

Accessing Bisphosphine Copper(I) Complexes with Recalcitrant Pterin–Phenanthroline Ligands Through Mechanochemistry


Abstract

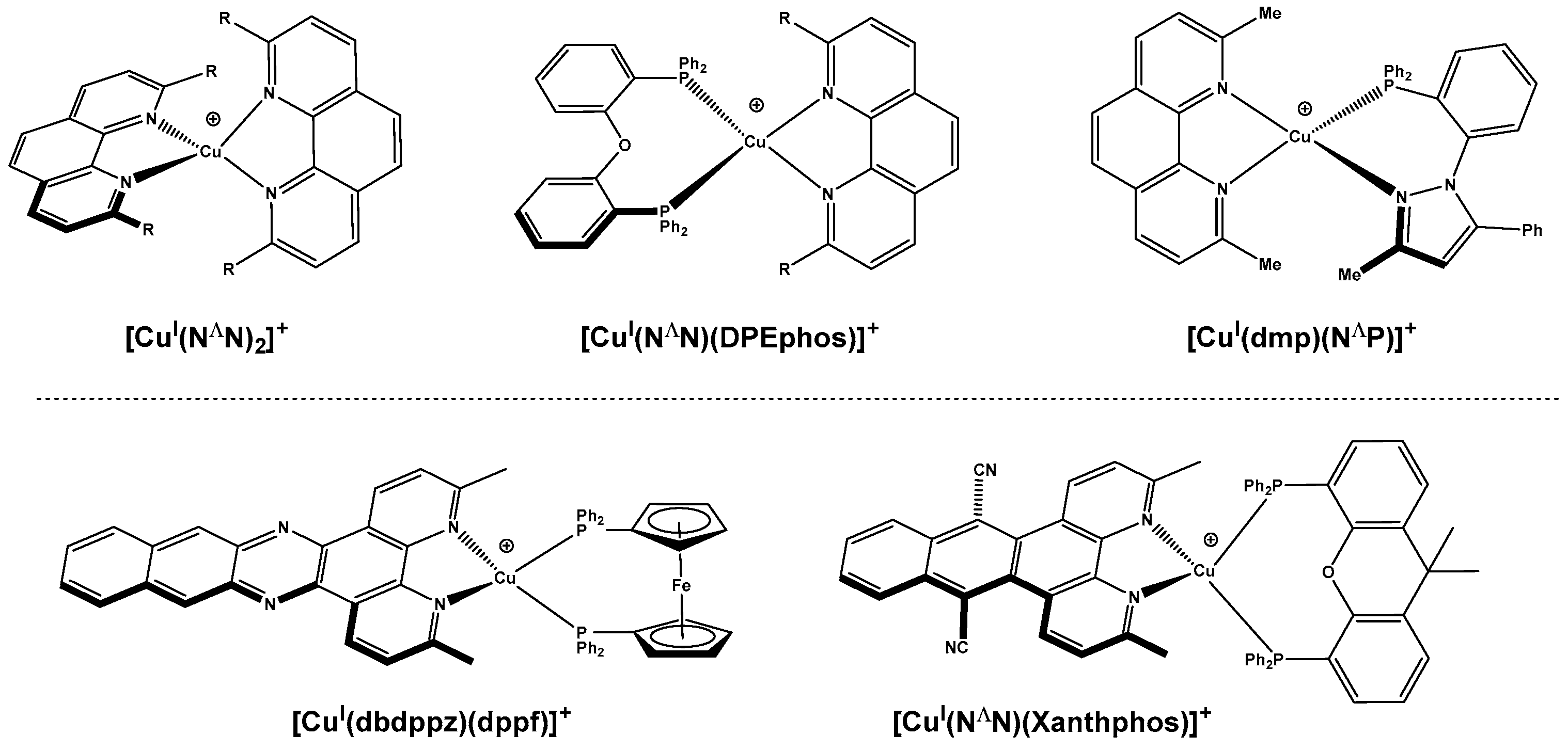
1. Introduction
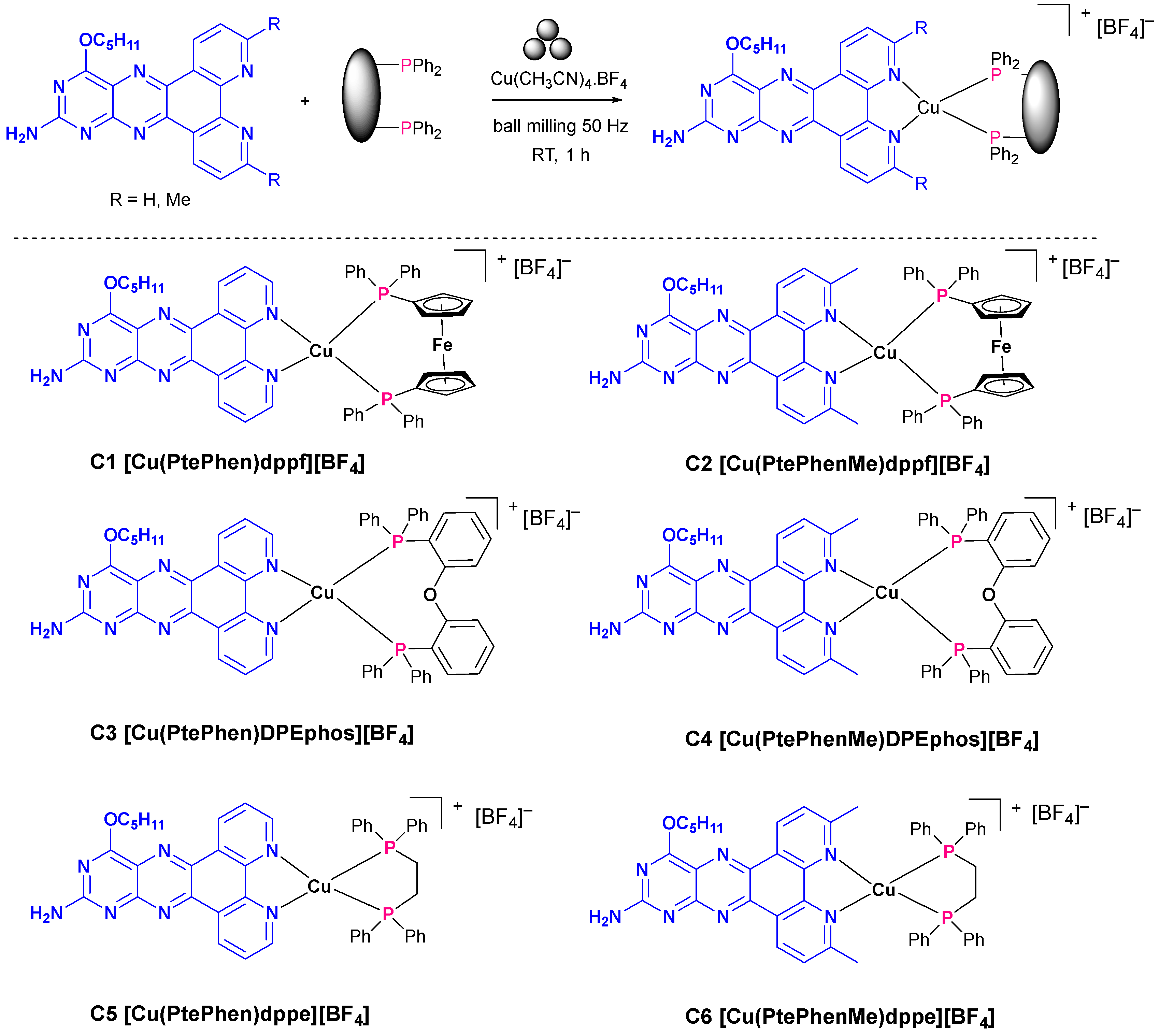
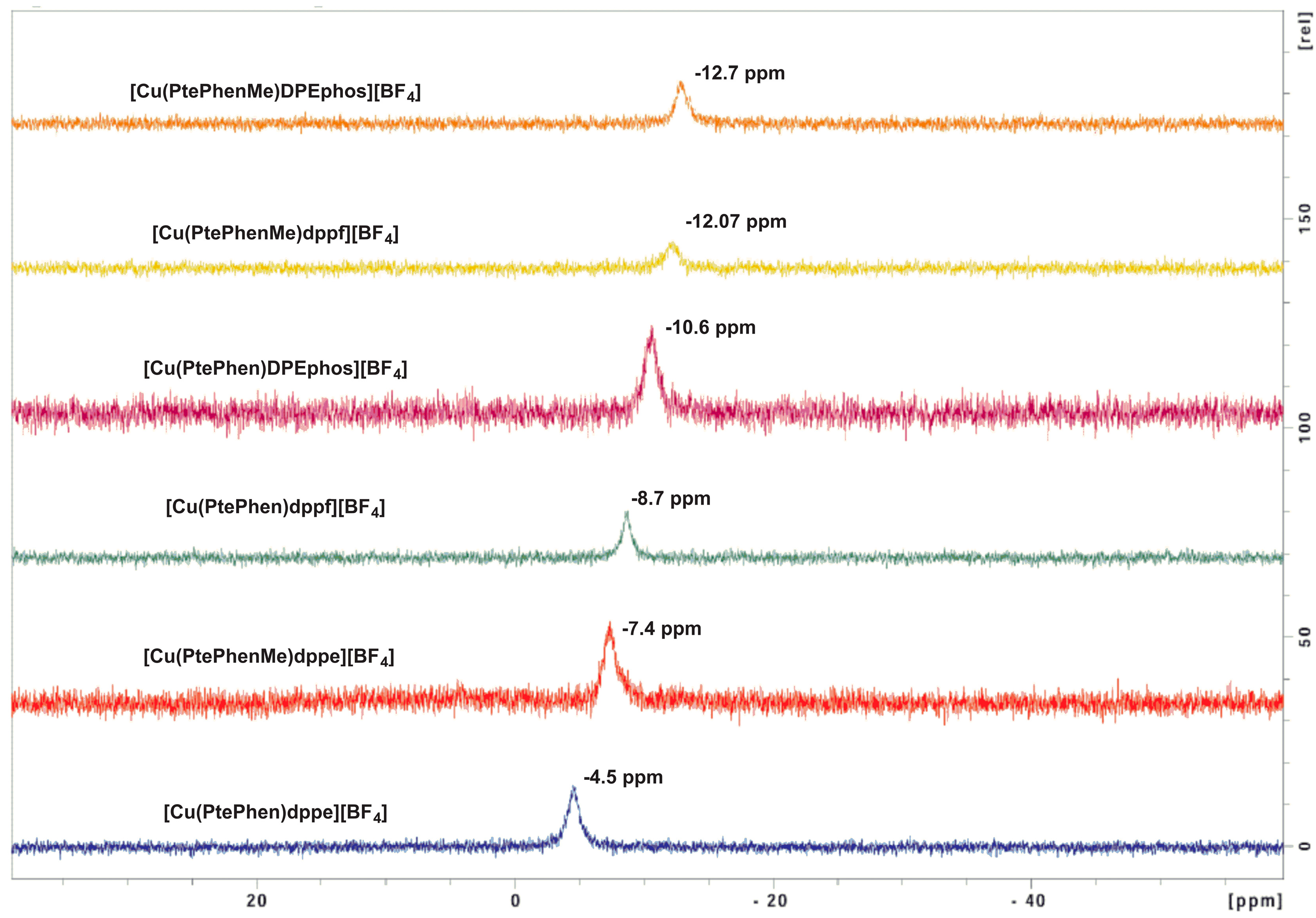
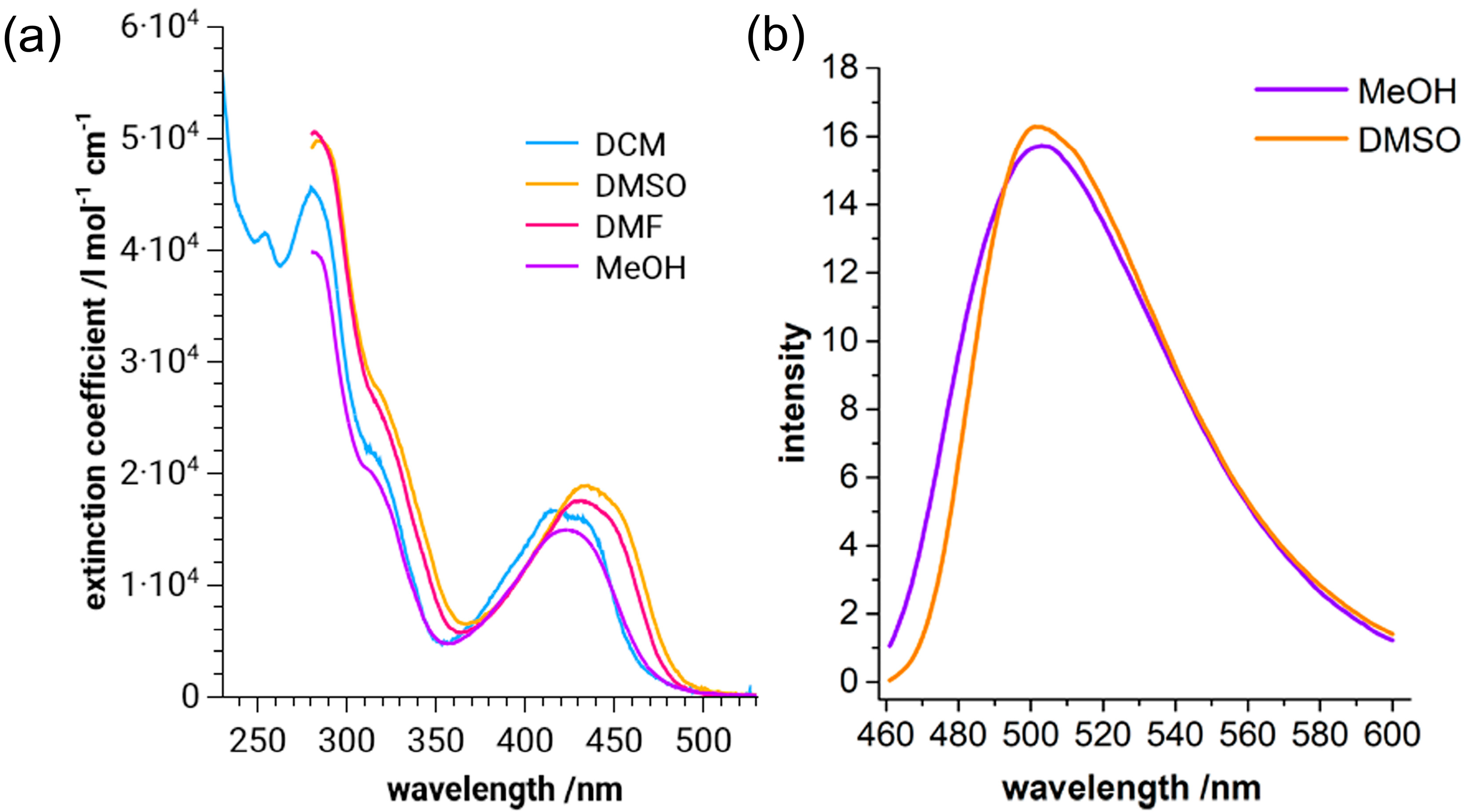
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis of N^N Chelate Ligands PtePhen (L1) and PtePhenMe (L2) by Mechanochemical Solid-State Ball Milling
3.2. Synthesis of [Cu(PtePhenR N˄N) (PP)][BF4] Complexes by Mechanochemical Soid-State Ball Milling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ATRA | Atom-transfer radical addition |
| D | Debye |
| DCM | Dichloromethane |
| DMF | Dimethylformamide |
| DMSO | Dimethylsulfoxide |
| eV | Electron Volt |
| FT-IR | Fourier Transform Infrared Spectroscopy |
| HER | Hydrogen Evolution Reaction |
| MeOH | Methanol |
| MLCT | Metal-to-ligand charge transfer |
| NMR | Nuclear Magnetic Resonance Spectroscopy |
| nm | Nanometre |
| UV-Vis | Ultraviolet–Visible Spectroscopy |
References
- Akita, M.; Ceroni, P.; Stephenson, C.R.J.; Masson, G. Progress in photocatalysis for organic chemistry. J. Org. Chem. 2023, 88, 6281–6283. [Google Scholar] [CrossRef] [PubMed]
- Beatty, J.W.; Stephenson, C.R.J. Amine functionalization via oxidative photoredox catalysis: Methodology development and complex molecule synthesis. Acc. Chem. Res. 2015, 48, 1474–1484. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Bawden, J.C.; Francis, P.S.; DiLuzio, S.; Hayne, D.J.; Doeven, E.H.; Truong, J.; Alexander, R.; Henderson, L.C.; Gómez, D.E.; Massi, M.; et al. Reinterpreting the fate of iridium(iii) photocatalysts─screening a combinatorial library to explore light-driven side-reactions. J. Am. Chem. Soc. 2022, 144, 11189–11202. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Tlahuext-Aca, A.; Glorius, F. Merging visible light photoredox and gold catalysis. Acc. Chem. Res. 2016, 49, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Teegardin, K.; Day, J.I.; Chan, J.; Weaver, J. Advances in photocatalysis: A microreview of visible light mediated ruthenium and iridium catalyzed organic transformations. Org. Process Res. Dev. 2016, 20, 1156–1163. [Google Scholar] [CrossRef]
- Hossain, A.; Bhattacharyya, A.; Reiser, O. Copper’s rapid ascent in visible-light photoredox catalysis. Science 2019, 364, eaav9713. [Google Scholar] [CrossRef]
