
The Synthesis of Tetrakis(N,N-dimethylaminomethyl)ferrocene and Its Bimetallic Nickel(II) Dichloride Complex: The Search for Precursors for Methoxycarbonylation Ligands

 ,
,  , , , and
, , , and
Abstract
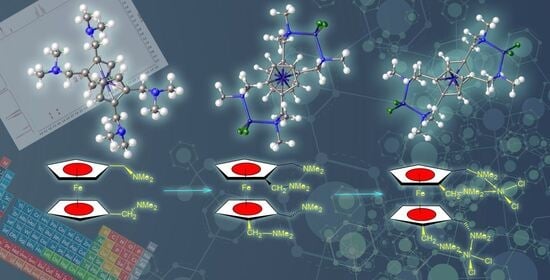
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
Additional Information and Future Pointers
3. Materials and Methods
3.1. 1,2-Bis-(N,N-Dimethylaminomethyl)ferrocene (6) [44,45]
3.2. 1,1′-Bis-(N,N-Dimethylaminomethyl)-ferrocene 5 (Prepared from 1,1′-Dilithioferrocene)
3.3. 1,2,1′,2′-Tetrakis(N,N-dimethylaminomethyl)ferrocene 15 (Original Method)
3.4. Preparation of Nickel Complex of 1,2,1′,2′-Tetra-(N,N-dimethylaminomethyl)ferrocene
3.5. Alternative Synthetic Method for Compound 15
3.6. General Synthetic Note
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jaouen, G.; Vessières, A.; Top, S. Ferrocifen type anti-cancer drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. [Google Scholar] [CrossRef]
- Neuse, E.W. Macromolecular ferrocene compounds as cancer drug models. J. Inorg. Organomet. Polym. Mater. 2005, 215, 3–31. [Google Scholar] [CrossRef]
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic Anticancer Compounds. J. Med. Chem. 2011, 54, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Gielen, M.; Tiekink, E.R. (Eds.) Metallotherapeutic Drugs and Metal-Based Diagnostic Agents: The Use of Metals in Medicine; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar] [CrossRef]
- Ornelas, C. Application of ferrocene and its derivatives in cancer research. New J. Chem. 2011, 35, 1973–1985. [Google Scholar] [CrossRef]
- Paprocka, R.; Wiese-Szadkowska, M.; Janciauskiene, S.; Kosmalski, T.; Kulik, M.; Helmin-Basa, A. Latest developments in metal complexes as anticancer agents. Coord. Chem. Rev. 2022, 452, 214307. [Google Scholar] [CrossRef]
- Meléndez, E. Metallocenes as Target Specific Drugs for Cancer Treatment. Inorg. Chim. Acta 2012, 393, 36–52. [Google Scholar] [CrossRef][Green Version]
- Dombrowski, K.E.; Baldwin, W.; Sheats, J.E. Metallocenes in biochemistry, microbiology & medicine. J. Organomet. Chem. 2020, 905, 281–306. [Google Scholar] [CrossRef]
- Sansook, S.; Storm, H.-H.; Ocasio, C.; Spencer, J. Ferrocenes in medicinal chemistry; a personal perspective. J. Organomet. Chem. 2020, 905, 121017. [Google Scholar] [CrossRef]
- Peter, S.; Aderibigbe, B.A. Ferrocene-Based Compounds with Antimalaria/Anticancer Activity. Molecules 2019, 24, 3604. [Google Scholar] [CrossRef]
- Blackie, M.A.L.; Chibale, K. Metallocene antimalarials: The continuing quest. In Metal-Based Drugs; Metal-Containing Proteins, Macrocycles, and Coordination Complexes in Therapeutic Applications and Disease; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar] [CrossRef]
- Anthony, M.P.; Burrows, J.N.; Duparc, S.; Moehrle, J.; Wells, T.N. The global pipeline of new medicines for the control and elimination of malaria. Malar. J. 2012, 11, 316. [Google Scholar] [CrossRef]
- Evans, D.M.; Hughes, D.D.; Murphy, P.J.; Horton, P.N.; Coles, S.J.; de Biani, F.F.; Corsini, M.; Butler, I.R. Synthetic Route to 1, 1′,2,2′-Tetraiodoferrocene That Avoids Isomerization and the Electrochemistry of Some Tetrahaloferrocenes. Organometallics 2021, 40, 2496–2503. [Google Scholar] [CrossRef]
- Butler, I.R.; Beaumont, M.; Bruce, M.I.; Zaitseva, N.N.; Iggo, J.A.; Robertson, C.; Horton, P.N.; Coles, S.J. Synthesis and Structures of 1, 1′, 2-Tribromoferrocene, 1, 1′, 2, 2′-Tetrabromoferrocene, 1,1′,2,2′-Tetrabromoruthenocene: Expanding the Range of Precursors for the Metallocene Chemist’s Toolkit. Aust. J. Chem. 2020, 74, 204–210. [Google Scholar] [CrossRef]
