
Insights into Pd-Nb@In2Se3 Electrocatalyst for High-Performance and Selective CO2 Reduction Reaction from DFT



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Geometric Structures of Nb-Pd@In2Se3
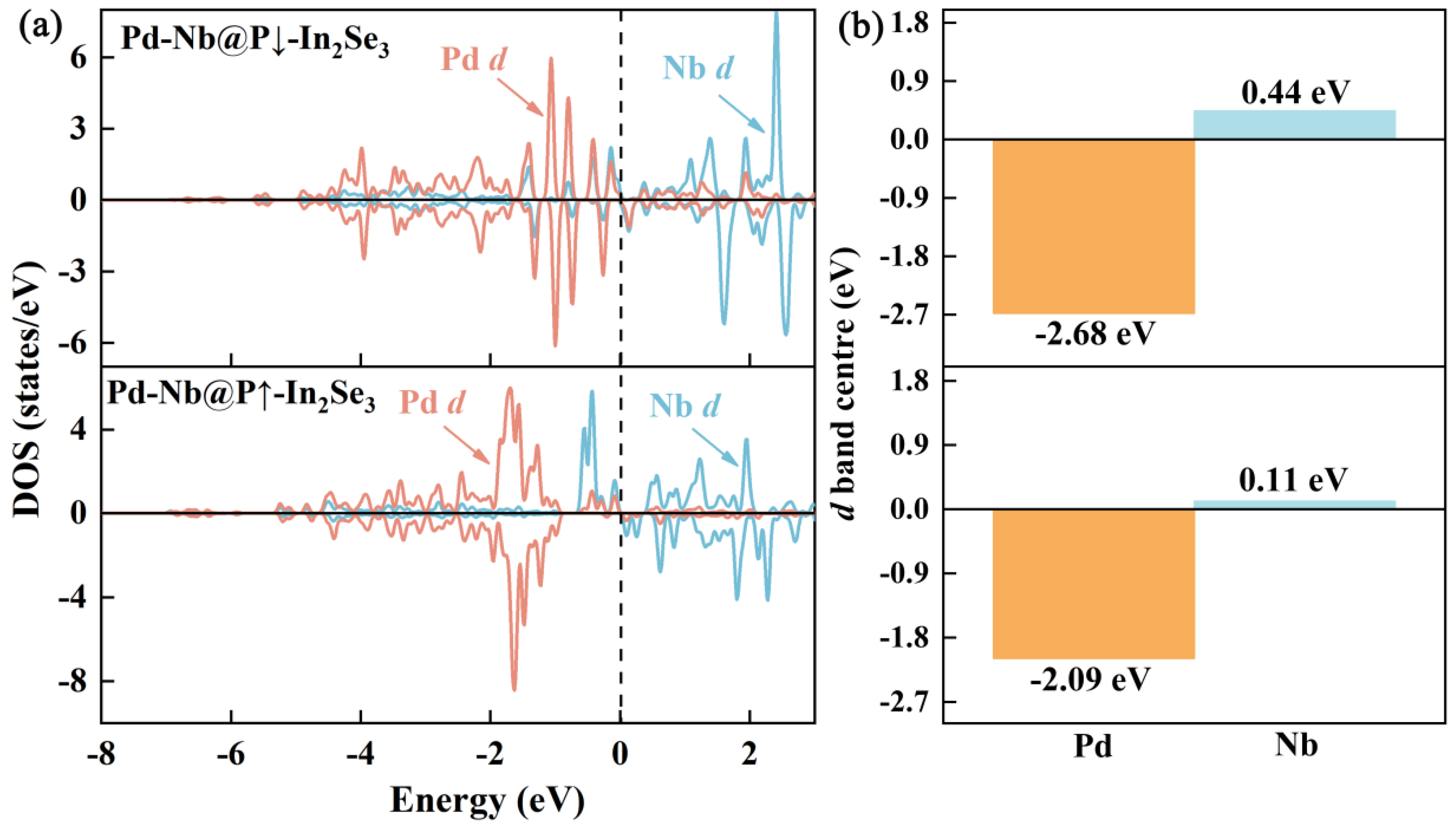
2.2. Activation of CO2 on Pd-Nb@In2Se3 DACs
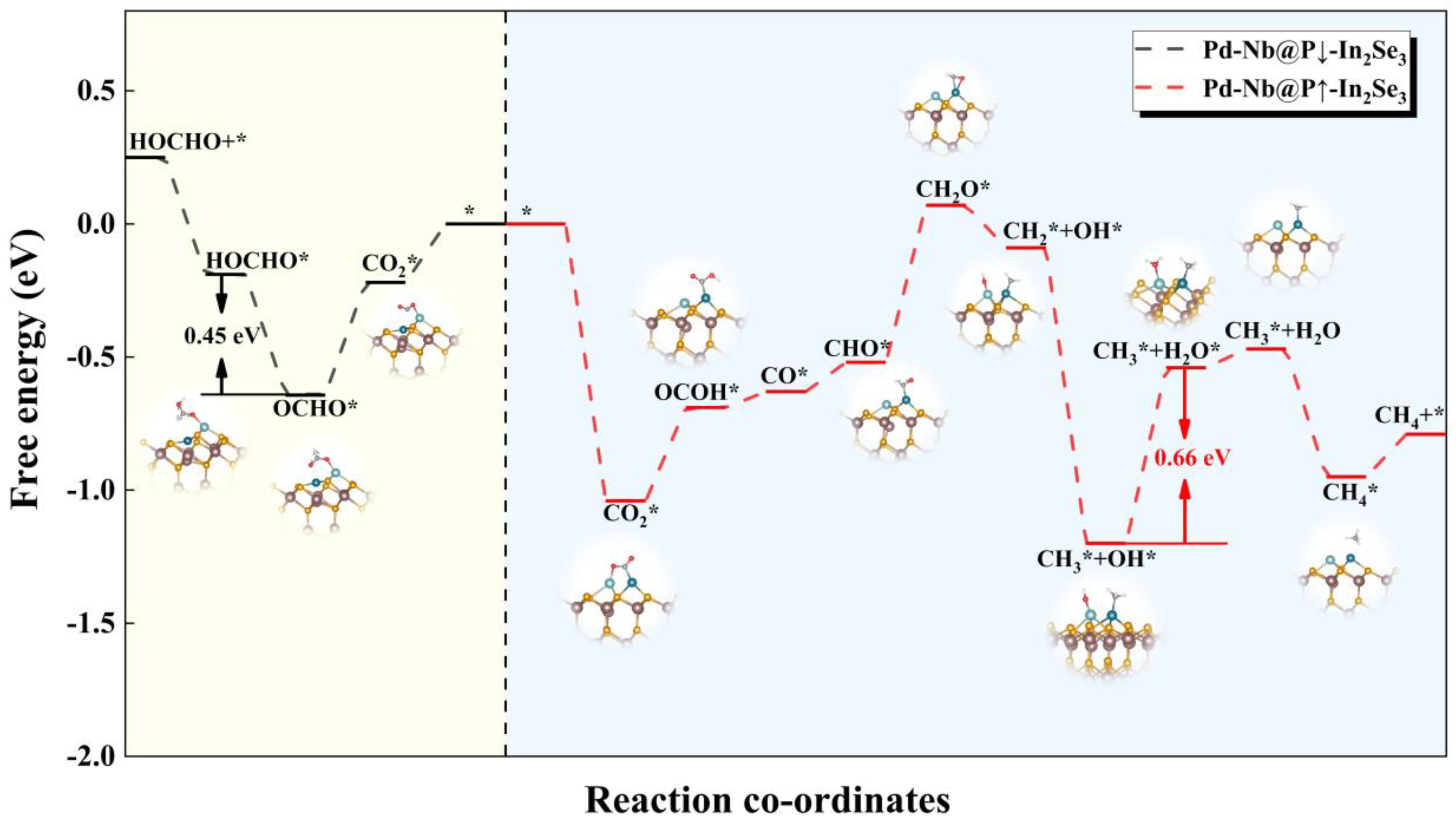
2.3. eCO2RR on Pd-Nb@In2Se3 DACs
2.4. Selectivity for HER vs. eCO2RR on Pd-Nb@In2Se3 DACs
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Obama, B. The irreversible momentum of clean energy. Science 2017, 355, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Lüthi, D.; Le Floch, M.; Bereiter, B.; Blunier, T.; Barnola, J.-M.; Siegenthaler, U.; Raynaud, D.; Jouzel, J.; Fischer, H.; Kawamura, K. High-resolution carbon dioxide concentration record 650,000–800,000 years before present. Nature 2008, 453, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Metz, B.; Davidson, O.; De Coninck, H.; Loos, M.; Meyer, L. Intergovernmental panel on climate change. In IPCC Special Report on Carbon Dioxide Capture and Storage; Cambridge University Press: New York, NY, USA, 2005; 442p. [Google Scholar]
- Chu, S.; Cui, Y.; Liu, N. The path towards sustainable energy. Nat. Mater. 2017, 16, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Zhu, X.; Jiang, Z. Recent Advances of Constructing Metal/Semiconductor Catalysts Designing for Photocatalytic CO2 Hydrogenation. Molecules 2023, 28, 5693. [Google Scholar] [CrossRef]
- Zhu, X.; Zong, H.; Pérez, C.J.V.; Miao, H.; Sun, W.; Yuan, Z.; Wang, S.; Zeng, G.; Xu, H.; Jiang, Z. Supercharged CO2 photothermal catalytic methanation: High conversion, rate, and selectivity. Angew. Chem. 2023, 135, e202218694. [Google Scholar] [CrossRef]
- Zhu, X.; Zhou, E.; Tai, X.; Zong, H.; Yi, J.; Yuan, Z.; Zhao, X.; Huang, P.; Xu, H.; Jiang, Z. g-C3N4 S-Scheme Homojunction through Van der Waals Interface Regulation by Intrinsic Polymerization Tailoring for Enhanced Photocatalytic H2 Evolution and CO2 Reduction. Angew. Chem. Int. Ed. 2025, 64, e202425439. [Google Scholar] [CrossRef]
- Yap, D.; Megaritis, A. Applying Forced Induction to Bioethanol HCCI Operation with Residual Gas Trapping. Energy Fuels 2005, 19, 1812–1821. [Google Scholar] [CrossRef]
- Kang, Z.; Wu, Z.; Zhang, Z.; Deng, J.; Hu, Z.; Li, L. Study of the Combustion Characteristics of a HCCI Engine Coupled with Oxy-Fuel Combustion Mode. SAE Int. J. Engines 2017, 10, 908–916. [Google Scholar] [CrossRef]