- Sandoval-Pauker, C.; Molina-Aguirre, G.; Pinter, B. Status report on copper (i) complexes in photoredox catalysis; photophysical and electrochemical properties and future prospects. Polyhedron 2021, 199, 115105. [Google Scholar] [CrossRef]
- McMillin, D.R.; McNett, K.M. Photoprocesses of copper complexes that bind to DNA. Chem. Rev. 1998, 98, 1201–1220. [Google Scholar] [CrossRef]
- McMillin, D.R.; Buckner, M.T.; Ahn, B.T. A light-induced redox reaction of bis(2,9-dimethyl-1,10-phenanthroline)copper(i). Inorg. Chem. 1977, 16, 943–945. [Google Scholar] [CrossRef]
- Dietrich-Buchecker, C.O.; Marnot, P.A.; Sauvage, J.-P.; Kirchhoff, J.R.; McMillin, D.R. Bis(2,9-diphenyl-1,10-phenanthroline)copper(i): A copper complex with a long-lived charge-transfer excited state. J. Chem. Soc. Chem. Commun. 1983, 513–515. [Google Scholar] [CrossRef]
- Kuang, S.-M.; Cuttell, D.G.; McMillin, D.R.; Fanwick, P.E.; Walton, R.A. Synthesis and structural characterization of cu(i) and ni(ii) complexes that contain the bis[2-(diphenylphosphino)phenyl]ether ligand. Novel emission properties for the cu(i) species. Inorg. Chem. 2002, 41, 3313–3322. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.T.; Gantzel, P.K.; Karpishin, T.B. Effects of sterics and electronic delocalization on the photophysical, structural, and electrochemical properties of 2,9-disubstituted 1,10-phenanthroline copper(i) complexes. Inorg. Chem. 1999, 38, 3414–3422. [Google Scholar] [CrossRef] [PubMed]
- Mara, M.W.; Fransted, K.A.; Chen, L.X. Interplays of excited state structures and dynamics in copper(i) diimine complexes: Implications and perspectives. Coord. Chem. Rev. 2015, 282–283, 2–18. [Google Scholar] [CrossRef]
- Zhang, Y.; Schulz, M.; Wächtler, M.; Karnahl, M.; Dietzek, B. Heteroleptic diimine–diphosphine cu(i) complexes as an alternative towards noble-metal based photosensitizers: Design strategies, photophysical properties and perspective applications. Coord. Chem. Rev. 2018, 356, 127–146. [Google Scholar] [CrossRef]
- Zhao, Y.-H.; Li, H.-Y.; Young, D.J.; Cao, X.; Zhu, D.-L.; Ren, Z.-G.; Li, H.-X. Heteroleptic copper(i) complexes [cu(dmp)(n^p)]bf4 for photoinduced atom-transfer radical addition reactions. Dalton Trans. 2023, 52, 8142–8154. [Google Scholar] [CrossRef]
