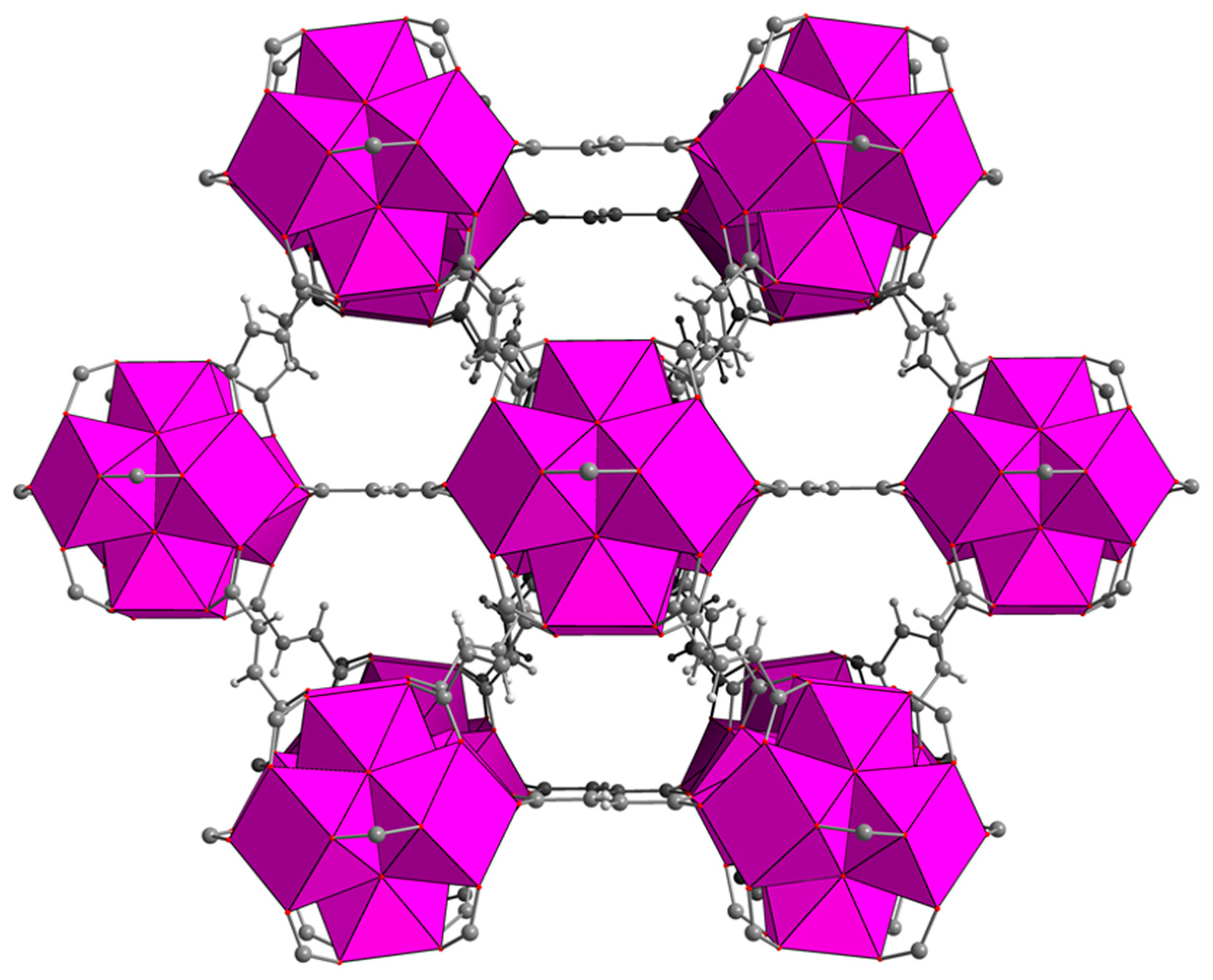
Zirconium-based MOFs are one of the most intensively investigated classes of MOFs, owing to their exceptional stability and topological diversity [
10]. MOF-801, based on fumaric acid as the organic linker, features twelve-connected [Zr
6O
4(OH)
4]
12+ clusters, giving rise to
fcu topology (
Figure 1) [
11,
12,
13]. Thanks to the high solubility of fumaric acid in water, MOF-801 can conveniently be synthesised in aqueous medium, with obvious benefits in terms of environmental impact [
12]. This aspect, combined with the commercial availability and very low cost of fumaric acid, makes MOF-801 a strong candidate for large scale production. In terms of applications, MOF-801 is widely recognised as one of the best MOFs for adsorbing water directly from the air [
13,
14], besides displaying good CO
2 uptake capacity [
15,
16]. To the best of our knowledge, there is no report of a systematic approach aimed at identifying the optimal conditions for compaction of MOF-801. Therefore, we set out to develop a compaction process potentially transferable to a real industrial setting and able to afford durable pellets retaining as much as possible of the original gas sorption performance of the MOF in powder form.
2. Results and Discussion
To start our investigation, we employed the conditions recently reported by Bambalaza et al. [
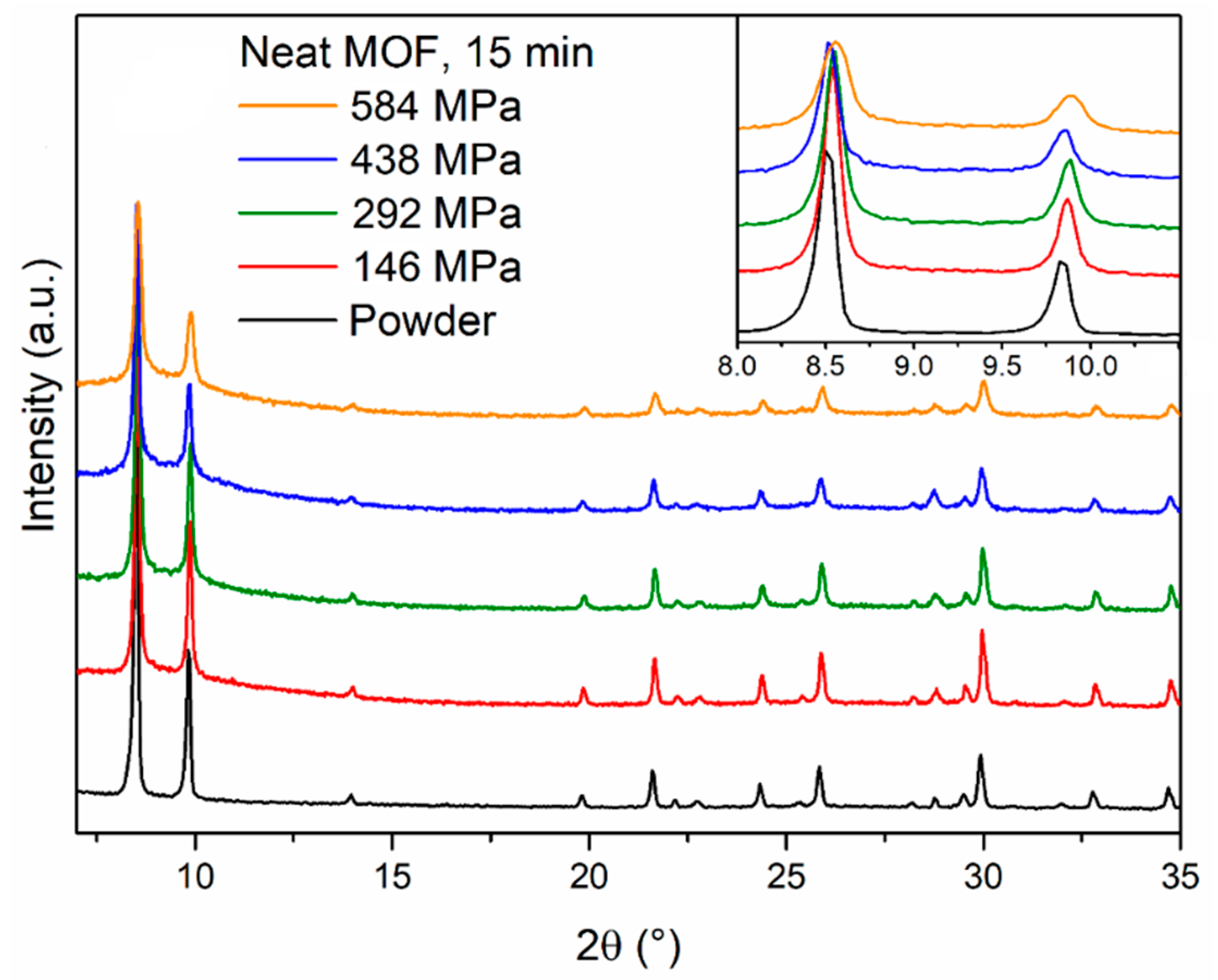
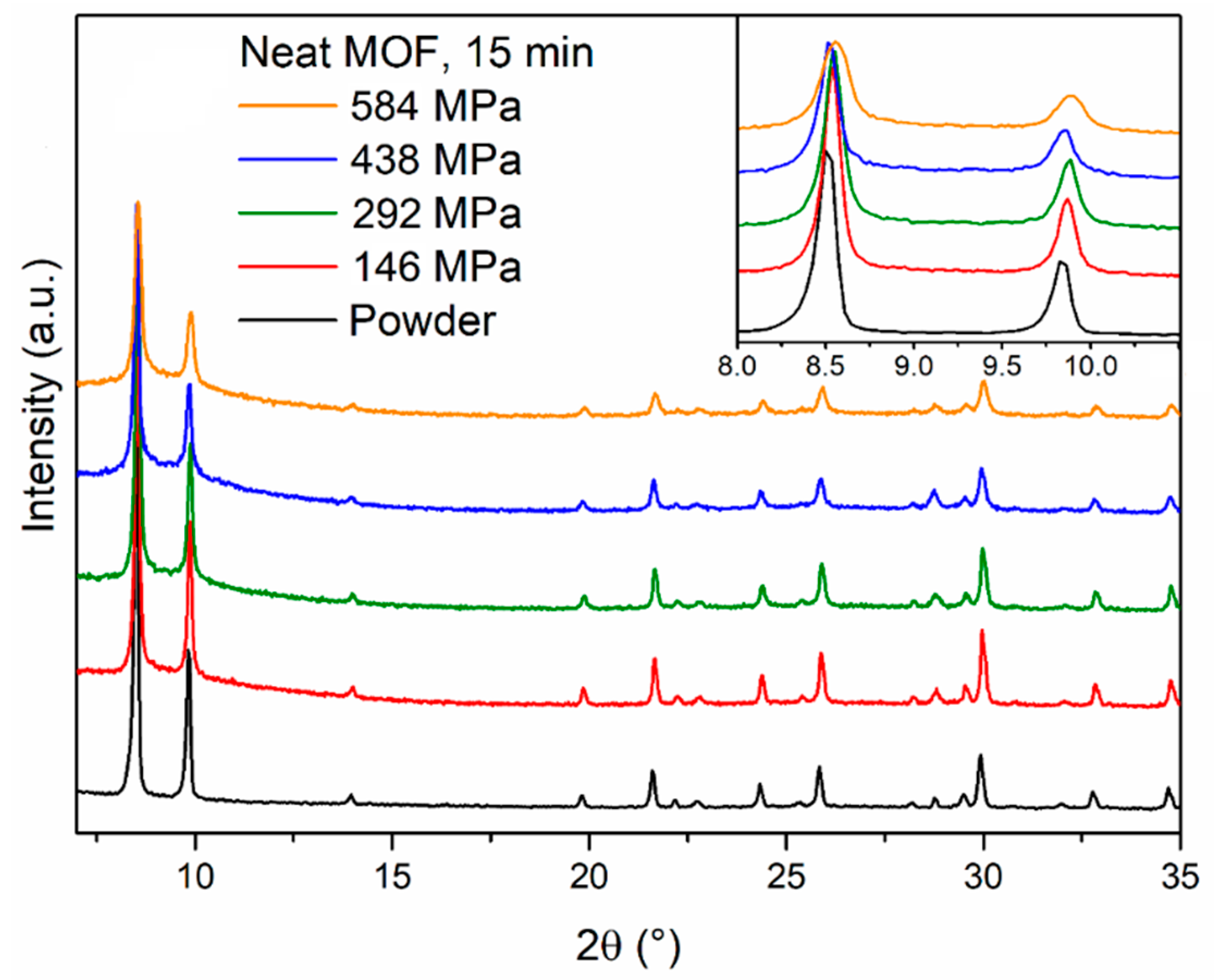
17] to produce pellets of UiO-66, which is closely related to MOF-801, having the same topology but a different linker (terephthalic acid). We prepared four MOF-801 pellets by compressing 400 mg of MOF for 15 min under four different pressures (146, 292, 438 and 584 MPa) using a Retsch PP25 pellet press with a 16-mm die. The pellets were 1.3-mm thick, which led to the calculation of an aspect ratio of 12.3. Powder X-ray diffraction (PXRD) analysis of the pellets (