- Berstad, D.; Straus, J.; Gundersen, T. CO2 capture and enhanced hydrogen production enabled by low-temperature separation of PSA tail gas: A detailed exergy analysis. Energies 2024, 17, 1072. [Google Scholar] [CrossRef]
- Reynolds, S.P.; Ebner, A.D.; Ritter, J.A. Stripping PSA cycles for CO2 recovery from flue gas at high temperature using a hydrotalcite-like adsorbent. Ind. Eng. Chem. Res. 2006, 45, 4278–4294. [Google Scholar] [CrossRef]
- Hoffert, M.I.; Caldeira, K.; Benford, G.; Criswell, D.R.; Green, C.; Herzog, H.; Jain, A.K.; Kheshgi, H.S.; Lackner, K.S.; Lewis, J.S. Advanced technology paths to global climate stability: Energy for a greenhouse planet. Science 2002, 298, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Xu, J.; Wei, Z.; Ma, J.; Guo, S.; Wang, S.; Liu, H.; Dou, S. Metal-free carbon materials for CO2 electrochemical reduction. Adv. Mater. 2017, 29, 1701784. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef]
- Centi, G.; Quadrelli, E.A.; Perathoner, S. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 2013, 6, 1711–1731. [Google Scholar] [CrossRef]
- Ross, M.B.; De Luna, P.; Li, Y.; Dinh, C.-T.; Kim, D.; Yang, P.; Sargent, E.H. Designing materials for electrochemical carbon dioxide recycling. Nat. Catal. 2019, 2, 648–658. [Google Scholar] [CrossRef]
- Wang, G.; Zhong, S.; Xiong, X.; Li, J.; Wang, F.; Huo, L.; Wu, D.; Han, X.; Wang, Z.; Chen, Q. Plasma induced grain boundaries to boost electrochemical reduction of CO2 to formate. J. Energy Chem. 2024, 95, 636–643. [Google Scholar] [CrossRef]
- Wei, Z.; Wang, W.; Shao, T.; Yang, S.; Liu, C.; Si, D.; Cao, R.; Cao, M. Constructing Ag/Cu2O interface for efficient neutral CO2 electroreduction to C2H4. Angew. Chem. Int. Ed. 2025, 64, e202417066. [Google Scholar] [CrossRef]
- Yue, M.; Xie, W.; Zhong, Z.; Li, M.; Zhang, T.; Shao, M.; Li, H.; Wang, Q. Isolating and stabilizing active copper species in layered double hydroxide to enhance electrocatalytic CO2 reduction to CH4. J. Catal. 2025, 443, 115959. [Google Scholar] [CrossRef]
- Dong, J.; Liu, Y.; Pei, J.; Li, H.; Ji, S.; Shi, L.; Zhang, Y.; Li, C.; Tang, C.; Liao, J. Continuous electroproduction of formate via CO2 reduction on local symmetry-broken single-atom catalysts. Nat. Commun. 2023, 14, 6849. [Google Scholar] [CrossRef]
- Chatterjee, S.; Dutta, I.; Lum, Y.; Lai, Z.; Huang, K.-W. Enabling storage and utilization of low-carbon electricity: Power to formic acid. Energy Environ. Sci. 2021, 14, 1194–1246. [Google Scholar] [CrossRef]