- Giereth, R.; Reim, I.; Frey, W.; Junge, H.; Tschierlei, S.; Karnahl, M. Remarkably long-lived excited states of copper photosensitizers containing an extended π-system based on an anthracene moiety. Sustain. Energy Fuels 2019, 3, 692–700. [Google Scholar] [CrossRef]
- Sosoe, J.; Cruché, C.; Morin, É.; Collins, S.K. Evaluating heteroleptic copper(i)-based complexes bearing π-extended diimines in different photocatalytic processes. Can. J. Chem. 2020, 98, 461–465. [Google Scholar] [CrossRef]
- Pfleiderer, W. Pteridines. Properties, reactivities, and biological significance. J. Heterocycl. Chem. 1992, 29, 583–605. [Google Scholar] [CrossRef]
- Pfleiderer, W.; Zondler, H.; Mengel, R. Pteridine, xxxix. Synthese und struktur von pterin-carbonsäure-(6) und -(7). Liebigs Ann. Chem. 1970, 741, 64–78. [Google Scholar] [CrossRef]
- Serrano, M.P.; Lorente, C.; Morán Vieyra, F.E.; Borsarelli, C.D.; Thomas, A.H. Photosensitizing properties of biopterin and its photoproducts using 2′-deoxyguanosine 5′-monophosphate as an oxidizable target. Phys. Chem. Chem. Phys. 2012, 14, 11657–11665. [Google Scholar] [CrossRef] [PubMed]
- Nichol, C.A.; Smith, G.K.; Duch, D.S. Biosynthesis and metabolism of tetrahydrobiopterin and molybdopterin. Annu. Rev. Biochem. 1985, 54, 729–764. [Google Scholar] [CrossRef] [PubMed]
- Lorente, C.; Thomas, A.H. Photophysics and photochemistry of pterins in aqueous solution. Acc. Chem. Res. 2006, 39, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Ortega, G.; Rodríguez-Muñiz, G.M.; Lhiaubet-Vallet, V.; Lorente, C.; Thomas, A.H. Pterin–thymidine adducts: From their photochemical synthesis to their photosensitizing properties. J. Phys. Chem. B 2025, 129, 3334–3344. [Google Scholar] [CrossRef]
- Choi, S.-W.; Mason, J.B. Folate and carcinogenesis: An integrated scheme. J. Nutr. 2000, 130, 129–132. [Google Scholar] [CrossRef]
- Colston, K.J.; Basu, P. Synthesis, redox and spectroscopic properties of pterin of molybdenum cofactors. Molecules 2022, 27, 3324. [Google Scholar] [CrossRef]
- Andrade, P.; Carneiro, M. Pterin-based pigmentation in animals. Biol. Lett. 2021, 17, 20210221. [Google Scholar] [CrossRef]
- Kosuge, T.; Tsuji, K.; Hirai, K.; Yamaguchi, K.; Okamoto, T.; Iitaka, Y. Isolation and structure determination of a new marine toxin, neosurugatoxin, from the japanese ivory shell, babylonia japonica. Tetrahedron Lett. 1981, 22, 3417–3420. [Google Scholar] [CrossRef]