Figure 2) showed that the reflections of MOF-801 underwent progressive broadening as the pressure increased, suggesting partial loss of long-range order. In addition, a diffuse scattering feature below 10° 2θ appeared in the pellets, suggesting that part of the solid might have undergone more severe amorphisation. Interestingly, we observed that, upon compression, the MOF released water which was originally trapped in its pores. Therefore, we treated 400 mg of MOF-801 at 120 °C for 2 h to remove most of the water present in the structure and prepared a pellet compressing the warm powder at 438 MPa for 15 min, observing a much larger drop in the intensity of MOF-801 reflections (
Figure S1). This suggests that the water present within the pores might act as a “cushion” and help to evenly dissipate the applied pressure, preventing the collapse of the framework during compression. Similar behaviour was previously reported for MOF-5 [
18] and HKUST-1 [
19,
20]. The durability of the pellets prepared using the solvated MOF was assessed with simple drop tests. The pellets were dropped from a height of 36 cm until they underwent severe damage, intended here as breakage into more than two fragments. This choice was justified by the fact that the shape of the pellets is highly anisotropic and the meridian is by far the weakest direction along which breakage would occur. Overall, the pellets were quite brittle, failing after five drops at most and cracking into several tiny fragments (
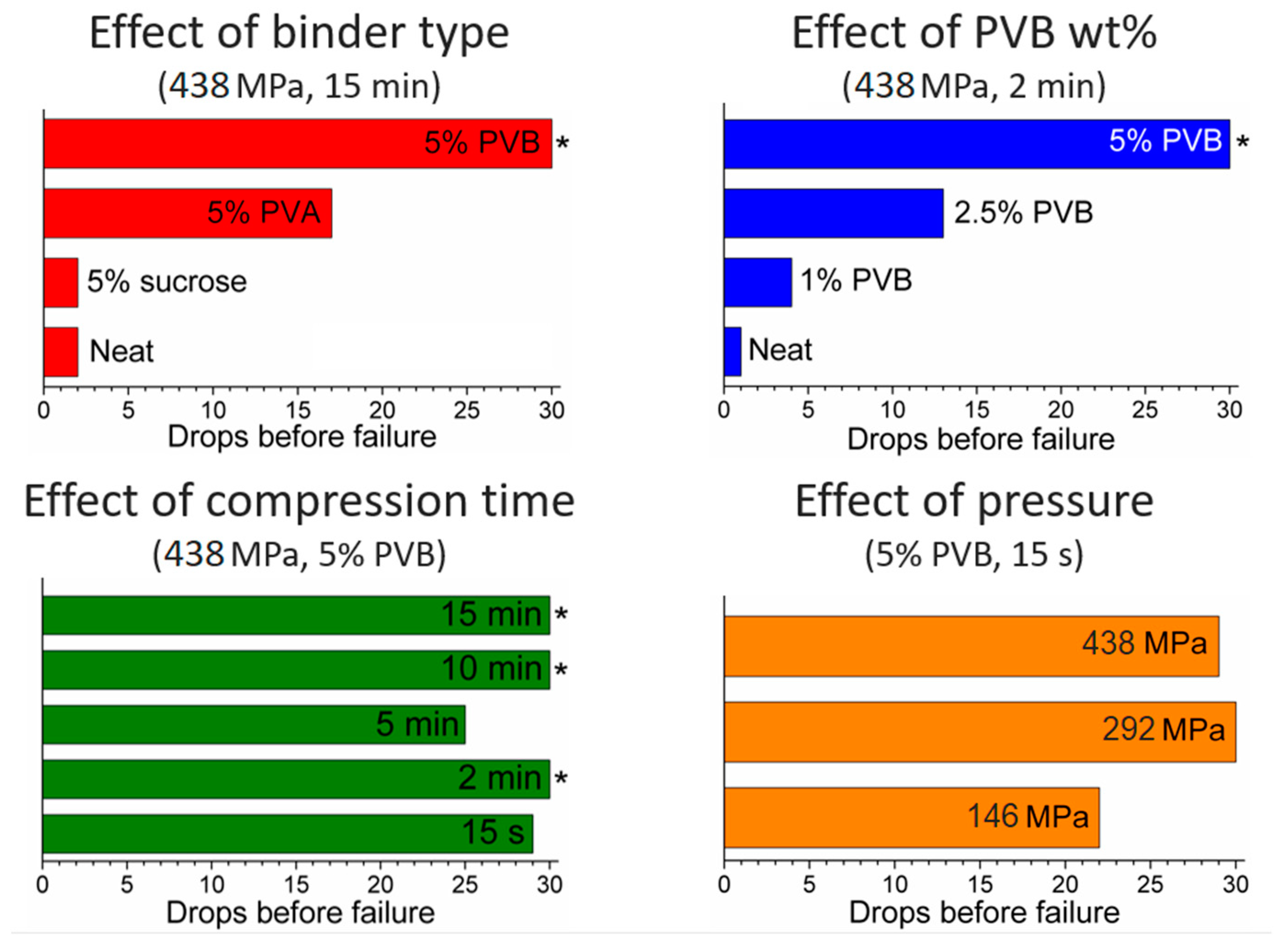
Figure S2). Increasing the pressure from 146 to 584 MPa yielded a little improvement in mechanical strength.
