

Cu@Pt Core–Shell Nanostructures for Ammonia Oxidation: Bridging Electrocatalysis and Electrochemical Sensing



Abstract
1. Introduction
2. Results and Discussion
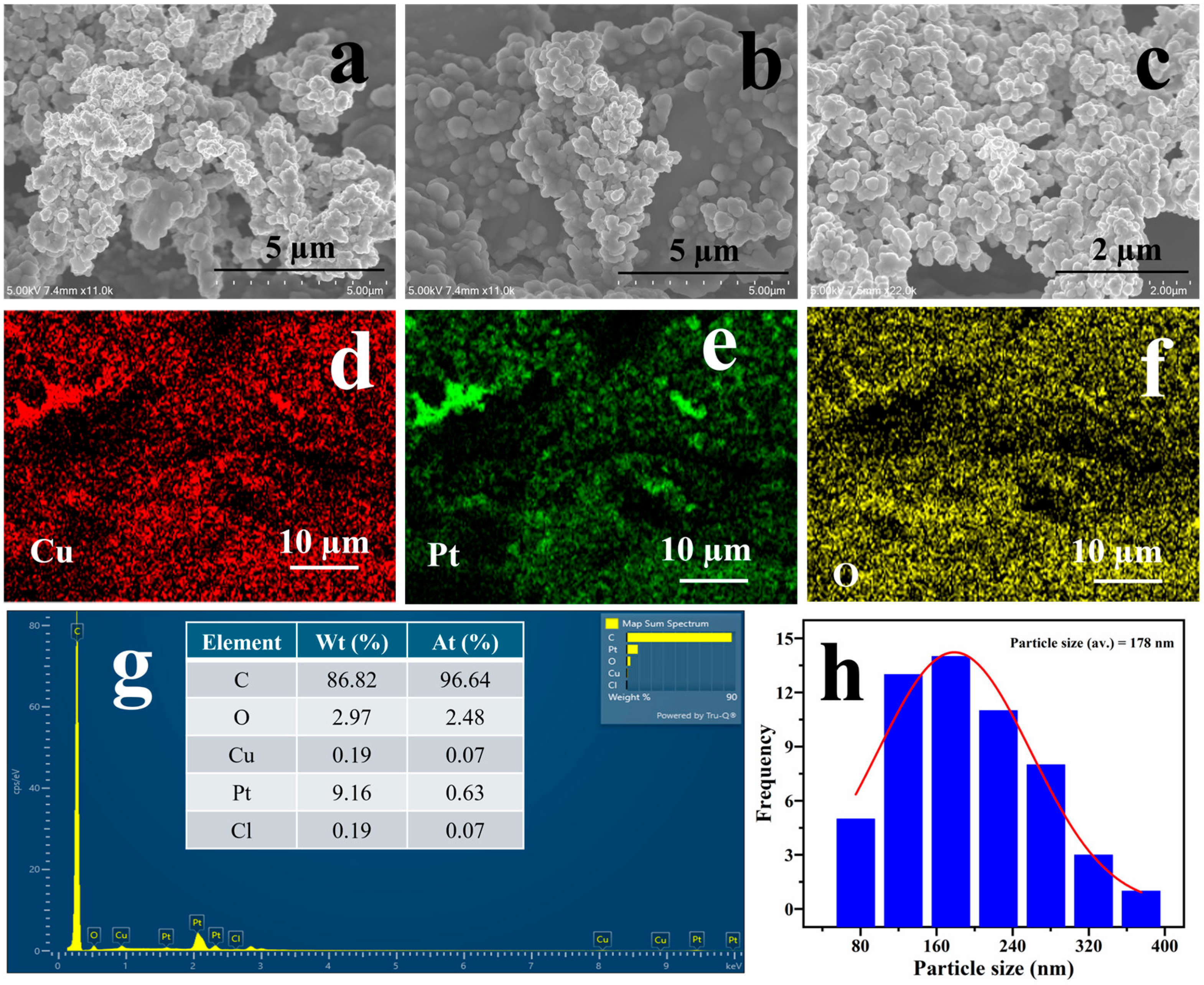
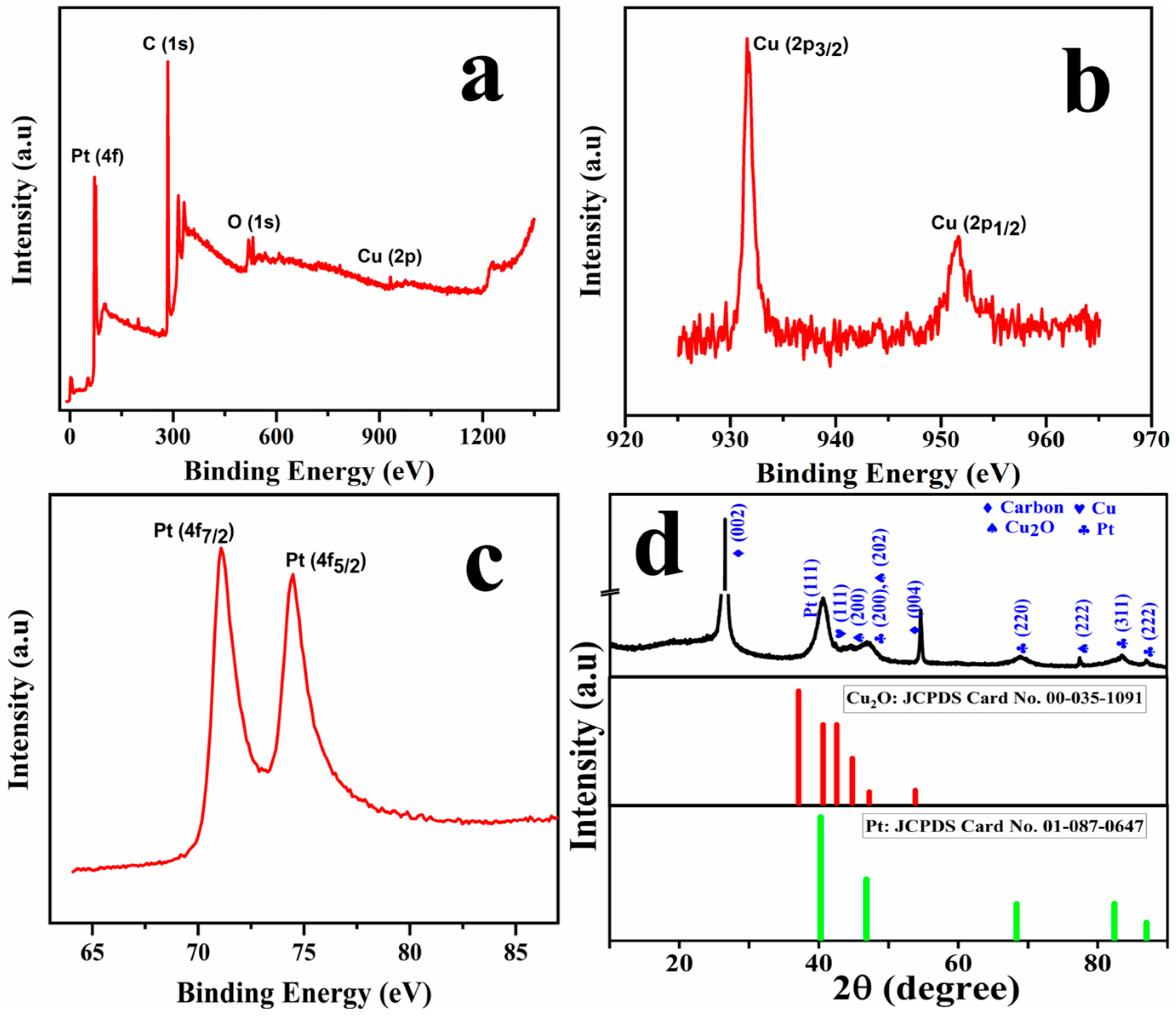
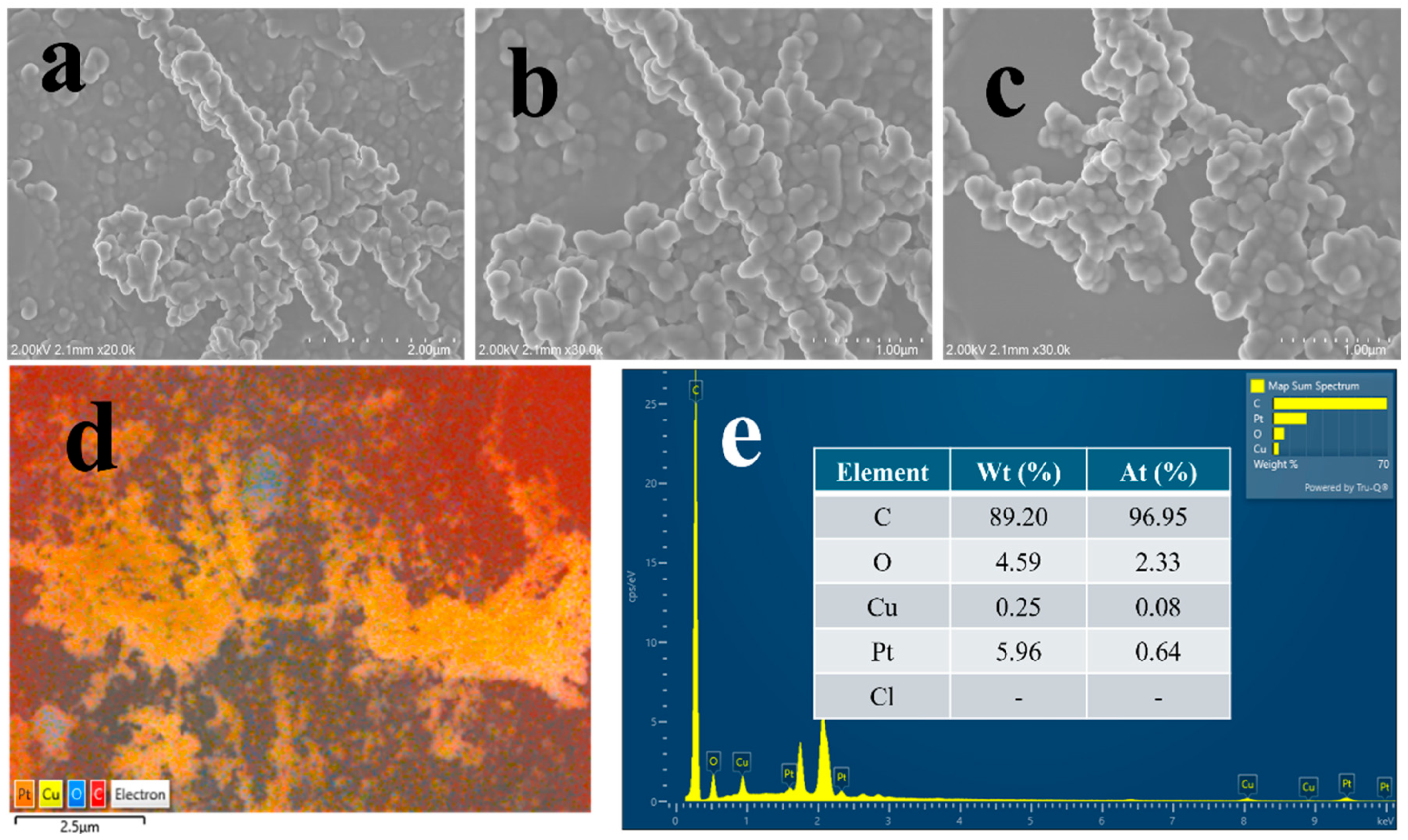
2.1. Physical Characterisation
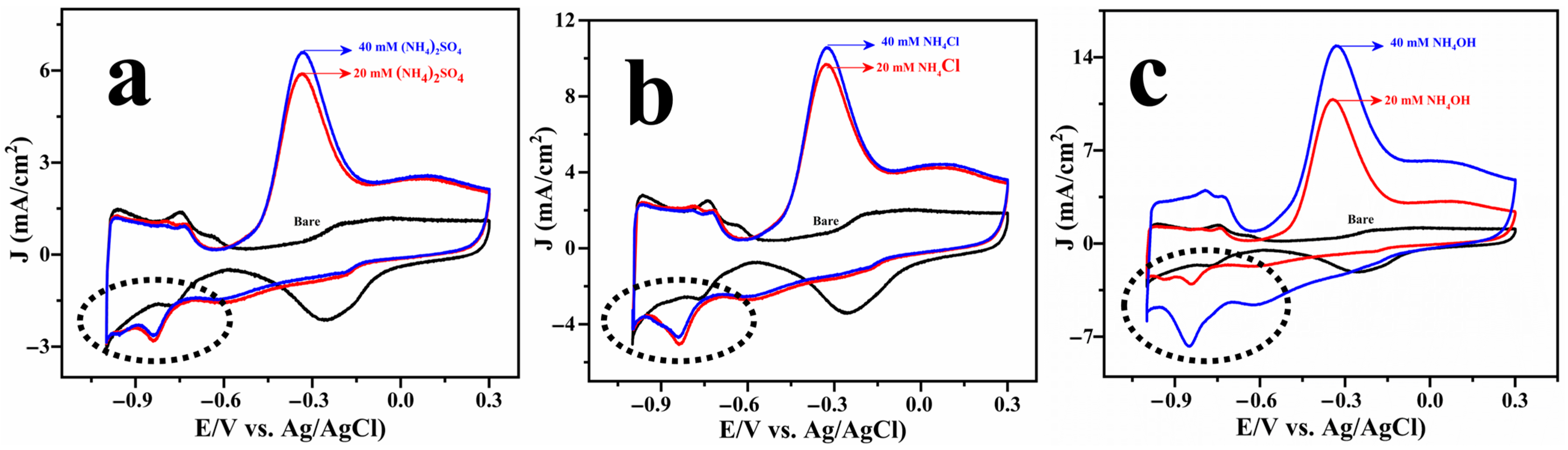
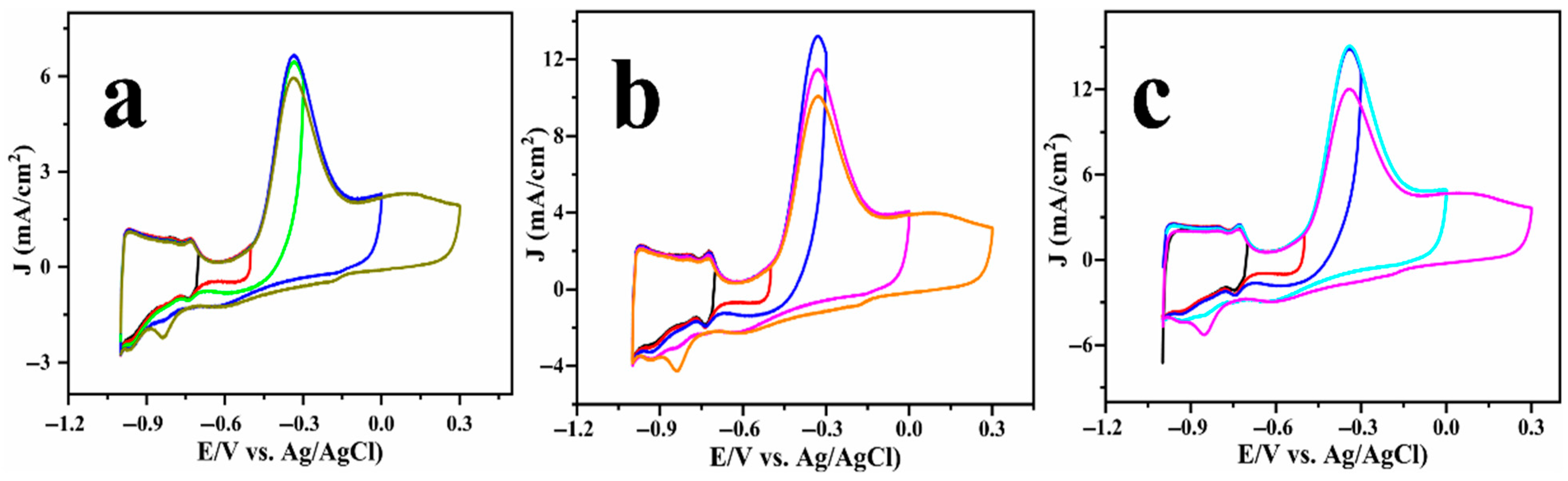
2.2. Electrocatalysis of Ammonia
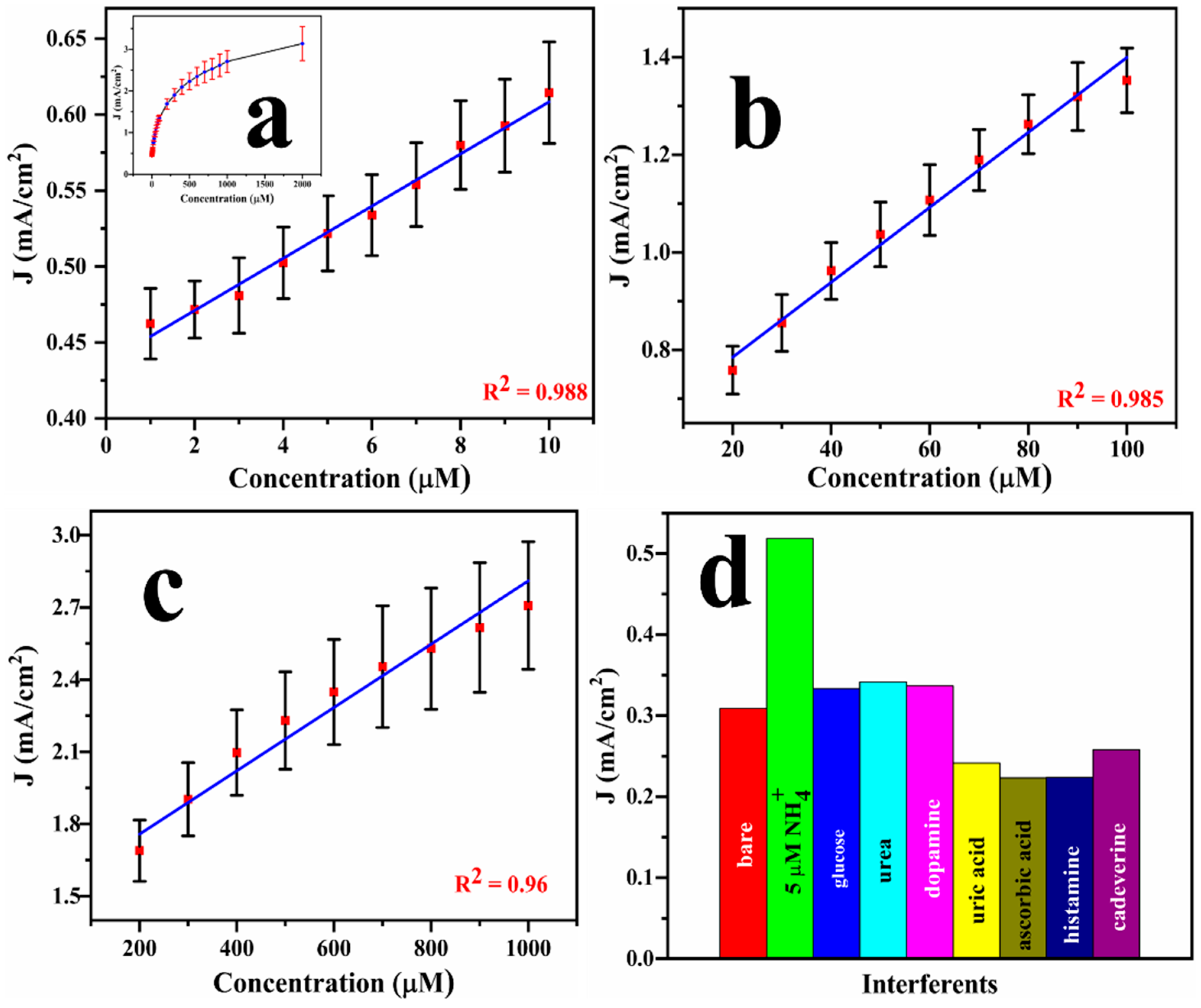
2.3. Electroanalysis of Ammonia
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, Z.; Xi, L.; Wang, Y.; Zhao, X. Chronic Exposure to Environmental Pollutant Ammonia Causes Damage to the Olfactory System and Behavioral Abnormalities in Mice. Environ. Sci. Technol. 2023, 57, 15412–15421. [Google Scholar] [CrossRef] [PubMed]
- Nayak-Luke, R.M.; Hatton, L.; Cesaro, Z.; Banares-Alcantara, R. Assessing the Viability of Decarbonising India’s Nitrogenous Fertiliser Consumption. J. Clean. Prod. 2022, 366, 132462. [Google Scholar] [CrossRef]
- Li, M.; Beko, G.; Wargocki, P.; Lucic, G.; Williams, J. Human Ammonia Emission Rates under Various Indoor Environmental Conditions. Environ. Sci. Technol. 2020, 54, 5419–5428. [Google Scholar] [CrossRef]
- Zhang, T.-X.; Li, M.-R.; Liu, C.; Wang, S.-P.; Yan, Z.-G. A Review of the Toxic Effects of Ammonia on Invertebrates in Aquatic Environments. Environ. Pollut. 2023, 336, 122374. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, G.; Qi, L.; Wang, H.; Xian, G. Chlorine-Mediated Electrochemical Advanced Oxidation Process for Ammonia Removal: Mechanisms, Characteristics and Expectation. Sci. Total Environ. 2023, 896, 165169. [Google Scholar] [CrossRef]
- Munir, N.; Javaid, A.; Abideen, Z.; Jarar, H.; El-Keblawy, A.; Sheteiwy, M.S. The Potential of Zeolite Nanocomposites in Removing Microplastics, Ammonia, and Trace Metals from Wastewater and Their Role in Phytoremediation. Environ. Sci. Pollut. Res. 2023, 31, 1695–1718. [Google Scholar] [CrossRef]