- Li, M.; Idros, M.N.; Wu, Y.; Burdyny, T.; Garg, S.; Zhao, X.S.; Wang, G.; Rufford, T.E. The role of electrode wettability in electrochemical reduction of carbon dioxide. J. Mater. Chem. A 2021, 9, 19369–19409. [Google Scholar] [CrossRef]
- Möller, T.; Ju, W.; Bagger, A.; Wang, X.; Luo, F.; Ngo Thanh, T.; Varela, A.S.; Rossmeisl, J.; Strasser, P. Efficient CO2 to CO electrolysis on solid Ni–N–C catalysts at industrial current densities. Energy Environ. Sci. 2019, 12, 640–647. [Google Scholar] [CrossRef]
- Guo, S.; Liu, Y.; Murphy, E.; Ly, A.; Xu, M.; Matanovic, I.; Pan, X.; Atanassov, P. Robust palladium hydride catalyst for electrocatalytic formate formation with high CO tolerance. Appl. Catal. B Environ. 2022, 316, 121659. [Google Scholar] [CrossRef]
- Detweiler, Z.M.; White, J.L.; Bernasek, S.L.; Bocarsly, A.B. Anodized indium metal electrodes for enhanced carbon dioxide reduction in aqueous electrolyte. Langmuir 2014, 30, 7593–7600. [Google Scholar] [CrossRef]
- Ikeda, S.; Takagi, T.; Ito, K. Selective formation of formic acid, oxalic acid, and carbon monoxide by electrochemical reduction of carbon dioxide. Bull. Chem. Soc. Jpn. 1987, 60, 2517–2522. [Google Scholar] [CrossRef]
- Hori, Y.; Wakebe, H.; Tsukamoto, T.; Koga, O. Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media. Electrochim. Acta 1994, 39, 1833–1839. [Google Scholar] [CrossRef]
- Ding, S.; Hülsey, M.J.; Pérez-Ramírez, J.; Yan, N. Transforming energy with single-atom catalysts. Joule 2019, 3, 2897–2929. [Google Scholar] [CrossRef]
- Su, X.; Yang, X.-F.; Huang, Y.; Liu, B.; Zhang, T. Single-atom catalysis toward efficient CO2 conversion to CO and formate products. Acc. Chem. Res. 2018, 52, 656–664. [Google Scholar] [CrossRef]
- Liu, K.; Fu, J.; Zhu, L.; Zhang, X.; Li, H.; Liu, H.; Hu, J.; Liu, M. Single-atom transition metals supported on black phosphorene for electrochemical nitrogen reduction. Nanoscale 2020, 12, 4903–4908. [Google Scholar] [CrossRef]
- Lu, F.; Yi, D.; Liu, S.; Zhan, F.; Zhou, B.; Gu, L.; Golberg, D.; Wang, X.; Yao, J. Engineering platinum–oxygen dual catalytic sites via charge transfer towards highly efficient hydrogen evolution. Angew. Chem. 2020, 132, 17865–17871. [Google Scholar] [CrossRef]
- Nguyen, T.; Salehi, M.; Le, Q.; Seifitokaldani, A.; Dinh, C. Fundamentals of electrochemical CO2 reduction on single-metal-atom catalysts. ACS Catal. 2020, 10, 10068–10095. [Google Scholar] [CrossRef]