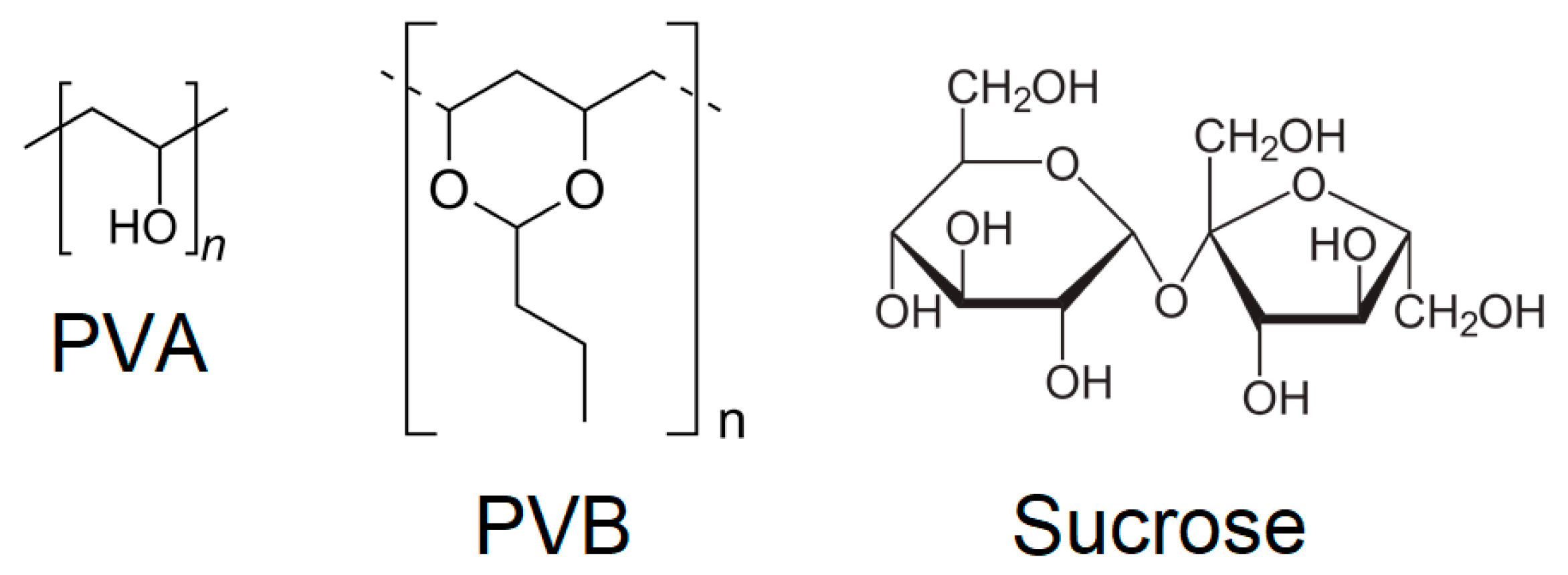
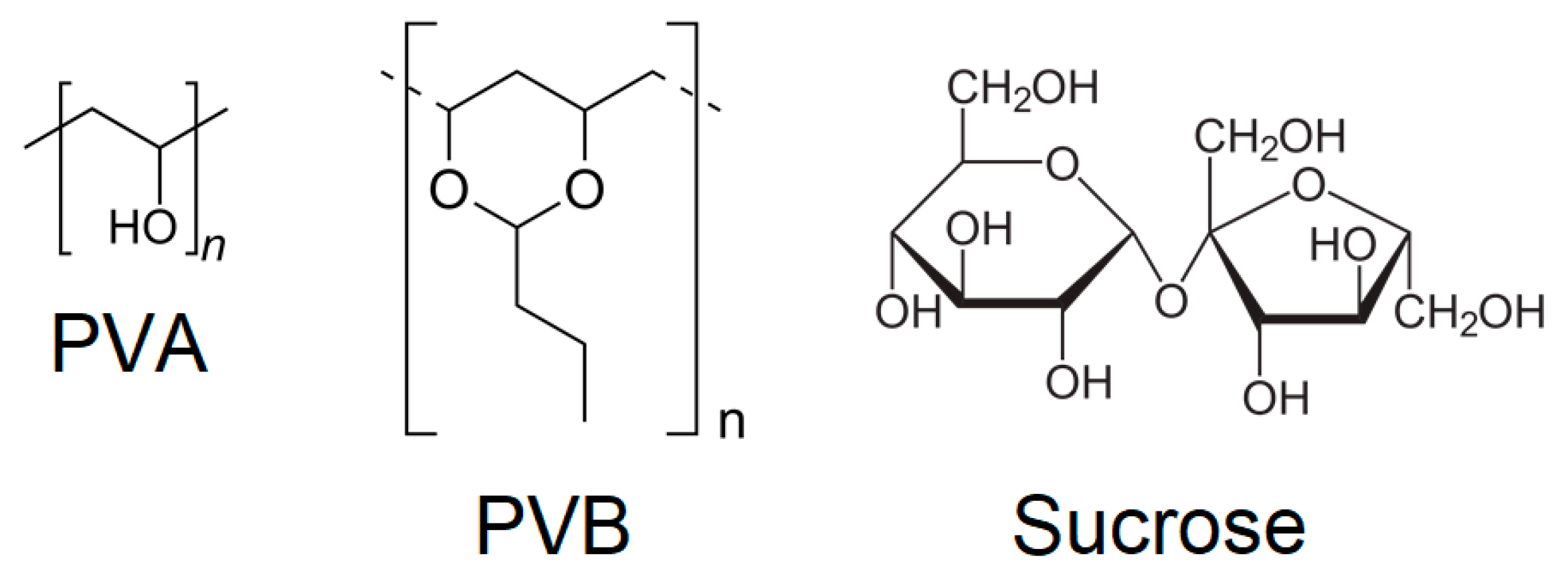
The addition of binders was then considered, initially adding 5 wt % of either polyvinylalcohol (PVA), polyvinylbutyral (PVB) or sucrose, whose structures are shown in
Figure 3, compressing at 438 MPa for 15 min. The choice of these binders was based on recent literature reports where they were employed to produce pellets of MOFs [
21,
22,
23]. Before characterisation, the pellets containing binders were treated at 120 °C for 2 h. This treatment was applied under the assumption that in a real gas separation/storage application, the pellets would need to be activated at high temperature to make their porosity accessible.
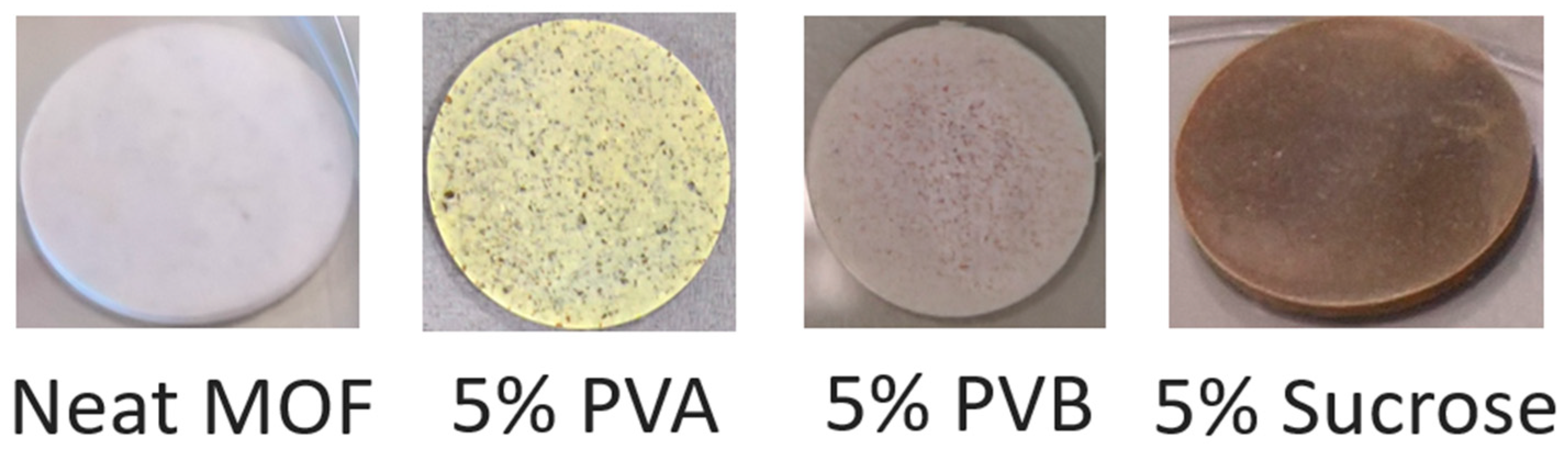
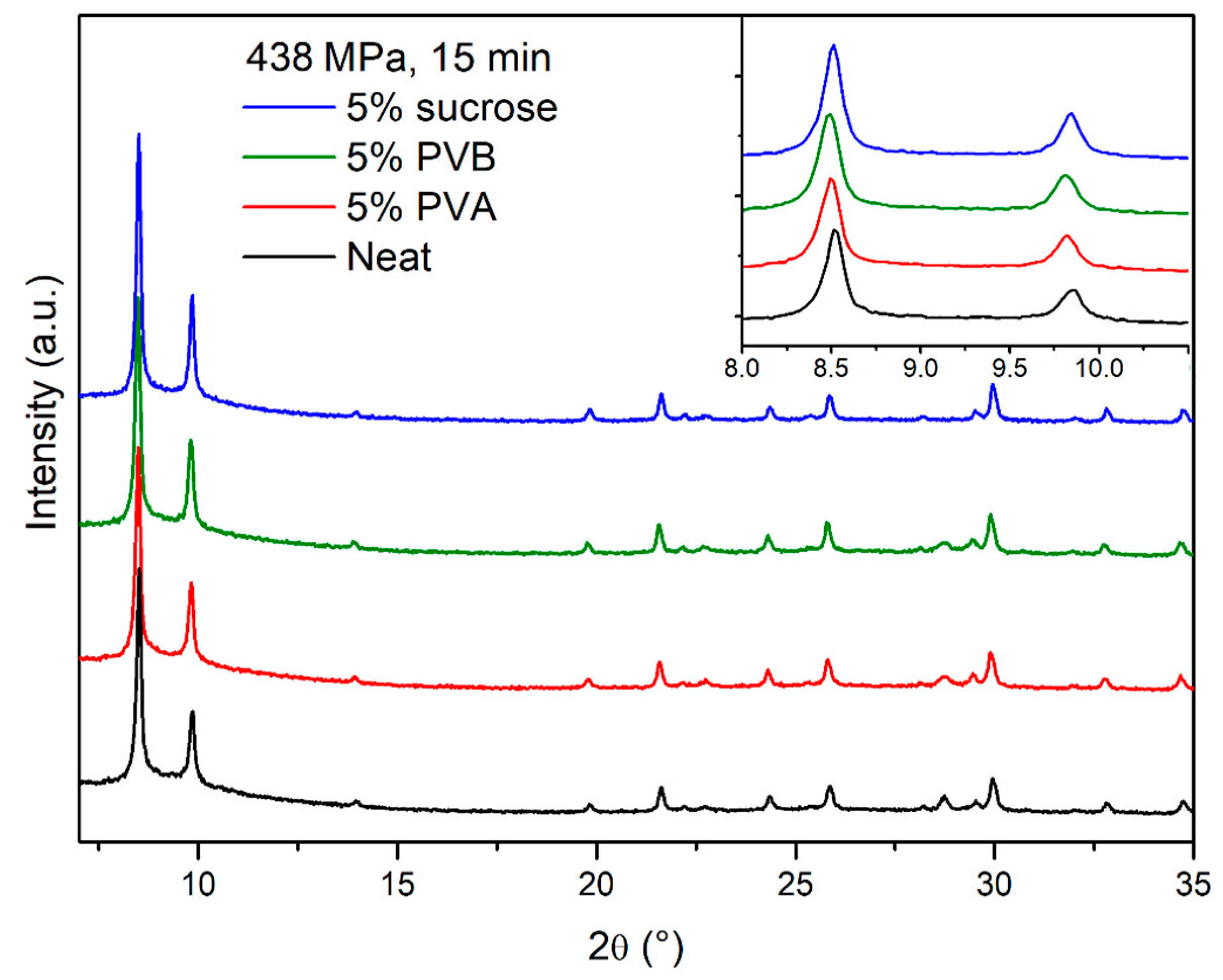
Figure 4 shows photographs of representative pellets prepared either with neat MOF-801 or with the addition of binders. All the pellets containing binders underwent colour change upon thermal treatment, with the ones containing PVA and sucrose showing the most evident effect, suggesting that these two binders might suffer from thermal degradation.