- Cao, Q.; Li, X.; Li, N.; Li, Z.; Liu, X.; Li, D. Ammonia Removal through Combined Methane Oxidation and Nitrification-Denitrification and the Interactions among Functional Microorganisms. Water Res. 2021, 188, 116555. [Google Scholar] [CrossRef]
- Hassan, N.S.; Jalil, A.A.; Saravanan, R.; Izzuddin, N.M.; Bahari, M.B.; Prasetyoko, D.; Nugraha, R.E. Recent Progress on Electrocatalysts in Ammonia Electrooxidation Reaction for Clean Hydrogen Production. J. Mater. Chem. A 2024, 12, 23202–23217. [Google Scholar] [CrossRef]
- Cechanaviciute, I.A.; Schuhmann, W. Electrocatalytic Ammonia Oxidation Reaction: Selective Formation of Nitrite and Nitrate as Value-Added Products. ChemSusChem 2025, 18, e202402516. [Google Scholar] [CrossRef]
- Kostopoulos, N.; Robert, M.; Sekretareva, A. The Potential of Molecular Electrocatalysis for Ammonia-to-Dinitrogen Conversion. ChemElectroChem 2024, 11, e202300462. [Google Scholar] [CrossRef]
- Shi, H.; Tang, J.; Yu, W.; Tadé, M.O.; Shao, Z. Advances in Power Generation from Ammonia via Electrocatalytic Oxidation in Direct Ammonia Fuel Cells. Chem. Eng. J. 2024, 488, 150896. [Google Scholar] [CrossRef]
- Liu, H.; Yang, C.; Shao, Z.; Huang, Z. Electrocatalytic Ammonia Oxidation to Nitrite and Nitrate with NiOOH-Ni. Adv. Energy Mater. 2024, 14, 2401675. [Google Scholar] [CrossRef]
- Naveen, B.; Reddy, B.P.; Palathedath, S.K. Dual Performing Copper–Platinum Core–Shell Nanozyme for Environmental Electrochemistry–Electrocatalytic Oxidation and Electroanalysis of Ammonia. Environ. Sci. Nano 2021, 8, 3603–3612. [Google Scholar] [CrossRef]
- Johnston, S.; Suryanto, B.H.R.; MacFarlane, D.R. Electro-Oxidation of Ammonia on Electrochemically Roughened Platinum Electrodes. Electrochim. Acta 2019, 297, 778–783. [Google Scholar] [CrossRef]
- Wallace, S.W.; McCrum, I.T.; Janik, M.J. Ammonia Electro-Oxidation Mechanism on the Platinum (100) Surface. Catal. Today 2021, 371, 50–57. [Google Scholar] [CrossRef]