- Black, K.J.; Huang, H.; High, S.; Starks, L.; Olson, M.; McGuire, M.E. Ruthenium(ii) complexes of a fused phenanthroline-pteridinedione ligand. Inorg. Chem. 1993, 32, 5591–5596. [Google Scholar] [CrossRef]
- Gao, F.; Chao, H.; Zhou, F.; Yuan, Y.-X.; Peng, B.; Ji, L.-N. DNA interactions of a functionalized ruthenium(ii) mixed-polypyridyl complex [ru(bpy)2ppd]2+. J. Inorg. Biochem. 2006, 100, 1487–1494. [Google Scholar] [CrossRef]
- Gao, F.; Chao, H.; Zhou, F.; Xu, L.-C.; Zheng, K.-C.; Ji, L.-N. Synthesis, characterization, and DNA-binding properties of the chiral ruthenium(ii) complexes δ- and λ-[ru(bpy)2(dmppd)]2+ (dmppd = 10,12-dimethylpteridino[6,7-f] [1,10]phenanthroline-11,13(10h,12h)-dione; bpy = 2,2′-bipyridine). Helv. Chim. Acta 2007, 90, 36–51. [Google Scholar] [CrossRef]
- Dalton, S.R.; Glazier, S.; Leung, B.; Win, S.; Megatulski, C.; Burgmayer, S.J.N. DNA binding by ru(ii)–bis(bipyridine)–pteridinyl complexes. J. Biol. Inorg. Chem. 2008, 13, 1133–1148. [Google Scholar] [CrossRef] [PubMed]
- Ragone, F.; Yañuk, J.G.; Cabrerizo, F.M.; Prieto, E.; Wolcan, E.; Ruiz, G.T. DNA structural changes (photo)induced by tricarbonyl (pterin)rhenium(i) complex. J. Inorg. Biochem. 2024, 252, 112471. [Google Scholar] [CrossRef]
- Forero Cortés, P.A.; Marx, M.; Trose, M.; Beller, M. Heteroleptic copper complexes with nitrogen and phosphorus ligands in photocatalysis: Overview and perspectives. Chem Catalysis 2021, 1, 298–338. [Google Scholar] [CrossRef]
- Henriquez, M.A.; Engl, S.; Jaque, P.; Gonzalez, I.A.; Natali, M.; Reiser, O.; Cabrera, A.R. Phosphine evaluation on a new series of heteroleptic copper(i) photocatalysts with dpa ligand [cu(dpa)(p,p)]bf4. Eur. J. Inorg. Chem. 2021, 2021, 4020–4029. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Zhang, J.; Fouassier, J.P.; Gigmes, D.; Lalevée, J. Copper complexes in radical photoinitiating systems: Applications to free radical and cationic polymerization upon visible leds. Macromolecules 2014, 47, 3837–3844. [Google Scholar] [CrossRef]
- Nonogawa, M.; Arai, T.; Endo, N.; Pack, S.P.; Kodaki, T.; Makino, K. Hydrogen bond removal of pterin derivative whose structure is similar to nucleic acid bases. Nucleic Acids Symp. Ser. 2005, 49, 311–312. [Google Scholar] [CrossRef]