- Butler, I.R. Sitting Out the Halogen Dance. Room-Temperature Formation of 2,2′-Dilithio-1,1′-dibromoferrocene. TMEDA and Related Lithium Complexes: A Synthetic Route to Multiply Substituted Ferrocenes. Organometallics 2021, 40, 3240–3244. [Google Scholar] [CrossRef]
- Butler, I.R.; Evans, D.M.; Horton, P.N.; Coles, S.J.; Murphy, P.J. 1, 1′,2,2′-Tetralithioferrocene and 1,1′ 2,2′ 3 3′-Hexalithioferrocene: Useful Additions to Ferrocene Precursor Compounds. Organometallics 2021, 40, 600–605. [Google Scholar] [CrossRef]
- Horton, P.N.; Coles, S.J.; Clegg, W.; Harrington, R.W.; Butler, I.R. A Rapid General Synthesis and the Spectroscopic Data of 2,2′-Bis-(di-isopropylphosphino)-1, 1′-dibromoferrocene,(bpdbf), 1,1′,2,2′-Tetrakis-(di-isopropylphosphino) Ferrocene, (tdipf) and Related Ligands: Taking dppf into the Future. Inorganics 2025, 13, 10. [Google Scholar] [CrossRef]
- Lindsay, J.K.; Hauser, C.R. Aminomethylation of Ferrocene to Form N, N-Dimethylaminomethylferrocene and Its Conversion to the Corresponding Alcohol and Aldehyde. J. Org. Chem. 1957, 22, 355–358. [Google Scholar] [CrossRef]
- Lednicer, D.; Hauser, C.R. N, N-Dimethylaminomethylferrocene Methiodide: Iron, cyclopentadienyl [(dimethylaminomethyl) cyclopentadienyl]-, methiodid. Org. Synth. 2003, 40, 31. [Google Scholar] [CrossRef]
- Gokel, G.W.; Ugi, I.K. Preparation and resolution of N, N-dimethyl-o-ferrocenylethylamine. An advanced organic experiment. J. Chem. Ed. 1972, 49, 294. [Google Scholar] [CrossRef]
- Herrmann, R.; Hübener, G.; Ugi, I. Chiral sulfoxides from (R)-α-dimethylaminoethylferrocene. Tetrahedron 1985, 41, 941–947. [Google Scholar] [CrossRef]
- Jain, S.C.; Rivest, R. Coordination complexes of some group (IV) halides: Preparation and ir spectra of the complexes of group (IV) halides with ferrocene acetonitrile and N, N-dimethylaminomethylferrocene as ligands. J. Inorg. Nucl. Chem. 1970, 32, 1579–1584. [Google Scholar] [CrossRef]
- Grelaud, G.; Roisnel, T.; Dorcet, V.; Humphrey, M.G.; Paul, F.; Argouarch, G. Synthesis, reactivity, and some photochemistry of ortho-N, N-dimethylaminomethyl substituted aryl and ferrocenyl pentamethylcyclopentadienyl dicarbonyl iron complexes. J. Organomet. Chem. 2013, 741, 47–58. [Google Scholar] [CrossRef]
- Picart-Goetgheluck, S.; Delacroix, O.; Maciejewski, L.; Brocard, J. High yield synthesis of 2-substituted (N, N-dimethylaminomethyl) ferrocenes. Synthesis 2000, 10, 1421–1426. [Google Scholar] [CrossRef]
- Butler, I.R.; Cullen, W.R. The synthesis of primary, secondary, and tertiary ferrocenylethylamines. Can. J. Chem. 1983, 61, 2354–2358. [Google Scholar] [CrossRef]
- Butler, I.R.; Cullen, W.R.; Herring, F.G.; Jagannathan, N.R. α-N, N-Dimethylaminoethylferrocene. A nuclear magnetic resonance study relating to stereoselective metalation. Can. J. Chem. 1986, 64, 667–669. [Google Scholar] [CrossRef]
- Colbert, M.C.; Lewis, J.; Long, N.J.; Raithby, P.R.; Bloor, D.A.; Cross, G.H. The synthesis of chiral ferrocene ligands and their metal complexes. J. Organomet. Chem. 1997, 531, 183–190. [Google Scholar] [CrossRef]
- Blaser, H.U.; Brieden, W.; Pugin, B.; Spindler, F.; Studer, M.; Togni, A. Solvias Josiphos ligands: From discovery to technical applications. Top. Catal. 2002, 19, 3–16. [Google Scholar] [CrossRef]