- Paliwal, A.; Assaf, L.; Long, H.; Haasch, R.T.; Gupta, J.K.; Reynolds, M.A.; Son, Y.J.; Zhang, K.; Kenis, P.J.A.; Gewirth, A.A. Mechanistic Insights into the Enhanced Ammonia Oxidation Activity of PtRh Electrodeposits. ACS Appl. Mater. Interfaces 2025, 17, 3383–3392. [Google Scholar] [CrossRef]
- Tang, H.; Gu, Y.; Luo, J.; Zhang, W.; Zhang, J.; Zhou, Y. Controlled Synthesis of Bead-like Pt as an Efficient Electrocatalyst for Electrocatalytic Ammonia Oxidation. ChemistrySelect 2024, 9, e202400482. [Google Scholar] [CrossRef]
- Pillai, H.S.; Li, Y.; Wang, S.-H.; Omidvar, N.; Mu, Q.; Achenie, L.E.K.; Abild-Pedersen, F.; Yang, J.; Wu, G.; Xin, H. Author Correction: Interpretable Design of Ir-Free Trimetallic Electrocatalysts for Ammonia Oxidation with Graph Neural Networks. Nat. Commun. 2023, 14, 950. [Google Scholar] [CrossRef]
- Naveen, B.; Lee, S.-W. Template-Assisted Electrodeposited Copper Nanostructres for Selective Detection of Hydrogen Peroxide. Molecules 2024, 29, 4571. [Google Scholar] [CrossRef]
- Shen, Z.; Liu, D.; Peng, G.; Peng, J.; Ding, L. Electrocatalytic Reduction of Nitrate in Water Using Cu/Pd Modified Ni Foam Cathode: High Nitrate Removal Efficiency and N2-Selectivity. Sep. Purif. Technol. 2020, 241, 116743. [Google Scholar] [CrossRef]
- Pinon-Espitia, M.; Lardizabal-Gutierrez, D.; Camacho-Rios, M.L.; Herrera-Perez, G.; Ochoa-Lara, M.T. Electronic Structure Comparison of Cu 2p and O 1s X-Ray Photoelectron Spectra for CuxO Nanofibers (x = 1, 2, i). Mater. Chem. Phys. 2021, 272, 124981. [Google Scholar] [CrossRef]
- Liu, X.; Song, X.; Jiang, G.; He, Y.; Dong, F. Pt Single-Atom Collaborate with Pt Atom-Clusters by an In-Situ Confined Strategy for Accelerating Electrocatalytic Hydrogen Evolution. Chem. Eng. J. 2024, 481, 148430. [Google Scholar] [CrossRef]
- Chen, L.; Kang, L.; Song, S.; Jia, H.; Wang, Y. Rational Design of Ultrafine Pt Nanoparticles with Strong Metal-Support Interaction for Efficient Hydrogen Evolution. Adv. Funct. Mater. 2024, 34, 2403467. [Google Scholar] [CrossRef]