- Seo, T.; Toyoshima, N.; Kubota, K.; Ito, H. Tackling solubility issues in organic synthesis: Solid-state cross-coupling of insoluble aryl halides. J. Am. Chem. Soc. 2021, 143, 6165–6175. [Google Scholar] [CrossRef]
- DeGroot, H.P.; Hanusa, T.P. Solvate-assisted grinding: Metal solvates as solvent sources in mechanochemically driven organometallic reactions. Organometallics 2021, 40, 3516–3525. [Google Scholar] [CrossRef]
- Gomollón-Bel, F. Ten chemical innovations that will change our world: Iupac identifies emerging technologies in chemistry with potential to make our planet more sustainable. Chem. Int. 2019, 41, 12–17. [Google Scholar] [CrossRef]
- Ferguson, M.; Giri, N.; Huang, X.; Apperley, D.; James, S.L. One-pot two-step mechanochemical synthesis: Ligand and complex preparation without isolating intermediates. Green Chem. 2014, 16, 1374–1382. [Google Scholar] [CrossRef]
- Cuccu, F.; De Luca, L.; Delogu, F.; Colacino, E.; Solin, N.; Mocci, R.; Porcheddu, A. Mechanochemistry: New tools to navigate the uncharted territory of “impossible” reactions. ChemSusChem 2022, 15, e202200362. [Google Scholar] [CrossRef]
- Al Jomeh, G.A.S.; McGown, A.; Richards, E.; Tizzard, G.J.; Coles, S.J.; González-Méndez, R.; Dadswell, C.; Spencer, J.; Kostakis, G.E. Mechanochemical cu(ii) complexes and propargylamine synthetic adventures. RSC Sustain. 2024, 2, 528–535. [Google Scholar] [CrossRef]
- Kazimi, S.G.T.; Iqbal, M.S.; Mulligan, C.C.; Baseer, M.; Rehman, A.U.; Farooqi, F.; Person, J.R. Mechanochemical synthesis of six cu(ii) complexes with selected thiols, their physicochemical characterization and interaction with DNA. J. Mol. Struct. 2022, 1265, 133436. [Google Scholar] [CrossRef]
- Remy-Speckmann, I.; Zimmermann, B.M.; Gorai, M.; Lerch, M.; Teichert, J.F. Mechanochemical solid state synthesis of copper(i)/nhc complexes with k3po4. Beilstein J. Org. Chem. 2023, 19, 440–447. [Google Scholar] [CrossRef]
- Liao, Q.; Su, K.; Cai, H.; Zhao, T.; Liu, F. Mechanochemical synthesis of carbene copper complexes for co2 hydrogenation to formate. J. CO2 Util. 2022, 59, 101963. [Google Scholar] [CrossRef]
- Jaros, S.W.; Sokolnicki, J.; Siczek, M.; Smoleński, P. Strategy for an effective eco-optimized design of heteroleptic cu(i) coordination polymers exhibiting thermally activated delayed fluorescence. Inorg. Chem. 2023, 62, 19898–19907. [Google Scholar] [CrossRef]