- Blaser, H.U.; Pugin, B.; Spindler, F.; Mejía, E.; Togni, A. Josiphos ligands: From discovery to technical applications. In Privileged Chiral Ligands and Catalysts; Wiley: Hoboken, NJ, USA, 2011; pp. 93–136. [Google Scholar] [CrossRef]
- Treiber, M.; Wernsdorfer, G.; Wiedermann, U.; Congpuong, K.; Sirichaisinthop, J.; Wernsdorfer, W.H. Sensibilität von Plasmodium vivax gegenüber Chloroquin, Mefloquin, Artemisinin und Atovaquon im Nordwesten Thailands. Wien. Klin. Wochenschr. 2011, 123, 20–25. [Google Scholar] [CrossRef]
- Bray, P.G.; Martin, R.E.; Tilley, L.; Ward, S.A.; Kirk, K.; Fidock, D.A. Defining the role of PfCRT in Plasmodium falciparum chloroquine resistance. Mol. Microbiol. 2005, 56, 323–333. [Google Scholar] [CrossRef]
- Held, J.; Supan, C.; Salazar, C.L.; Tinto, H.; Bonkian, L.N.; Nahum, A.; Moulero, B.; Sié, A.; Coulibaly, B.; Sirima, S.B.; et al. Ferroquine and artesunate in African adults and children with Plasmodium falciparum malaria: A phase 2, multicentre, randomised, double-blind, dose-ranging, non-inferiority study. Lancet Infect. Dis. 2015, 15, 1409–1419. [Google Scholar] [CrossRef]
- Ecker, A.; Lehane, A.M.; Clain, J.; Fidock, D.A. PfCRT and its role in antimalarial drug resistance. Trends Parasitol. 2012, 28, 504–514. [Google Scholar] [CrossRef]
- Biot, C.; Castro, W.; Botté, C.Y.; Navarro, M. The therapeutic potential of metal-based antimalarial agents: Implications for the mechanism of action. Dalton Trans. 2012, 41, 6335–6349. [Google Scholar] [CrossRef]
- Dive, D.; Biot, C. Ferrocene conjugates of chloroquine and other antimalarials: The development of ferroquine, a new antimalarial. ChemMedChem 2007, 3, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Biot, C.; Taramelli, D.; Forfar-Bares, I.; Maciejewski, L.A.; Boyce, M.; Nowogrocki, G.; Brocard, J.S.; Basilico, N.; Olliaro, P.; Egan, T.J. Insights into the mechanism of action of ferroquine. Relationship between physicochemical properties and antiplasmodial activity. Mol. Pharm. 2005, 2, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Faustine, D.; Sylvain Bohic, S.; Slomianny, C.; Morin, J.-C.; Thomas, P.; Kalamou, H.; Guérardel, Y.; Cloetens, P.; Jamal Khalife, J.; Biot, C. In situ nanochemical imaging of label-free drugs: A case study of antimalarials in Plasmodium falciparum-infected erythrocytes. Chem. Commun. 2012, 48, 910–912. [Google Scholar] [CrossRef]
- Biot, C.; Nosten, F.; Fraisse, L.; Ter-Minassian, D.; Khalife, J.; Dive, D. The antimalarial ferroquine: From bench to clinic. Parasite J. Société Française Parasitol. 2011, 18, 207–214. [Google Scholar] [CrossRef]
- Delhaes, L.; Biot, C.; Berry, L.; Delcourt, P.; Maciejewski, L.A.; Camus, D.; Brocard, J.S.; Dive, D. Synthesis of ferroquine enantiomers: First investigation of effects of metallocenic chirality upon antimalarial activity and cytotoxicity. ChemBioChem 2002, 3, 418–423. [Google Scholar] [CrossRef]
- Keiser, J.; Vargas, M.; Rubbiani, R.; Gasser, G.; Biot, C. In vitro and in vivo antischistosomal activity of ferroquine derivatives. Parasites Vectors 2014, 7, 424. [Google Scholar] [CrossRef]
- Wani, W.A.; Jameel, E.; Baig, U.; Mumtazuddin, S.; Hun, L.T. Ferroquine and its derivatives: New generation of antimalarial agents. Eur. J. Med. Chem. 2015, 101, 534–551. [Google Scholar] [CrossRef]
- Meo, S.A.; Klonoff, D.C.; Akram, J. Efficacy of chloroquine and hydroxychloroquine in the treatment of COVID-19. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4539–4547. Available online: https://www.talkingaboutthescience.com/studies/HCQ/Meo2020.pdf (accessed on 11 January 2026).