- Matsui, T.; Suzuki, S.; Katayama, Y.; Yamauchi, K.; Okanishi, T.; Muroyama, H.; Eguchi, K. In Situ Attenuated Total Reflection Infrared Spectroscopy on Electrochemical Ammonia Oxidation over Pt Electrode in Alkaline Aqueous Solutions. Langmuir 2015, 31, 11717–11723. [Google Scholar] [CrossRef]
- Li, Y.; Pillai, H.S.; Wang, T.; Hwang, S.; Su, D.; Xin, H.; Yan, Y.; Wu, G. High-Performance Ammonia Oxidation Catalysts for Anion-Exchange Membrane Direct Ammonia Fuel Cells. Energy Environ. Sci. 2021, 14, 1449–1460. [Google Scholar] [CrossRef]
- Oswin, H.G.; Salomon, M. The Anodic Oxidation of Ammonia at Platinum Black Electrodes in Aqueous Koh Electrolyte. Can. J. Chem. 1963, 41, 1686–1694. [Google Scholar] [CrossRef]
- Gerischer, H.; Mauerer, A. Untersuchungen Zur Anodischen Oxidation von Ammoniak an Platin-Elektroden. J. Electroanal. Chem. Interfacial Electrochem. 1970, 25, 421–433. [Google Scholar] [CrossRef]
- Wang, H.; Dekel, D.R.; Abruña, H.D. Unraveling the Mechanism of Ammonia Electrooxidation by Coupled Differential Electrochemical Mass Spectrometry and Surface-Enhanced Infrared Absorption Spectroscopic Studies. J. Am. Chem. Soc. 2024, 146, 15926–15940. [Google Scholar] [CrossRef]
- Endo, K.; Katayama, Y.; Miura, T. A Rotating Disk Electrode Study on the Ammonia Oxidation. Electrochim. Acta 2005, 50, 2181–2185. [Google Scholar] [CrossRef]
- Vidal-Iglesias, F.J.; Solla-Gullón, J.; Pérez, J.M.; Aldaz, A. Evidence by SERS of Azide Anion Participation in Ammonia Electrooxidation in Alkaline Medium on Nanostructured Pt Electrodes. Electrochem. Commun. 2006, 8, 102–106. [Google Scholar] [CrossRef]
- Jaksic, M.; Johansen, B.; Tunold, R. Electrochemical Behaviour of Platinum in Alkaline and Acidic Solutions of Heavy and Regular Water. Int. J. Hydrogen Energy 1993, 18, 817–837. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods Fundamentals and Applications, 2nd ed.; John Wiley and Sons: Noida, India, 2004. [Google Scholar]