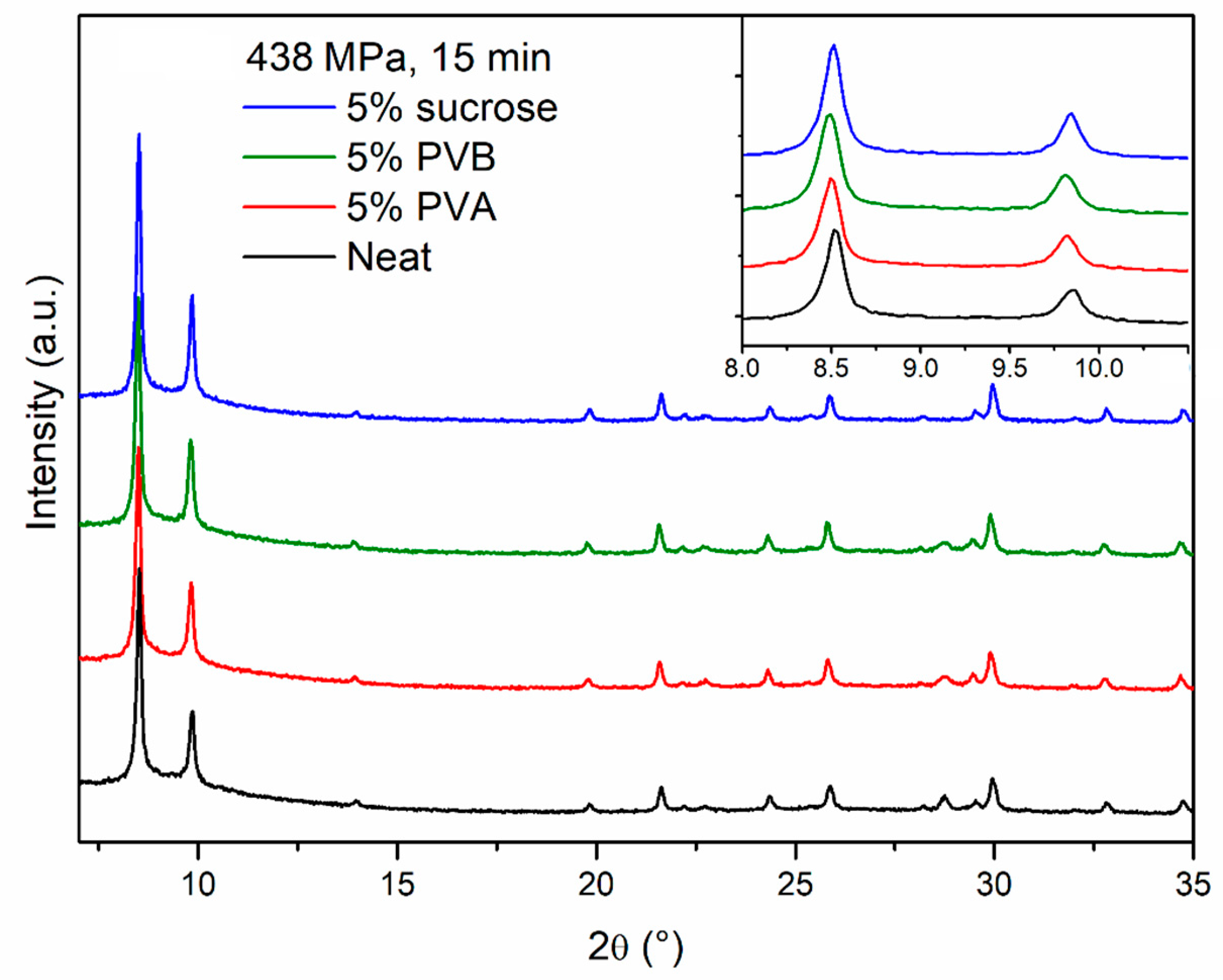
PXRD analysis does not evidence any relevant difference between the neat and the binder-containing pellets (
Figure 5). In terms of durability, sucrose did not have any benefit and the pellets were as brittle as those made of neat MOF, whereas PVA and PVB had a clear strengthening effect and the pellets cracked into larger fragments, with no release of fine powder (
Figure 6,
Figures S3–S5). Scanning electron microscopy (SEM) images of the section of cracked pellets, obtained by compressing MOF-801 at 438 MPa for 2 min either neat or blended with 5% PVB, were acquired using backscattered electrons to enhance the contrast between the MOF and the binder. The neat pellet appears homogeneously compacted and relatively smooth (
Figure 7a,c), whereas the one containing the binder displayed a rougher texture and dark spots of binder with size <10 µm dispersed throughout the MOF matrix (
Figure 7b,d). Since PVB displayed the highest durability, we decided to carry out additional screening focusing on this binder.
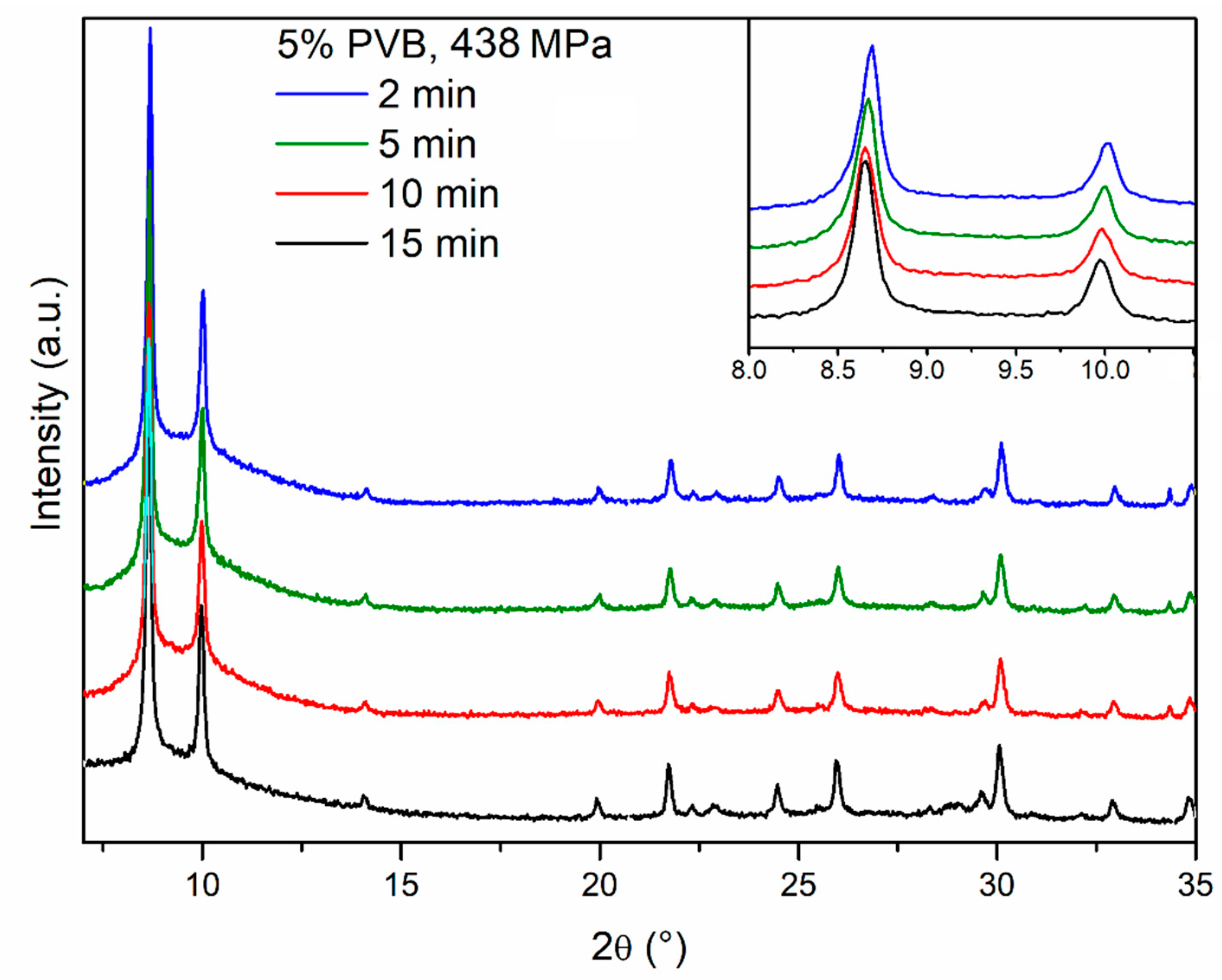
We then moved on to investigate the effect of compression time, with the aim of reducing this to a minimum. This is indeed important in a real compaction process, e.g., using a rotary pellet press, where it is not practical to keep the powder under pressure for as long as 15 min because of the low throughput of pellets produced in such conditions. We progressively decreased the compression time down to 10 min, 5 min, 2 min and 15 s, while keeping pressure to 438 MPa, observing no evident difference in the PXRD patterns (
Figure 8). Drop tests for these pellets did not show any evidence of obvious dependence of mechanical strength on the compression time, suggesting that 15 s was a viable compression time in terms of durability, while enabling a much higher throughput than the initial 15 min (
Figure 6). Using a hand-operated pellet press does not allow the further decrease of compression time in an accurate manner, but we speculate that time could be further decreased in a real, continuous compaction process without compromising the durability of the pellets.