- Kobayashi, A.; Hasegawa, T.; Yoshida, M.; Kato, M. Environmentally friendly mechanochemical syntheses and conversions of highly luminescent cu(i) dinuclear complexes. Inorg. Chem. 2016, 55, 1978–1985. [Google Scholar] [CrossRef]
- Garra, P.; Dumur, F.; Al Mousawi, A.; Graff, B.; Gigmes, D.; Morlet-Savary, F.; Dietlin, C.; Fouassier, J.P.; Lalevée, J. Mechanosynthesized copper(i) complex based initiating systems for redox polymerization: Towards upgraded oxidizing and reducing agents. Polym. Chem. 2017, 8, 5884–5896. [Google Scholar] [CrossRef]
- Zeng, C.; Wang, N.; Peng, T.; Wang, S. Copper(i) complexes bearing 1,2-phenyl-bridged p∧n, p∧n∧p, and n∧p∧n chelate ligands: Structures and phosphorescence. Inorg. Chem. 2017, 56, 1616–1625. [Google Scholar] [CrossRef]
- Garra, P.; Dumur, F.; Morlet-Savary, F.; Dietlin, C.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Mechanosynthesis of a copper complex for redox initiating systems with a unique near infrared light activation. J. Polym. Sci. Polym. Chem. 2017, 55, 3646–3655. [Google Scholar] [CrossRef]
- Garra, P.; Dumur, F.; Mokbel, H.; Monnier, V.; Morlet-Savary, F.; Dietlin, C.; Gigmes, D.; Fouassier, J.-P.; Lalevée, J. New synthetic route to an highly efficient photoredox catalyst by mechanosynthesis. ACS Omega 2018, 3, 10938–10944. [Google Scholar] [CrossRef]
- Correia, J.V.; Bandaru, S.S.M.; Schulzke, C. Pushing at the boundaries of pterin chemistry. Molecules 2024, 29, 4587. [Google Scholar] [CrossRef]
- Kaeser, A.; Mohankumar, M.; Mohanraj, J.; Monti, F.; Holler, M.; Cid, J.-J.; Moudam, O.; Nierengarten, I.; Karmazin-Brelot, L.; Duhayon, C.; et al. Heteroleptic copper(i) complexes prepared from phenanthroline and bis-phosphine ligands. Inorg. Chem. 2013, 52, 12140–12151. [Google Scholar] [CrossRef]
- Pianet, I.; Vincent, J.-M. Isomerization dynamics in homo- and heterochiral atropoisomer copper(i) diimine complexes: A 2d exsy nmr study. Inorg. Chem. 2004, 43, 2947–2953. [Google Scholar] [CrossRef]
- Comba, P.; Katsichtis, C.; Nuber, B.; Pritzkow, H. Solid-state and solution structural properties of copper(i) compounds with bidentate phosphane ligands. Eur. J. Inorg. Chem. 1999, 1999, 777–783. [Google Scholar] [CrossRef]
- Listorti, A.; Accorsi, G.; Rio, Y.; Armaroli, N.; Moudam, O.; Gégout, A.; Delavaux-Nicot, B.; Holler, M.; Nierengarten, J.-F. Heteroleptic copper(i) complexes coupled with methano[60]fullerene: Synthesis, electrochemistry, and photophysics. Inorg. Chem. 2008, 47, 6254–6261. [Google Scholar] [CrossRef]