- Nakata, K.; Okubo, A.; Shimazu, K.; Yamakata, A.; Ye, S.; Osawa, M. Surface-Enhanced Infrared Absorption Spectroscopic Studies of Adsorbed Nitrate, Nitric Oxide, and Related Compounds 1: Reduction of Adsorbed NO on a Platinum Electrode. Langmuir 2008, 24, 4352–4357. [Google Scholar] [CrossRef]
- Nakata, K.; Kayama, Y.; Shimazu, K.; Yamakata, A.; Ye, S.; Osawa, M. Surface-Enhanced Infrared Absorption Spectroscopic Studies of Adsorbed Nitrate, Nitric Oxide, and Related Compounds 2: Nitrate Ion Adsorption at a Platinum Electrode. Langmuir 2008, 24, 4358–4363. [Google Scholar] [CrossRef] [PubMed]
- Rima, F.R.; Nakata, K.; Shimazu, K.; Osawa, M. Surface-Enhanced Infrared Absorption Spectroscopic Studies of Adsorbed Nitrate, Nitric Oxide, and Related Compounds. 3. Formation and Reduction of Adsorbed Nitrite at a Platinum Electrode. J. Phys. Chem. C 2010, 114, 6011–6018. [Google Scholar] [CrossRef]
- Shinagawa, T.; Garcia-Esparza, A.T.; Takanabe, K. Insight on Tafel Slopes from a Microkinetic Analysis of Aqueous Electrocatalysis for Energy Conversion. Sci. Rep. 2015, 5, 13801. [Google Scholar] [CrossRef]
- van der Heijden, O.; Park, S.; Vos, R.E.; Eggebeen, J.J.J.; Koper, M.T.M. Tafel Slope Plot as a Tool to Analyze Electrocatalytic Reactions. ACS Energy Lett. 2024, 9, 1871–1879. [Google Scholar] [CrossRef]
- Asghari, S.; Mokmeli, A.; Samavati, M. Study of PEM Fuel Cell Performance by Electrochemical Impedance Spectroscopy. Int. J. Hydrogen Energy 2010, 35, 9283–9290. [Google Scholar] [CrossRef]
- Shen, J.; Wang, J. Analysis of Dc Link Oscillations in a Hybrid Fuel Cell Powertrain Brought by in Situ Converter Based Electrochemical Impedance Spectroscopy. Int. J. Hydrogen Energy 2020, 45, 31080–31090. [Google Scholar] [CrossRef]
- Adli, N.M.; Zhang, H.; Mukherjee, S.; Wu, G. Review—Ammonia Oxidation Electrocatalysis for Hydrogen Generation and Fuel Cells. J. Electrochem. Soc. 2018, 165, J3130–J3147. [Google Scholar] [CrossRef]