The next parameter we screened was the amount of binder added, preparing pellets containing 2.5 and 1% of PVB and compressing at 438 MPa for 2 min. Drop tests showed that the durability scales almost linearly with the amount of binder (
Figure 6), suggesting that our initial choice of using 5% PVB was a good compromise, able to provide durable pellets while keeping the binder loading to a relatively low value, thus limiting its effect on the adsorption performance of the pellets.
Finally, we decreased the pressure while keeping the compression time fixed at 15 s. This is an important parameter in a production process, since the materials constituting the pellet press must be able to withstand the repeated stress induced during compression over a long period of time. Reducing the pressure needed to obtain pellets is therefore crucial in limiting the equipment capital cost. We observed that the pellets compressed at 146 MPa displayed lower mechanical strength than those at 292 and 438 MPa, but their durability was still reasonable (more than 20 drops were needed for severe damage, with no release of fine powder). In order to assess the effect of aspect ratio on the durability of pellets, a 5-mm diameter die was employed, compressing 70 mg of MOF blended with 5% PVB at 146 MPa for 15 s. Pellets with a thickness of 2 mm and an aspect ratio of 2.5 were obtained. These pellets proved to be extremely tough, showing no signs of damage after 30 drops. In addition, we tested the resistance of our 16 mm pellets to attrition by loading them in a capped glass vial and using a vortex mixer to shake it for 30 s up to four times. After each cycle, the pellets were weighed to determine how much mass had been lost to fine powder, observing a less than 4% decrease for the PVB-containing pellets compressed at 146 and 263 MPa (
Table 1).
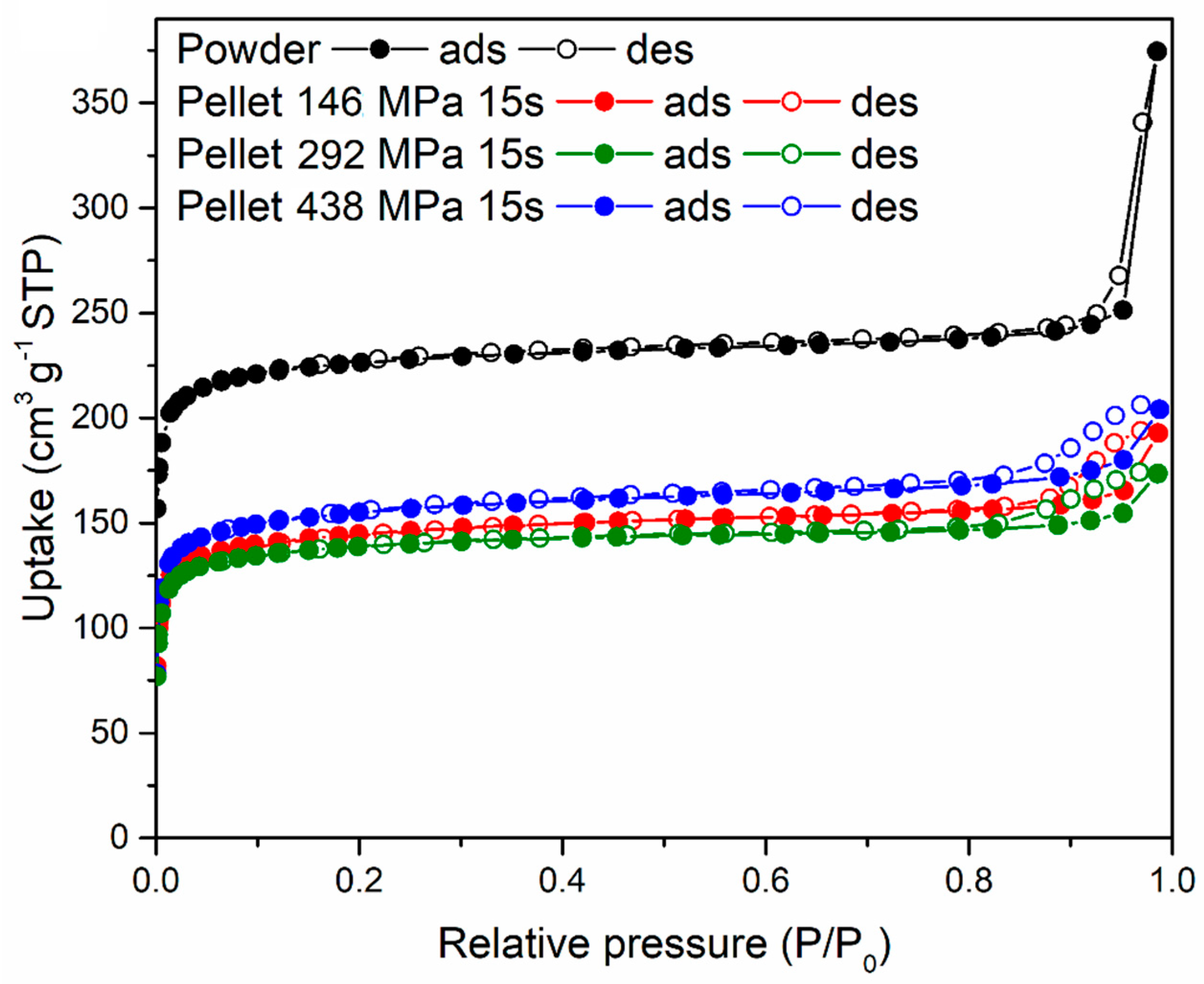
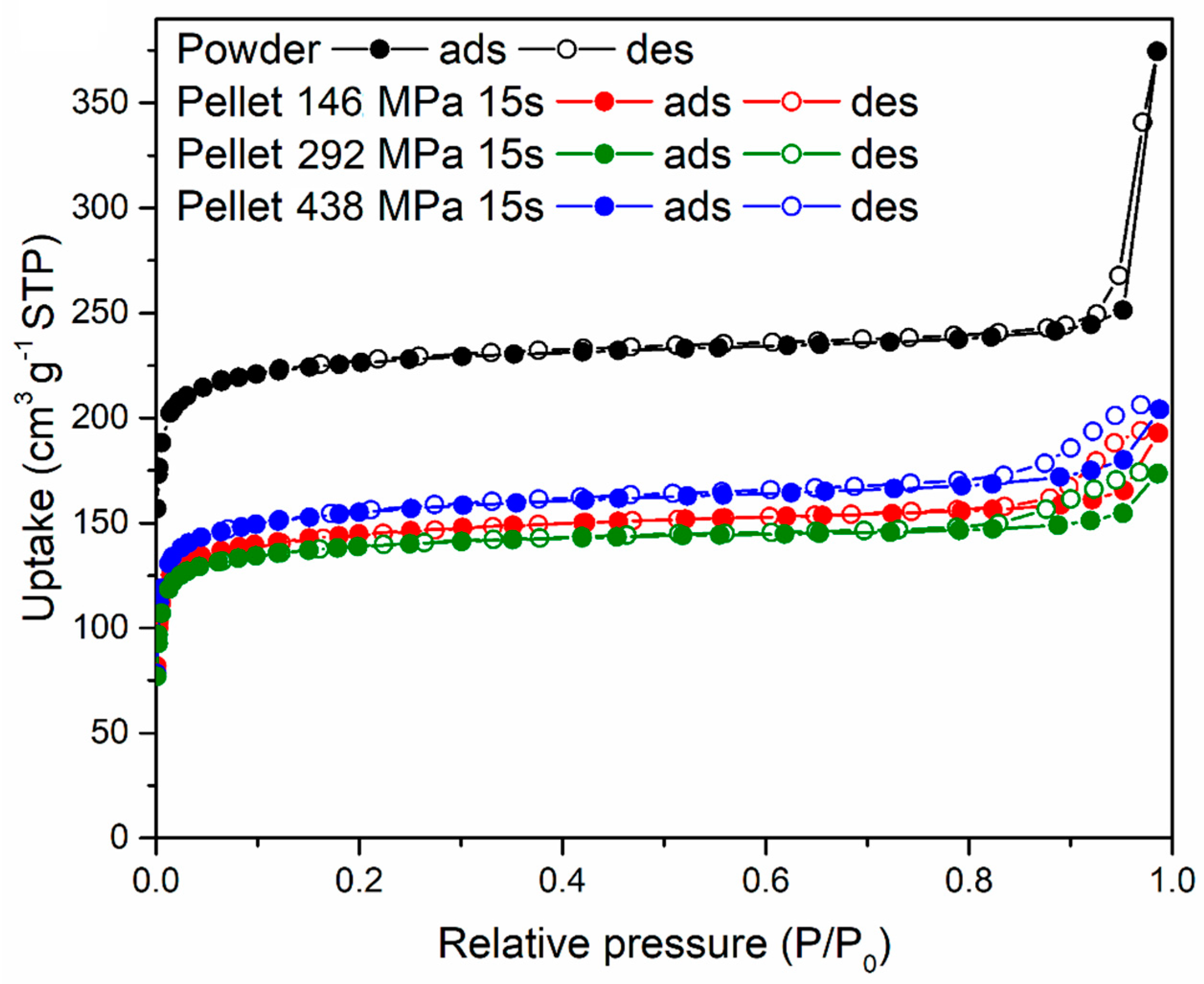
The three 16-mm diameter pellets prepared using 5% PVB compressing for 15 s at 146, 292 and 438 MPa, respectively, were characterised for their gas sorption properties and compared with the original MOF-801 powder. N
2 sorption isotherms at 77 K, shown in
Figure 9, display a decrease in uptake when the powder was compressed into pellets, with no major differences between different pressures. The shape of the isotherms suggests that the MOF retained its microporosity when compressed and that compaction reduced interparticular porosity (as indicated by the reduction of condensation occurring at high relative pressure). The BET surface area of the MOF-801 powder was 899 m
2 g
−1, whereas the pellets displayed 569 m
2 g
−1 (146 MPa), 546 m
2 g
−1 (292 MPa) and 605 m
2 g
−1 (438 MPa).