- Sun, J.-F.; Wu, J.-T.; Xu, Q.-Q.; Zhou, D.; Yin, J.-Z. CO2 electrochemical reduction using single-atom catalysts. Preparation, characterization and anchoring strategies: A review. Environ. Chem. Lett. 2020, 18, 1593–1623. [Google Scholar] [CrossRef]
- Asset, T.; Garcia, S.T.; Herrera, S.; Andersen, N.; Chen, Y.; Peterson, E.J.; Matanovic, I.; Artyushkova, K.; Lee, J.; Minteer, S.D. Investigating the nature of the active sites for the CO2 reduction reaction on carbon-based electrocatalysts. ACS Catal. 2019, 9, 7668–7678. [Google Scholar] [CrossRef]
- Zhang, Q.; Guan, J. Single-atom catalysts for electrocatalytic applications. Adv. Funct. Mater. 2020, 30, 2000768. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, B.; Ran, J.; Davey, K.; Qiao, S.Z. Atomic-level reactive sites for semiconductor-based photocatalytic CO2 reduction. Adv. Energy Mater. 2020, 10, 1903879. [Google Scholar] [CrossRef]
- Luo, G.; Jing, Y.; Li, Y. Rational design of dual-metal-site catalysts for electroreduction of carbon dioxide. J. Mater. Chem. A 2020, 8, 15809–15815. [Google Scholar] [CrossRef]
- Ouyang, Y.; Shi, L.; Bai, X.; Li, Q.; Wang, J. Breaking scaling relations for efficient CO2 electrochemical reduction through dual-atom catalysts. Chem. Sci. 2020, 11, 1807–1813. [Google Scholar] [CrossRef]
- Li, Y.; Chen, C.; Cao, R.; Pan, Z.; He, H.; Zhou, K. Dual-atom Ag2/graphene catalyst for efficient electroreduction of CO2 to CO. Appl. Catal. B Environ. 2020, 268, 118747. [Google Scholar] [CrossRef]
- Li, Y.; Su, H.; Chan, S.H.; Sun, Q. CO2 Electroreduction Performance of Transition Metal Dimers Supported on Graphene: A Theoretical Study. ACS Catal. 2015, 5, 6658–6664. [Google Scholar] [CrossRef]
- Li, X.; Sun, Y.; Xu, J.; Shao, Y.; Wu, J.; Xu, X.; Pan, Y.; Ju, H.; Zhu, J.; Xie, Y. Selective visible-light-driven photocatalytic CO2 reduction to CH4 mediated by atomically thin CuIn5S8 layers. Nat. Energy 2019, 4, 690–699. [Google Scholar] [CrossRef]
- Li, S.; Nagarajan, A.V.; Alfonso, D.R.; Sun, M.; Kauffman, D.R.; Mpourmpakis, G.; Jin, R. Boosting CO2 electrochemical reduction with atomically precise surface modification on gold nanoclusters. Angew. Chem. Int. Ed. 2021, 60, 6351–6356. [Google Scholar] [CrossRef] [PubMed]
- Wen, G.; Lee, D.U.; Ren, B.; Hassan, F.M.; Jiang, G.; Cano, Z.P.; Gostick, J.; Croiset, E.; Bai, Z.; Yang, L. Orbital interactions in Bi-Sn bimetallic electrocatalysts for highly selective electrochemical CO2 reduction toward formate production. Adv. Energy Mater. 2018, 8, 1802427. [Google Scholar] [CrossRef]