- Gasmi, A.; Peana, M.; Noor, S.; Lysiuk, R.; Menzel, A.; Gasmi Benahmed, A.; Bjørklund, G. Chloroquine and hydroxychloroquine in the treatment of COVID-19: The never-ending story. Appl. Microbiol. Biotech. 2021, 105, 1333–1343. [Google Scholar] [CrossRef]
- Butler, I.R.; Baker, P.K.; Eastham, G.R.; Fortune, K.M.; Horton, P.N.; Hursthouse, M.B. Ferrocenylmethylphosphines ligands in the palladium-catalysed synthesis of methyl propionate. Inorg. Chem. Commun. 2004, 7, 1049–1052. [Google Scholar] [CrossRef]
- Fortune, K.M.; Castel, C.; Robertson, C.M.; Horton, P.N.; Light, M.E.; Coles, S.J.; Waugh, M.; Clegg, W.; Harrington, R.W.; Butler, I.R. Ferrocenylmethylphosphanes and the Alpha Process for Methoxycarbonylation: The Original Story. Inorganics 2021, 9, 57. [Google Scholar] [CrossRef]
- Morris, K. An Investigation into the Synthesis of Phosphine-Based Ligands and Their Application in Pd-Catalysed Processes in the Production of Polymethylmethacrylate. Ph.D. Thesis, Bangor University, Bangor, UK, 2008. [Google Scholar]
- Glidewell, C.; Royles, B.J.; Smith, D.M. A simple high-yielding synthesis of ferrocene-1,1′-diylbis-(methyltrimethylammonium iodide). J. Organomet. Chem. 1997, 527, 259–261. [Google Scholar] [CrossRef]
- Fortune, K.M. Nitrogen Donor Complexes of Molybdenum and Tungsten and New Routes to bis-1,2 & tris-1,2,3 Substituted Ferrocenes. Ph.D. Thesis, Bangor University, Gwynedd, UK, 2004. [Google Scholar]
- Butler, I.R.; Horton, P.N.; Fortune, K.M.; Morris, K.; Greenwell, C.H.; Eastham, G.R.; Hursthouse, M.B. The first 1, 2, 3-tris (phosphinomethyl) ferrocene. Inorg. Chem. Commun. 2004, 7, 923–928. [Google Scholar] [CrossRef]
- Rausch, M.D.; Ciappenelli, D.J. Organometallic π-complexes XII. The metalation of benzene and ferrocene by n-butyllithium-N, N, N′, N′-tetramethylethylenediamine. J. Organomet. Chem. 1967, 10, 127–136. [Google Scholar] [CrossRef]
- Meijboom, R.; Beagley, P.; Moss, J.R.; Roodt, A. Lithiated dimethylaminomethyl ferrocenes and ruthenocenes. J. Organomet. Chem. 2006, 691, 916–920. [Google Scholar] [CrossRef]
- Bolton, E.S.; Pauson, P.L.; Sandhu, M.D.; Watts, W.E. Ferrocene derivatives. Part XXI. Lithiation of 1,1′-bis-(NN-dimethylaminomethyl)ferrocene. J. Chem. Soc. C Org. 1969, 2260–2263. [Google Scholar] [CrossRef]
- Butler, I.R.; Cullen, W.R.; Rettig, S.J. Synthesis of derivatives of [.alpha. (dimethylamino) ethyl] ferrocene via lithiation reactions and the structure of 2-[.alpha.-(dimethylamino) ethyl]-1, 1’, 3-tris (trimethylsilyl) ferrocene. Organometallics 1986, 5, 1320–1328. [Google Scholar] [CrossRef]
- Butler, I.R.; Cullen, W.R.; Reglinski, J.; Rettig, S.J. Ferrocenyllithium derivatives: Lithiation of α-N, N-deimethylaminoethylferrocene and the single crystal X-ray structure of [(η5-C5H4Li) Fe (η5-C5H3LiCH (Me) NMe2)] 4 [LiOEt] 2 (TMED) 2. J. Organomet. Chem. 1983, 249, 183–194. [Google Scholar] [CrossRef]