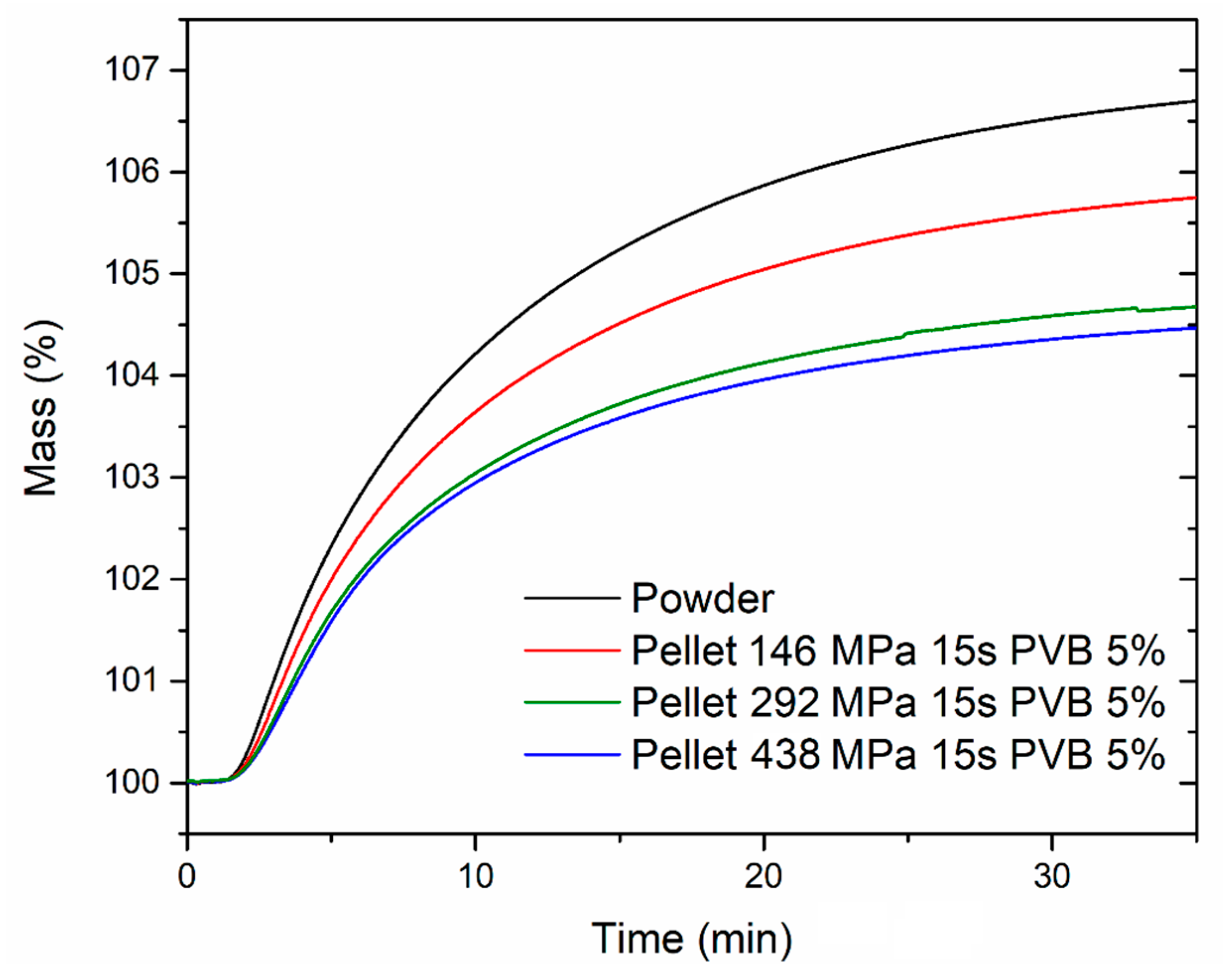
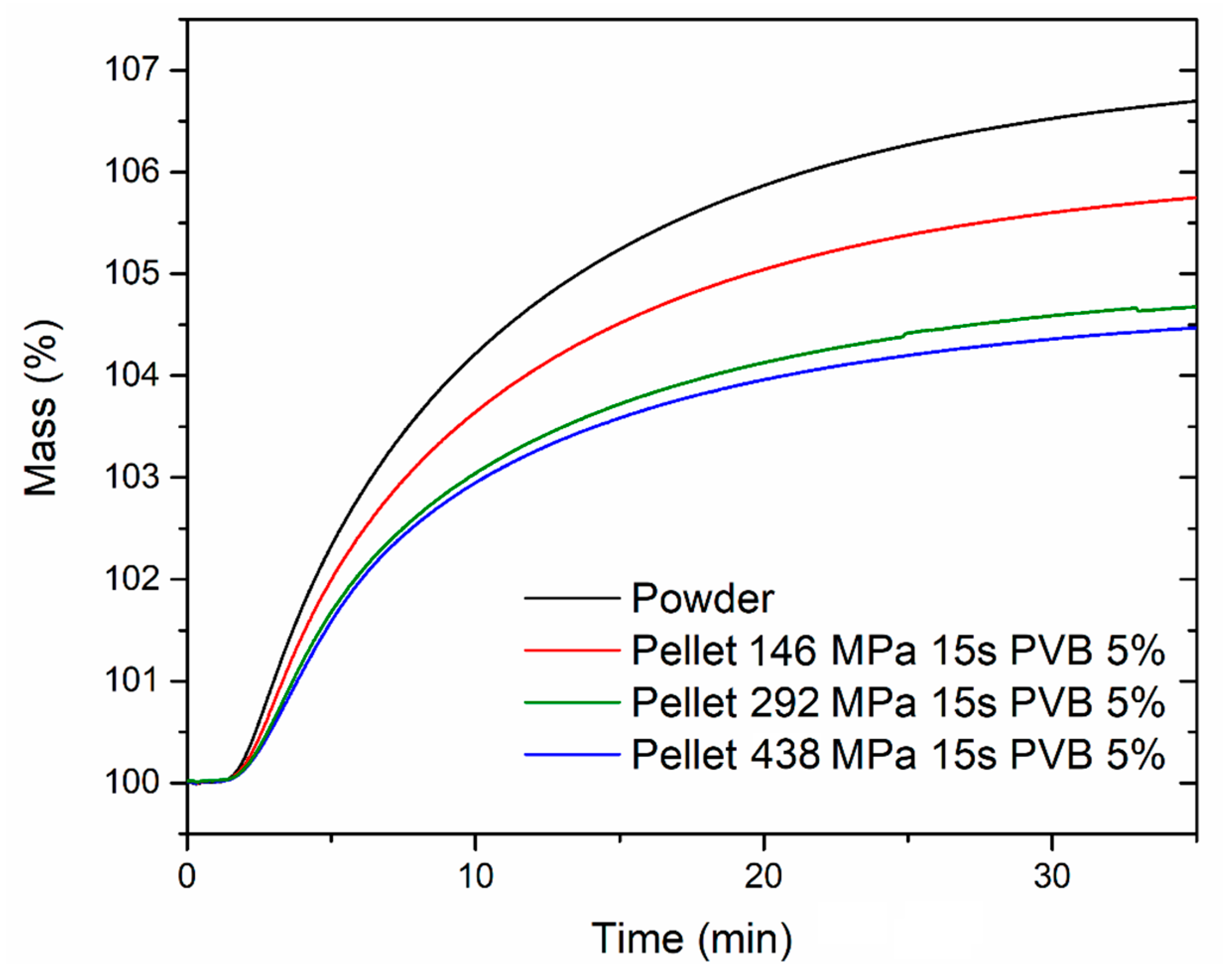
The CO
2 uptake capacity in dynamic conditions was also evaluated, exposing both the powder and the pellets to 1 atm CO
2 at 40 °C in a thermogravimetric analyzer (
Figure 10). The samples were previously activated by heating at 120 °C for 2 h under an Ar atmosphere, followed by cooling to 40 °C and switch to a CO
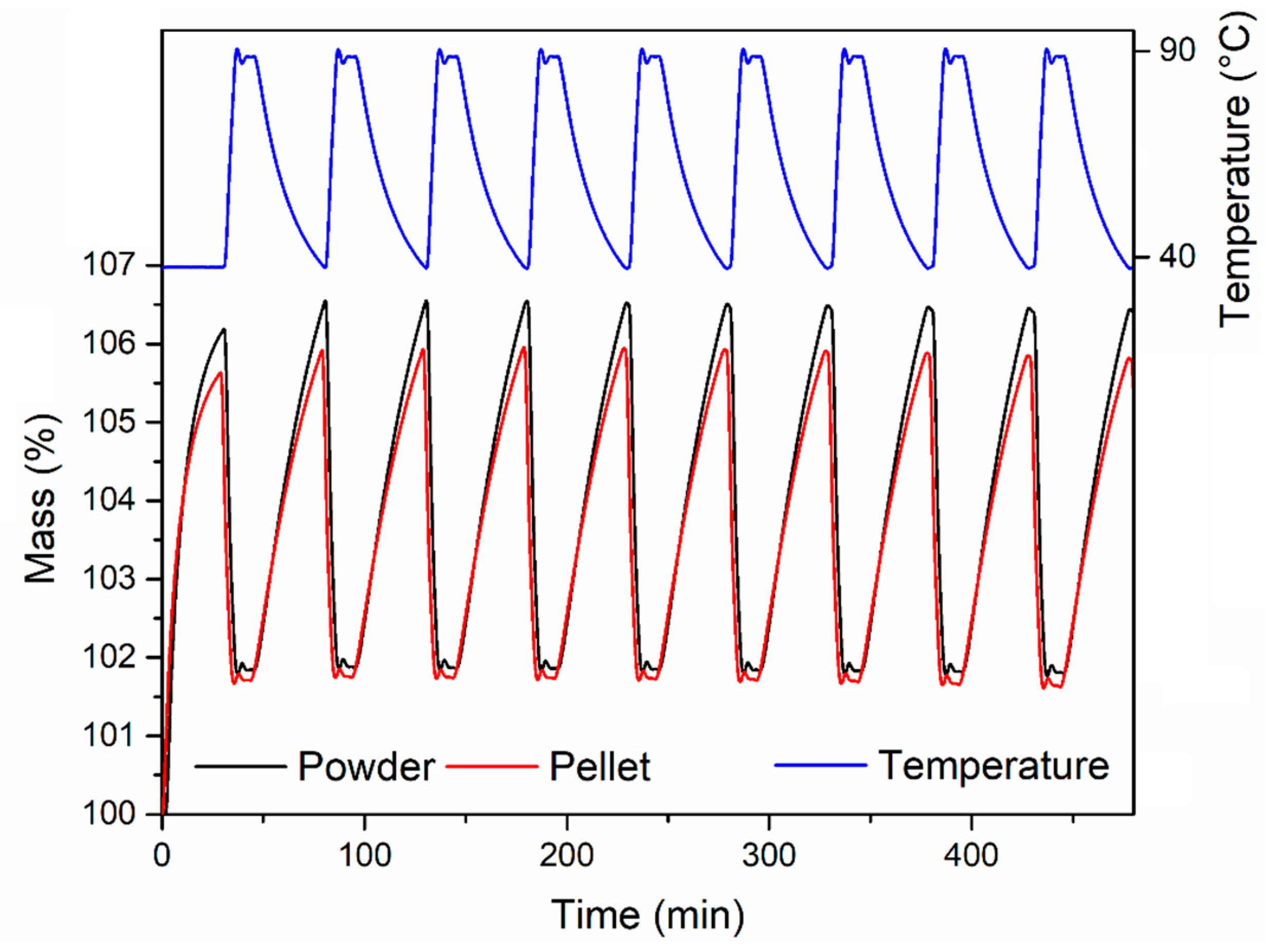
2 flow. The powder reached an uptake of 6.70 wt % after 35 min exposure, whereas the pellets reached 5.74 wt % (corresponding to 86% of the powder uptake; 146 MPa), 4.67 wt % (corresponding to 70% of the powder uptake; 292 MPa) and 4.47 wt % (corresponding to 67% of the powder uptake; 438 MPa), respectively. The adsorption kinetics were not significantly influenced upon compression of the powder, with all the samples reaching 50% and 90% of their maximum