- Lin, L.; Li, H.; Yan, C.; Li, H.; Si, R.; Li, M.; Xiao, J.; Wang, G.; Bao, X. Synergistic catalysis over iron-nitrogen sites anchored with cobalt phthalocyanine for efficient CO2 electroreduction. Adv. Mater. 2019, 31, 1903470. [Google Scholar] [CrossRef]
- Hu, K.; Li, Z.; Bai, L.; Yang, F.; Chu, X.; Bian, J.; Zhang, Z.; Xu, H.; Jing, L. Synergetic subnano Ni-and Mn-Oxo clusters anchored by chitosan oligomers on 2D g-C3N4 boost photocatalytic CO2 reduction. Sol. Rrl 2021, 5, 2000472. [Google Scholar] [CrossRef]
- Bok, J.; Lee, S.Y.; Lee, B.-H.; Kim, C.; Nguyen, D.L.T.; Kim, J.W.; Jung, E.; Lee, C.W.; Jung, Y.; Lee, H.S. Designing atomically dispersed Au on tensile-strained Pd for efficient CO2 electroreduction to formate. J. Am. Chem. Soc. 2021, 143, 5386–5395. [Google Scholar] [CrossRef]
- Ding, T.; Liu, X.; Tao, Z.; Liu, T.; Chen, T.; Zhang, W.; Shen, X.; Liu, D.; Wang, S.; Pang, B. Atomically precise dinuclear site active toward electrocatalytic CO2 reduction. J. Am. Chem. Soc. 2021, 143, 11317–11324. [Google Scholar] [CrossRef]
- He, Q.; Liu, D.; Lee, J.H.; Liu, Y.; Xie, Z.; Hwang, S.; Kattel, S.; Song, L.; Chen, J.G. Electrochemical conversion of CO2 to syngas with controllable CO/H2 ratios over Co and Ni single-atom catalysts. Angew. Chem. Int. Ed. 2020, 59, 3033–3037. [Google Scholar] [CrossRef]
- Darif, B.; Ojala, S.; Pirault-Roy, L.; Bensitel, M.; Brahmi, R.; Keiski, R.L. Study on the catalytic oxidation of DMDS over Pt-Cu catalysts supported on Al2O3, AlSi20 and SiO2. Appl. Catal. B Environ. 2016, 181, 24–33. [Google Scholar] [CrossRef]
- Winter, L.R.; Gomez, E.; Yan, B.; Yao, S.; Chen, J.G. Tuning Ni-catalyzed CO2 hydrogenation selectivity via Ni-ceria support interactions and Ni-Fe bimetallic formation. Appl. Catal. B Environ. 2018, 224, 442–450. [Google Scholar] [CrossRef]
- Wang, J.; Huang, Z.; Liu, W.; Chang, C.; Tang, H.; Li, Z.; Chen, W.; Jia, C.; Yao, T.; Wei, S.; et al. Design of N-Coordinated Dual-Metal Sites: A Stable and Active Pt-Free Catalyst for Acidic Oxygen Reduction Reaction. J. Am. Chem. Soc. 2017, 139, 17281–17284. [Google Scholar] [CrossRef]