- Steffen, P.; Unkelbach, C.; Christmann, M.; Hiller, W.; Strohmann, C. Catalytic and stereoselective ortho-lithiation of a ferrocene derivative. Angew. Chem. Int. Ed. 2013, 52, 9836–9840. [Google Scholar] [CrossRef]
- Krupp, A.; Wegge, J.; Otte, F.; Kleinheider, J.; Wall, H.; Strohmann, C. Crystal structures of [(N,N-dimethylamino) methyl] ferrocene and (Rp, Rp)-bis {2-[(dimethylamino) methyl] ferrocenyl} dimethylsilane. Acta Crystallogr. Sect. E Crystallogr. Commun. 2020, 76, 1437–1441. [Google Scholar] [CrossRef]
- Klotz, F.D.; Felipe, C.A.; Villafañe, F.; Strohmann, C. Synthesis and crystal structure study of (R,R)-TMCDA ethanol derivatives doubly protonated with FeCl4− and Cl− as counter-ions. Acta Crystallogr. 2025, E81, 372–376. [Google Scholar] [CrossRef]
- Butler, I.R.; Williams, R.M.; Heeroma, A.; Horton, P.N.; Coles, S.J.; Jones, L.F. The Effect of Localized Magnetic Fields on the Spatially Controlled Crystallization of Transition Metal Complexes. Inorganics 2025, 13, 117. [Google Scholar] [CrossRef]
- Boussandel, S.; Erb, W.; Roisnel, T.; Blot, M.; Hurvois, J.-P.; Butler, I.; Samarat, A.; Mongin, F. Hexafluoroisopropanol-Promoted Substitution Toward the Synthesis of Enantiopure Ferrocene Phosphines. Synthesis 2025, 57, 3211–3226. [Google Scholar] [CrossRef]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Butler, I.R.; Horton, P.N.; Clegg, W.; Coles, S.J.; Murphy, L.; Elliott, S. The Synthesis of Tetrakis(N,N-dimethylaminomethyl)ferrocene and Its Bimetallic Nickel(II) Dichloride Complex: The Search for Precursors for Methoxycarbonylation Ligands. Inorganics 2026, 14, 37. https://doi.org/10.3390/inorganics14020037
Butler IR, Horton PN, Clegg W, Coles SJ, Murphy L, Elliott S. The Synthesis of Tetrakis(N,N-dimethylaminomethyl)ferrocene and Its Bimetallic Nickel(II) Dichloride Complex: The Search for Precursors for Methoxycarbonylation Ligands. Inorganics. 2026; 14(2):37. https://doi.org/10.3390/inorganics14020037
Chicago/Turabian StyleButler, Ian R., Peter N. Horton, William Clegg, Simon J. Coles, Loretta Murphy, and Steven Elliott. 2026. "The Synthesis of Tetrakis(N,N-dimethylaminomethyl)ferrocene and Its Bimetallic Nickel(II) Dichloride Complex: The Search for Precursors for Methoxycarbonylation Ligands" Inorganics 14, no. 2: 37. https://doi.org/10.3390/inorganics14020037
APA StyleButler, I. R., Horton, P. N., Clegg, W., Coles, S. J., Murphy, L., & Elliott, S. (2026). The Synthesis of Tetrakis(N,N-dimethylaminomethyl)ferrocene and Its Bimetallic Nickel(II) Dichloride Complex: The Search for Precursors for Methoxycarbonylation Ligands. Inorganics, 14(2), 37. https://doi.org/10.3390/inorganics14020037