- Moudam, O.; Kaeser, A.; Delavaux-Nicot, B.; Duhayon, C.; Holler, M.; Accorsi, G.; Armaroli, N.; Séguy, I.; Navarro, J.; Destruel, P.; et al. Electrophosphorescent homo- and heteroleptic copper(i) complexes prepared from various bis-phosphine ligands. Chem. Commun. 2007, 3077–3079. [Google Scholar] [CrossRef]
- Sandroni, M.; Kayanuma, M.; Rebarz, M.; Akdas-Kilig, H.; Pellegrin, Y.; Blart, E.; Le Bozec, H.; Daniel, C.; Odobel, F. Heteroleptic diimine copper(i) complexes with large extinction coefficients: Synthesis, quantum chemistry calculations and physico-chemical properties. Dalton Trans. 2013, 42, 14628–14638. [Google Scholar] [CrossRef]
- Leoni, E.; Mohanraj, J.; Holler, M.; Mohankumar, M.; Nierengarten, I.; Monti, F.; Sournia-Saquet, A.; Delavaux-Nicot, B.; Nierengarten, J.-F.i.; Armaroli, N. Heteroleptic copper(i) complexes prepared from phenanthroline and bis-phosphine ligands: Rationalization of the photophysical and electrochemical properties. Inorg. Chem. 2018, 57, 15537–15549. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, D. Synthesis and study on a series of phosphorescent cu(i) complexes having sterically blocking ligands. Spectrochim. Acta Part A-Mol. Biomol. Spectrosc. 2014, 124, 341–348. [Google Scholar] [CrossRef]
- Alconchel, A.; Crespo, O.; Gimeno, M.C. Thermally activated delayed fluorescence in neutral and cationic copper(i) complexes with the 2-(4-thiazolyl)benzimidazole ligand. Inorg. Chem. 2023, 62, 10431–10439. [Google Scholar] [CrossRef]
- Siddique, Z.A.; Yamamoto, Y.; Ohno, T.; Nozaki, K. Structure-dependent photophysical properties of singlet and triplet metal-to-ligand charge transfer states in copper(i) bis(diimine) compounds. Inorg. Chem. 2003, 42, 6366–6378. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.K.; Becker, E.D.; Cabral De Menezes, S.M.; Goodfellow, R.; Granger, P. Nmr nomenclature: Nuclear spin properties and conventions for chemical shifts (iupac recommendations 2001). Concepts Magn. Reson. 2002, 14, 326–346. [Google Scholar] [CrossRef]