- Ren, W.; Tan, X.; Yang, W.; Jia, C.; Xu, S.; Wang, K.; Smith, S.C.; Zhao, C. Isolated Diatomic Ni-Fe Metal–Nitrogen Sites for Synergistic Electroreduction of CO2. Angew. Chem. Int. Ed. 2019, 58, 6972–6976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fischer, J.M.T.A.; Jia, Y.; Yan, X.; Xu, W.; Wang, X.; Chen, J.; Yang, D.; Liu, H.; Zhuang, L.; et al. Coordination of Atomic Co–Pt Coupling Species at Carbon Defects as Active Sites for Oxygen Reduction Reaction. J. Am. Chem. Soc. 2018, 140, 10757–10763. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhao, M.H.; Garra, J.; Kolpak, A.M.; Rappe, A.M.; Bonnell, D.A.; Vohs, J.M. Direct in situ determination of the polarization dependence of physisorption on ferroelectric surfaces. Nat. Mater. 2008, 7, 473–477. [Google Scholar] [CrossRef]
- Ju, L.; Shang, J.; Tang, X.; Kou, L. Tunable Photocatalytic Water Splitting by the Ferroelectric Switch in a 2D AgBiP2Se6 Monolayer. J. Am. Chem. Soc. 2019, 142, 1492–1500. [Google Scholar] [CrossRef]
- Ju, L.; Tan, X.; Mao, X.; Gu, Y.; Smith, S.; Du, A.; Chen, Z.; Chen, C.; Kou, L. Controllable CO2 electrocatalytic reduction via ferroelectric switching on single atom anchored In2Se3 monolayer. Nat. Commun. 2021, 12, 5128. [Google Scholar] [CrossRef]
- Ding, W.; Zhu, J.; Wang, Z.; Gao, Y.; Xiao, D.; Gu, Y.; Zhang, Z.; Zhu, W. Prediction of intrinsic two-dimensional ferroelectrics in In2Se3 and other III2-VI3 van der Waals materials. Nat. Commun. 2017, 8, 14956. [Google Scholar] [CrossRef]
- Cui, C.; Hu, W.J.; Yan, X.; Addiego, C.; Gao, W.; Wang, Y.; Wang, Z.; Li, L.; Cheng, Y.; Li, P.; et al. Intercorrelated In-Plane and Out-of-Plane Ferroelectricity in Ultrathin Two-Dimensional Layered Semiconductor In2Se3. Nano Lett. 2018, 18, 1253–1258. [Google Scholar] [CrossRef]
- Gao, G.; Jiao, Y.; Waclawik, E.R.; Du, A. Single Atom (Pd/Pt) Supported on Graphitic Carbon Nitride as an Efficient Photocatalyst for Visible-Light Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2016, 138, 6292–6297. [Google Scholar] [CrossRef]
- Li, X.; Bi, W.; Zhang, L.; Tao, S.; Chu, W.; Zhang, Q.; Luo, Y.; Wu, C.; Xie, Y. Single-Atom Pt as Co-Catalyst for Enhanced Photocatalytic H2 Evolution. Adv. Mater. 2016, 28, 2427–2431. [Google Scholar] [CrossRef]
- Vile, G.; Albani, D.; Nachtegaal, M.; Chen, Z.; Dontsova, D.; Antonietti, M.; Lopez, N.; Perez-Ramirez, J. A stable single-site palladium catalyst for hydrogenations. Angew. Chem. Int. Ed. 2015, 54, 11265–11269. [Google Scholar] [CrossRef]
- Khatun, R.; Siddiqui, N.; Pal, R.S.; Bhandari, S.; Khan, T.S.; Singh, S.; Poddar, M.K.; Samanta, C.; Bal, R. Low temperature reforming of methane with CO2 over Pt/CeO2, Ni/CeO2 and Pt–Ni/CeO2 catalysts prepared by a solution-combustion method. Catal. Sci. Technol. 2023, 13, 6431–6445. [Google Scholar] [CrossRef]