uptake within 6.8–7.3 min and 21.1–21.8 min, respectively. The stability over ten adsorption-desorption cycles was also tested for the powder and the pellet compressed at 146 MPa (
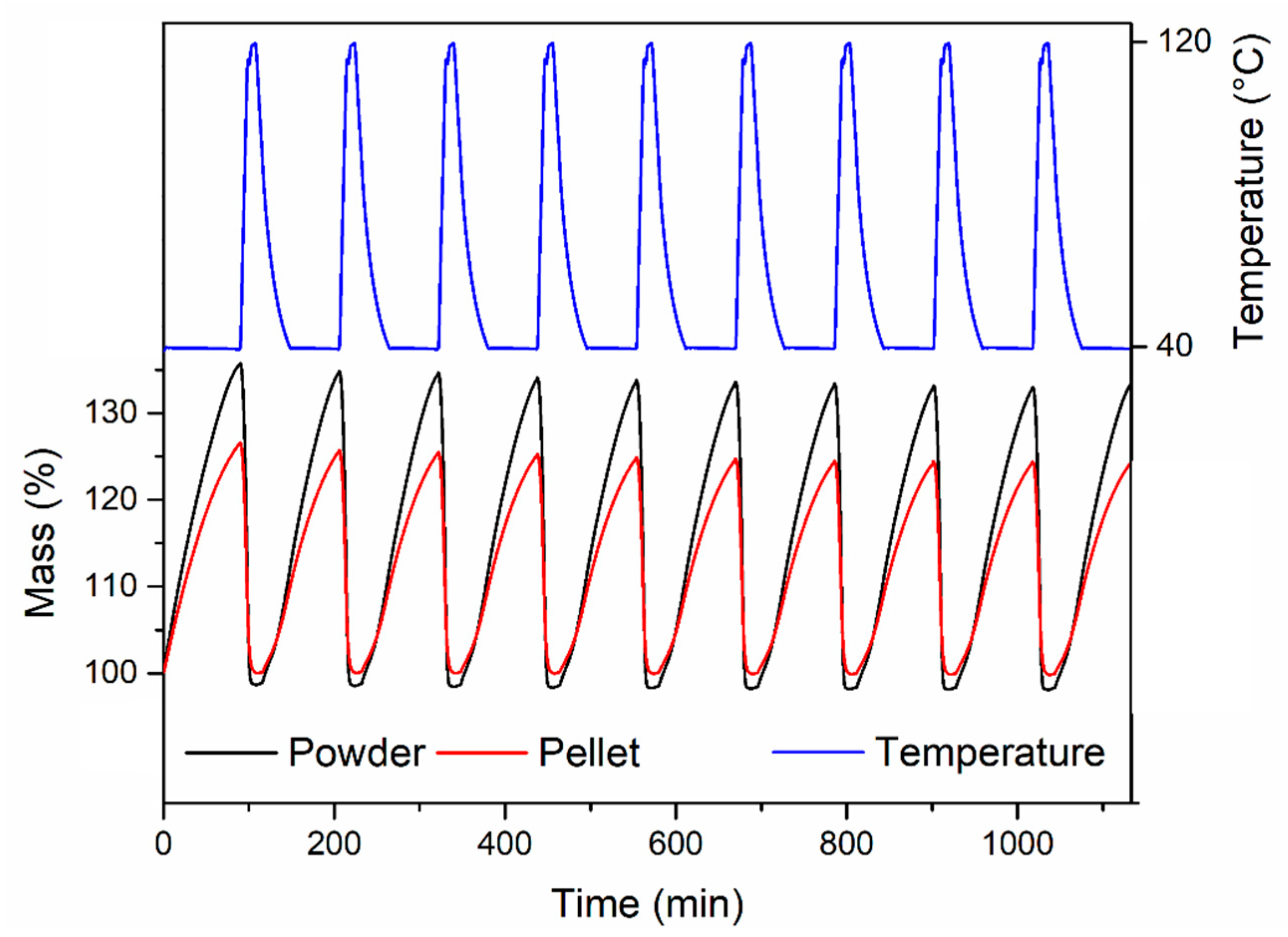
Figure 11). The activated samples were exposed to 1 atm CO
2, swinging the temperature between 40 °C and 90 °C. In these conditions, both samples displayed consistent working capacity over ten cycles: 4.64 wt % for the powder and 4.18 wt % for the pellet (corresponding to 90% of the working capacity of the powder). We note that the baseline gain after the first adsorption cycle is due to the fact that, during the first adsorption cycle, the MOF was completely desolvated and able to adsorb about 6 wt % of CO
2 at 40 °C. Heating the MOF to 90 °C under an atmosphere of CO
2 did not completely remove all the adsorbed CO
2, leading to the working capacities reported above.