- Li, Y.; Pillai, H.S.; Lattimer, J.; Mohd Adli, N.; Karakalos, S.; Xin, H.; Wu, G. Ternary PtIrNi Catalysts for Efficient Electrochemical Ammonia Oxidation. ACS Catal. 2020, 10, 3945–3957. [Google Scholar] [CrossRef]
- Xu, W.; Walker, M.; Wu, Z.; Wang, H.; Tao, S. Directly Growing Hierarchical Nickel-Copper Hydroxide Nanowires on Carbon Fibre Cloth for Efficient Electrooxidation of Ammonia. Appl. Catal. B Environ. 2017, 218, 470–479. [Google Scholar] [CrossRef]
- Xu, W.; Du, D.; Lan, R.; Wu, Z.; Irvine, J.T.S.; Tao, S. Electrodeposited NiCu Bimetal on Carbon Paper as Stable Non-Noble Anode for Efficient Electrooxidation of Ammonia. Appl. Catal. B Environ. 2018, 237, 1101–1109. [Google Scholar] [CrossRef]
- Almomani, F.; Bhosale, R.; Khraisheh, M.; Kumar, A.; Tawalbeh, M. Electrochemical Oxidation of Ammonia on Nickel Oxide Nanoparticles. Int. J. Hydrogen Energy 2020, 45, 10398–10408. [Google Scholar] [CrossRef]
- Chan, Y.T.; Siddharth, K.; Shao, M. Investigation of Cubic Pt Alloys for Ammonia Oxidation Reaction. Nano Res. 2020, 13, 1920–1927. [Google Scholar] [CrossRef]
- Matin, M.A.; Kim, S.; Byun, S.; Kim, H.-C. PtZn Alloy Nanoparticles with Enhanced Activity and Stability Synthesized by a Simplified Polyol Process for Electro-Oxidation of Ammonia. J. Power Sources 2024, 591, 233885. [Google Scholar] [CrossRef]
- Xue, Q.; Zhao, Y.; Zhu, J.; Ding, Y.; Jin, P.; Yin, S.; Chen, Y. PtRu Nanocubes as Bifunctional Electrocatalysts for Ammonia Electrolysis. J. Mater. Chem. A 2021, 9, 8444–8451. [Google Scholar] [CrossRef]
- Karakuş, S.; Baytemir, G.; Özeroğlu, C.; Taşaltın, N. An Ultra-Sensitive Smartphone-Integrated Digital Colorimetric and Electrochemical Camellia Sinensis Polyphenols Encapsulated CuO Nanoparticles-Based Ammonia Biosensor. Inorg. Chem. Commun. 2022, 143, 109733. [Google Scholar] [CrossRef]