- Ma, D.; Zeng, Z.; Liu, L.; Huang, X.; Jia, Y. Computational Evaluation of Electrocatalytic Nitrogen Reduction on TM Single-, Double-, and Triple-Atom Catalysts (TM = Mn, Fe, Co, Ni) Based on Graphdiyne Monolayers. J. Phys. Chem. C 2019, 123, 19066–19076. [Google Scholar] [CrossRef]
- Back, S.; Kim, H.; Jung, Y. Selective Heterogeneous CO2 Electroreduction to Methanol. ACS Catal. 2015, 5, 965–971. [Google Scholar] [CrossRef]
- Zhao, Z.; Lu, G. Cu-Based Single-Atom Catalysts Boost Electroreduction of CO2 to CH3OH: First-Principles Predictions. J. Phys. Chem. C 2019, 123, 4380–4387. [Google Scholar] [CrossRef]
- Wannakao, S.; Artrith, N.; Limtrakul, J.; Kolpak, A.M. Engineering Transition-Metal-Coated Tungsten Carbides for Efficient and Selective Electrochemical Reduction of CO2 to Methane. ChemSusChem 2015, 8, 2745–2751. [Google Scholar] [CrossRef]
- Elhenawy, S.E.M.; Khraisheh, M.; AlMomani, F.; Walker, G. Metal-organic frameworks as a platform for CO2 capture and chemical processes: Adsorption, membrane separation, catalytic-conversion, and electrochemical reduction of CO2. Catalysts 2020, 10, 1293. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, X.; Kang, Y.; Ye, C.; Jin, R.; Yan, H.; Lin, R.; Yang, J.; Xu, Q.; Wang, Y.; et al. A Supported Pd2 Dual-Atom Site Catalyst for Efficient Electrochemical CO2 Reduction. Angew. Chem. Int. Ed. 2021, 60, 13388–13393. [Google Scholar] [CrossRef]
- Hao, Q.; Zhong, H.; Wang, J.; Liu, K.; Yan, J.; Ren, Z.; Zhou, N.; Zhao, X.; Zhang, H.; Liu, D.; et al. Nickel dual-atom sites for electrochemical carbon dioxide reduction. Nat. Synth. 2022, 1, 719–728. [Google Scholar] [CrossRef]
- Huang, K.; Li, R.; Qi, H.; Yang, S.; An, S.; Lian, C.; Xu, Q.; Liu, H.; Hu, J. Regulating Adsorption of Intermediates via the Sulfur Modulating Dual-Atomic Sites for Boosting CO2RR. ACS Catal. 2024, 14, 8889–8898. [Google Scholar] [CrossRef]
- Chen, B.; Shi, D.; Deng, R.; Xu, X.; Liu, W.; Wei, Y.; Liu, Z.; Zhong, S.; Huang, J.; Yu, Y. Leveraging Atomic-Scale Synergy for Selective CO2 Electrocatalysis to CO over CuNi Dual-Atom Catalysts. ACS Catal. 2024, 14, 16224–16233. [Google Scholar] [CrossRef]