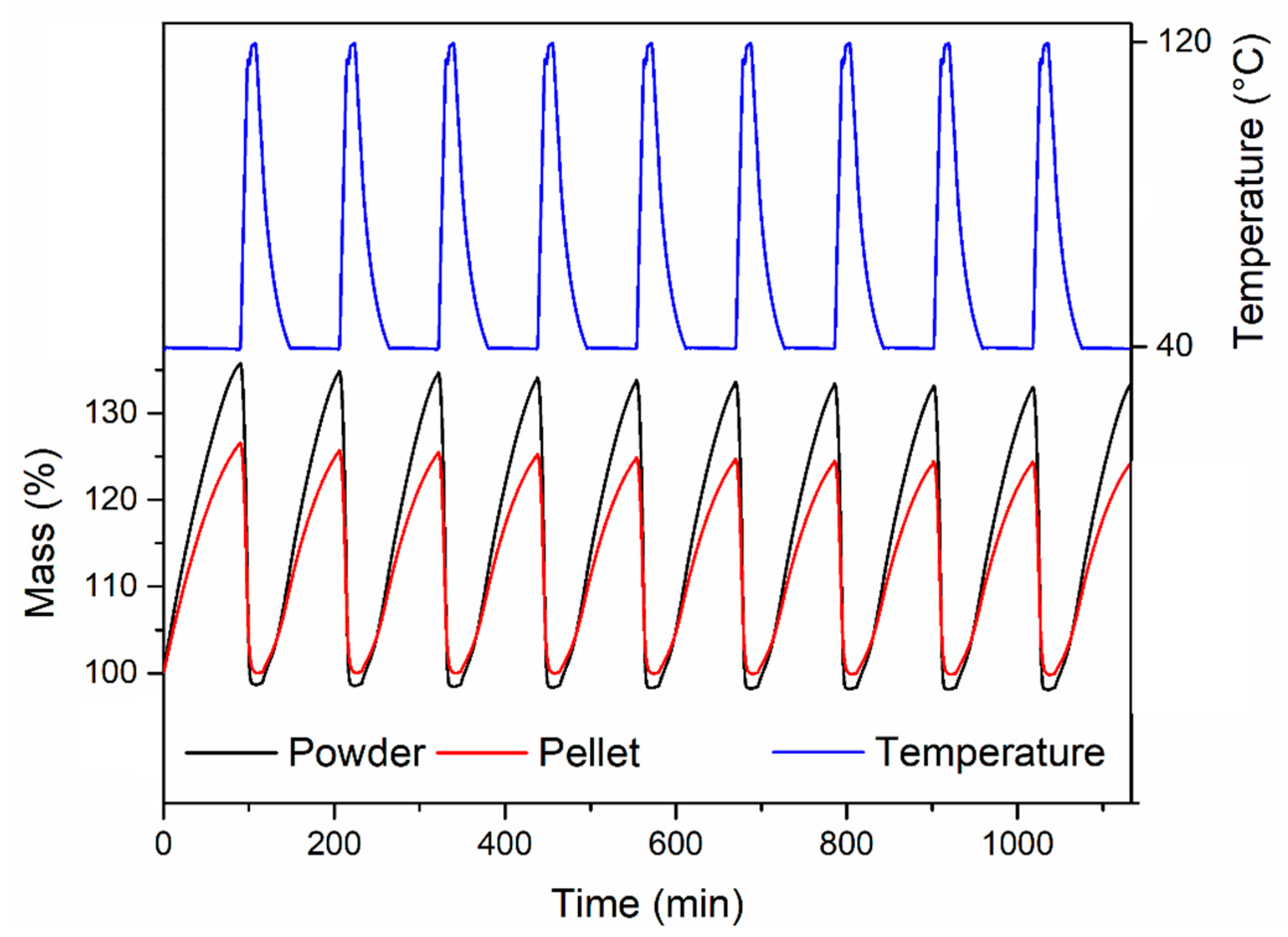
Finally, we compared the cyclic H
2O adsorption behaviour, exposing the powder and the pellet compressed at 146 MPa to a stream of humid air at 40 °C, regenerating by heating under Ar at 120 °C (
Figure 12). The powder displayed a constant working capacity of 35.5% over ten cycles. The pellet displayed a lower initial working capacity of 26.4% (corresponding to 74% of the working capacity of the powder), which slightly reduced to 24.1% over ten cycles.
3. Materials and Methods
3.1. Chemicals
Zirconium(IV) chloride (98%, anhydrous, Acros Organics, Geel, Belgium), formic acid (≥98%, Sigma Aldrich, Darmstadt, Germany), fumaric acid (≥99.0%, Sigma Aldrich), Polyvinyl butyral (Sigma-Aldrich), Poly(vinyl alcohol) (Mw 31,000–50,000, 98–99% hydrolysed, Sigma Aldrich), Sucrose (Sigma-Aldrich, ≥99.5%).
3.2. Synthesis of MOF-801
In a 500-mL round-bottom flask, water (377 mL) and formic acid (78 mL, 2.1 mol) were combined with stirring. To this mixture, zirconium chloride (4.8 g, 2.1 mmol) was added in portions and the solution stirred until clear. Fumaric acid (7.2 g, 6.2 mmol) was then added and the solution stirred until clear. The mixture was then heated to reflux and allowed to reflux for 72 h. Then, the solution was cooled and the solid recovered by dividing the reaction mixture into centrifuge tubes and centrifuging (5000 rpm, 5 min) until supernatant solution was clear. The solid was then redispersed in 50:50 water:ethanol mixture (50 mL per tube) while being agitated with a tilt table for 1 h. This process was repeated once and then the solid was dispersed in acetone (50 mL per tube) and agitated in the same manner for 1 h, repeating once. The solid was then allowed to dry at 65 °C overnight. Yield: 4.0 g.
3.3. Pellet Preparation
A Retsch PP25 pellet press was employed for compaction of MOF-801 powders. For pellets of 16 mm diameter, either 400 mg of neat MOF powder or 380 mg of MOF powder blended in an agate mortar with 20 mg of binder was introduced in the die and compressed under 3, 6, 9 or 12 metric tonnes (corresponding to pressures of 146, 292, 438 or 524 MPa, respectively) for 15 s, 30 s, 2 min, 5 min, 10 min or 15 min. For pellets with a 5-mm diameter, 190 mg of MOF powder blended in an agate mortar with 10 mg of binder was split into three equal parts and compressed under 0.3 metric tonnes (corresponding to a pressure of 146 MPa) for 15 s. The pellets were then treated at 120 °C for 2 h and exposed to air for 24 h before characterisation and testing.
3.4. Durability Tests
The durability of pellets was tested by repeatedly dropping them inside a 250-mL measuring cylinder having height of 36 cm. After each drop, the undamaged pellets were recovered by carefully turning the cylinder upside down and letting the pellet fall on a soft surface. Attrition resistance was tested by introducing about ¼ of a pellet in a 20 mL glass vial equipped with a screw cap and using a Hati Rotamixer 50 Hz vortex mixer at max power (200 W) in cycles of 30 s. After each cycle, the pellet fragment was recovered with tweezers and weighed to determine the mass loss due to attrition.
3.5. Analytical Procedures
Powder X-ray diffraction (PXRD) patterns were collected in the 4–30° 2θ range with a Bruker D8 Avance diffractometer working in reflection geometry and equipped with a LYNXEYE XE detector, using the Cu Kα radiation (Bruker, Karlsruhe, Germany). The X-ray tube was operated at 40 kV and 40 mA.
Scanning electron microscopy (SEM) micrographs of pellets were acquired with a Hitachi TM3030Plus microscope using an acceleration voltage of 5 kV (Hitachi High-Technologies, Krefeld, Germany). The samples were analysed uncoated.
N2 sorption isotherms at 77 K were measured with a Quantachrome Nova 2000e analyzer (Quantachrome, Boynton Beach, FL, USA). The samples (about 30–50 mg) were activated for four hours under vacuum at 120 °C prior to analysis. BET surface areas were calculated in the 0.001–0.043 P/P0 range.
3.6. CO2 and H2O Adsorption Tests
Dynamic CO2 adsorption experiments and cyclic CO2 and H2O adsorption experiments were performed with a TA Instruments SDT-Q600 instrument (TA Instruments, New Castle, DE, USA). The samples were loaded in alumina cups and activated at 120 °C under a 100 mL min−1 Ar flow for 2 h. The temperature was then decreased to 40 °C and kept at this value for 30 min.
In dynamic CO2 adsorption experiments, the gas flow was switched to CO2 at the same flow rate and adsorption started with a little delay. In the CO2 adsorption-desorption experiments, CO2 was constantly flown at 100 mL min−1 and the temperature was swung between 40 °C and 90 °C. Each adsorption cycle lasted 40 min, whereas desorption lasted 10 min.
In H2O adsorption-desorption experiments, a humidified air stream was achieved by bubbling dry air at 80 mL min−1 in H2O at room temperature. This stream was flown during adsorption, which took place at 40 °C, whereas desorption was performed by heating to 120 °C under a 100-mL min−1 Ar flow. Each adsorption cycle lasted 90 min, whereas desorption took 10 min.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}