- Bandaru, S.S.M.; Bhilare, S.; Chrysochos, N.; Gayakhe, V.; Trentin, I.; Schulzke, C.; Kapdi, A.R. Pd/ptabs: Catalyst for room temperature amination of heteroarenes. Org. Lett. 2018, 20, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Sokolovs, I.; Suna, E. Para-selective cu-catalyzed c–h aryloxylation of electron-rich arenes and heteroarenes. J. Org. Chem. 2016, 81, 371–379. [Google Scholar] [CrossRef]
- da Silva Miranda, F.; Signori, A.M.; Vicente, J.; de Souza, B.; Priebe, J.P.; Szpoganicz, B.; Gonçalves, N.S.; Neves, A. Synthesis of substituted dipyrido[3,2-a:2′,3′-c]phenazines and a new heterocyclic dipyrido[3,2-f:2′,3′-h]quinoxalino[2,3-b]quinoxaline. Tetrahedron 2008, 64, 5410–5415. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | DCM | MeOH | DMF | DMSO | Emission Profile (λ nm) a | |
|---|---|---|---|---|---|---|
| λmax nm [ε 104 M−1·cm−1] | DMSO | MeOH | ||||
| C1 | 410 (1.6) 320 (2.2) 285 (4.6) | 425 (1.4) 320 (1.8) 290 (3.8) | 430 (1.6) 330 (2.6) 300 (5.0) | 440 (1.8) 330 (2.6) 290 (5.0) | 497 | 504 |
| C2 | 410 (1.6) 320 (1.4) 290 (4.6) | 430 (1.4) 320 (2.0) 300 (4.0) | 430 (1.4) 300 (4.6) | 450 (1.6) 310 (4.8) | 512 | 497 |
| C3 | 410 (1.8) 290 (5.1) | 420 (2.0) 280 (5.8) | 420 (1.4) 290 (4.6) | 440 (1.6) 290 (4.6) | 502 | 494 |
| C4 | 410 (1.6) 300 (5.0) | 420 (1.6) 300 (4.6) | 440 (1.6) 300 (4.8) | 440 (1.6) 300 (4.0) | 504 | 502 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bandaru, S.S.M.; Fischer, C.; Correia, J.V.; Land, A.-L.; Schulzke, C. Accessing Bisphosphine Copper(I) Complexes with Recalcitrant Pterin–Phenanthroline Ligands Through Mechanochemistry. Inorganics 2025, 13, 175. https://doi.org/10.3390/inorganics13060175
Bandaru SSM, Fischer C, Correia JV, Land A-L, Schulzke C. Accessing Bisphosphine Copper(I) Complexes with Recalcitrant Pterin–Phenanthroline Ligands Through Mechanochemistry. Inorganics. 2025; 13(6):175. https://doi.org/10.3390/inorganics13060175
Chicago/Turabian StyleBandaru, Siva S. M., Christian Fischer, Jevy V. Correia, Anna-Lena Land, and Carola Schulzke. 2025. "Accessing Bisphosphine Copper(I) Complexes with Recalcitrant Pterin–Phenanthroline Ligands Through Mechanochemistry" Inorganics 13, no. 6: 175. https://doi.org/10.3390/inorganics13060175
APA StyleBandaru, S. S. M., Fischer, C., Correia, J. V., Land, A.-L., & Schulzke, C. (2025). Accessing Bisphosphine Copper(I) Complexes with Recalcitrant Pterin–Phenanthroline Ligands Through Mechanochemistry. Inorganics, 13(6), 175. https://doi.org/10.3390/inorganics13060175