- Roy, S.; Li, Z.; Chen, Z.; Mata, A.C.; Kumar, P.; Sarma, S.C.; Teixeira, I.F.; Silva, I.F.; Gao, G.; Tarakina, N.V.; et al. Cooperative Copper Single-Atom Catalyst in 2D Carbon Nitride for Enhanced CO2 Electrolysis to Methane. Adv. Mater. 2024, 36, e2300713. [Google Scholar] [CrossRef]
- Xue, H.; Zhao, Z.H.; Liao, P.Q.; Chen, X.M. “Ship-in-a-Bottle” Integration of Ditin(IV) Sites into a Metal-Organic Framework for Boosting Electroreduction of CO2 in Acidic Electrolyte. J. Am. Chem. Soc. 2023, 145, 16978–16982. [Google Scholar] [CrossRef]
- Monteiro, M.C.O.; Dattila, F.; Hagedoorn, B.; García-Muelas, R.; López, N.; Koper, M.T.M. Absence of CO2 electroreduction on copper, gold and silver electrodes without metal cations in solution. Nat. Catal. 2021, 4, 654–662. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Mao, X.; Kour, G.; Zhang, L.; He, T.; Wang, S.; Yan, C.; Zhu, Z.; Du, A. Silicon-doped graphene edges: An efficient metal-free catalyst for the reduction of CO2 into methanol and ethanol. Catal. Sci. Technol. 2019, 9, 6800–6807. [Google Scholar] [CrossRef]
- Cui, X.; An, W.; Liu, X.; Wang, H.; Men, Y.; Wang, J. C2N-graphene supported single-atom catalysts for CO2 electrochemical reduction reaction: Mechanistic insight and catalyst screening. Nanoscale 2018, 10, 15262–15272. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.K.; Studt, F.; Abild-Pedersen, F.; Bligaard, T. Fundamental Concepts in Heterogeneous Catalysis. Focus Catal. 2015, 2015, 7. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energ. Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ju, L.; Tang, X.; Zhang, Y.; Chen, M.; Liu, S.; Long, C. Insights into Pd-Nb@In2Se3 Electrocatalyst for High-Performance and Selective CO2 Reduction Reaction from DFT. Inorganics 2025, 13, 146. https://doi.org/10.3390/inorganics13050146
Ju L, Tang X, Zhang Y, Chen M, Liu S, Long C. Insights into Pd-Nb@In2Se3 Electrocatalyst for High-Performance and Selective CO2 Reduction Reaction from DFT. Inorganics. 2025; 13(5):146. https://doi.org/10.3390/inorganics13050146
Chicago/Turabian StyleJu, Lin, Xiao Tang, Yixin Zhang, Mengya Chen, Shuli Liu, and Chen Long. 2025. "Insights into Pd-Nb@In2Se3 Electrocatalyst for High-Performance and Selective CO2 Reduction Reaction from DFT" Inorganics 13, no. 5: 146. https://doi.org/10.3390/inorganics13050146
APA StyleJu, L., Tang, X., Zhang, Y., Chen, M., Liu, S., & Long, C. (2025). Insights into Pd-Nb@In2Se3 Electrocatalyst for High-Performance and Selective CO2 Reduction Reaction from DFT. Inorganics, 13(5), 146. https://doi.org/10.3390/inorganics13050146