- Yang, S.; Zang, G.; Zhang, G.; Zhao, Y.; Li, H.; Zhang, Y. In-Situ Growth of 3D Rosette-like Copper Nanoparticles on Carbon Cloth for Enhanced Sensing of Ammonia Based on Copper Electrodissolution. Anal. Chim. Acta 2020, 1104, 60–68. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, L.; Liu, J.; Gu, J.; Suo, H.; Zhao, C. One Step and in Situ Synthesis of Ni Foam-Supported Pt-Ni(OH)2 Nanosheets as Electrochemical Sensor for Ammonia–Nitrogen Detection. Mater. Lett. 2022, 318, 132197. [Google Scholar] [CrossRef]
- Lu, H.; Hu, J.; Zhang, S.; Long, M.; Tang, A. Cuprous Oxide and Ag Modified Titanium Dioxide Electrode for Ultra-Sensitive Detection of Ammonia Nitrogen. J. Electroanal. Chem. 2023, 949, 117842. [Google Scholar] [CrossRef]
- Zhao, H.; Li, Y.; Cong, A.; Tong, J.; Bian, C. Ultramicro Interdigitated Array Electrode Chip with Optimized Construction for Detection of Ammonia Nitrogen in Water. Micromachines 2023, 14, 629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, L.; Wei, L.; Cai, H.; Gu, J.; Wang, X. Anchoring Platinum Nanofibers on Nickel Hydroxide Nanosheets for Electrochemical Detection of Ammonia-Nitrogen in the Aqueous Environment. Mater. Lett. 2023, 336, 133868. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, J.; Peng, X.; Cui, Q.; He, D.; Zhao, C.; Suo, H. Fabrication of a Ni Foam-Supported Platinum Nanoparticles-Silver/Polypyrrole Electrode for Aqueous Ammonia Sensing. Synth. Met. 2020, 259, 116257. [Google Scholar] [CrossRef]
- Naveen, B.; Lee, S.-W. Surfactant Mediated Electrodeposition of Cu-Ag Nanostructures on Pencil Graphite Electrodes for Amperometric Detection of Nitrate. Microchem. J. 2025, 214, 114090. [Google Scholar] [CrossRef]
- Sharma, R.; Gyergyek, S.; Andersen, S.M. Critical Thinking on Baseline Corrections for Electrochemical Surface Area (ECSA) Determination of Pt/C through H-Adsorption/H-Desorption Regions of a Cyclic Voltammogram. Appl. Catal. B Environ. 2022, 311, 121351. [Google Scholar] [CrossRef]
- Shao, M.; Odell, J.H.; Choi, S.-I.; Xia, Y. Electrochemical Surface Area Measurements of Platinum- and Palladium-Based Nanoparticles. Electrochem. Commun. 2013, 31, 46–48. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode Material | Ammonia Concentration (mM) | Onset Potential (V) * | J (max) mA/cm2 | Ref. |
|---|---|---|---|---|
| PtIrNi | 100 | 0.20 | - | [41] |
| Ni0.8Cu0.2 LHs | 55 | 0.43 | 35 | [42] |
| NiCu/CP | 55 | 0.47 | - | [43] |
| NiO-TiO2 | 500 | 1.22 | 3.7 | [44] |
| Pt-Zn nanocube | 100 | −0.19 | - | [45] |
| PtZn | 20 | −0.62 | 7.5 | [46] |
| PtxRu nanocube | 100 | −0.19 | - | [47] |
| Cu@Pt/PGE | 20 | −0.63 | 11.2 | Present work |
| Electrode Material | Method | Linear Range (μM) | Sensitivity (μA·μM−1) | LOD | Ref. |
|---|---|---|---|---|---|
| PPS-CuO NPs | CV | 12.5–100 | 8.48 | 40.6 nM | [48] |
| Cu NPs/CC | i-t | 5–9425 | 0.0062 | 1.25 μM | [49] |
| Pt-Ni(OH)2 | DPV | 0.05–600 | 0.191 | 39.2 nM | [50] |
| Ag/Cu2O/TNTs | i-t | 0.1–101 | 4.940 | 74 nM | [51] |
| UME/MWCNT | i-t | 10–60 | 0.049 | 8.69 μM | [52] |
| Pt-Ni(OH)2-NF | DPV | 5–500 | 12.27 | 2.74 μM | [53] |
| Pt-Ag/PPY | CV | 0.05–50 | 8.9 | 37 nM | [54] |
| Cu@Pt/PGE | DPV | 200–1000 20–100 1–10 | 1.65 | 780 nM | Present work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naveen, B.; Lee, S.-W. Cu@Pt Core–Shell Nanostructures for Ammonia Oxidation: Bridging Electrocatalysis and Electrochemical Sensing. Inorganics 2025, 13, 241. https://doi.org/10.3390/inorganics13070241
Naveen B, Lee S-W. Cu@Pt Core–Shell Nanostructures for Ammonia Oxidation: Bridging Electrocatalysis and Electrochemical Sensing. Inorganics. 2025; 13(7):241. https://doi.org/10.3390/inorganics13070241
Chicago/Turabian StyleNaveen, Bommireddy, and Sang-Wha Lee. 2025. "Cu@Pt Core–Shell Nanostructures for Ammonia Oxidation: Bridging Electrocatalysis and Electrochemical Sensing" Inorganics 13, no. 7: 241. https://doi.org/10.3390/inorganics13070241
APA StyleNaveen, B., & Lee, S.-W. (2025). Cu@Pt Core–Shell Nanostructures for Ammonia Oxidation: Bridging Electrocatalysis and Electrochemical Sensing. Inorganics, 13(7), 241. https://doi.org/10.3390/inorganics13070241