1. Introduction
With climate change becoming increasingly dominant, switching from fossil fuels to sustainable energy sources and reducing global inefficiencies are main priorities. Zintl phases typically contain electropositive cations (alkali, alkaline earth, or rare earth metals) and electronegative early p-block metals and semimetals from groups 13 to 15, which are the (poly)anions [
1]. Given that many Zintl phases are intrinsic semiconductors, they could be the bases for materials with applications in solid-state energy conversion and/or storage [
2,
3,
4,
5,
6,
7,
8,
9,
10], which further sustainability.
In recent years, the lithiation behavior of Si-, Ge-, and Sn-based clathrates, many of which can be viewed as Zintl phases [
11], has become the subject of increased research interest [
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22]. The undertaken fundamental studies, either by us or by several other teams, have been carried out in the hopes of adapting the clathrates’ desirable characteristics for prospective high-capacity Li-ion battery applications [
23,
24]. In particular, over the course of our work with the type-I clathrates Ba
8Al
xSi
46−x and Ba
8Al
xGe
46−x (0 <
x < 16) [
21,
22], we observed the presence of some unknown phases as reaction byproducts. The latter were presumed to be new quaternary compounds with hitherto unknown structures and compositions. These observations were the motivation to create the following research plan for the summer internship; and so, the goal of this research was the rational synthesis of new Zintl phases comprising lithium and silicon or germanium. Hence, we set out to explore the
A–Li–Al–
Tt systems (
A = Mg, Ca, Sr, Ba;
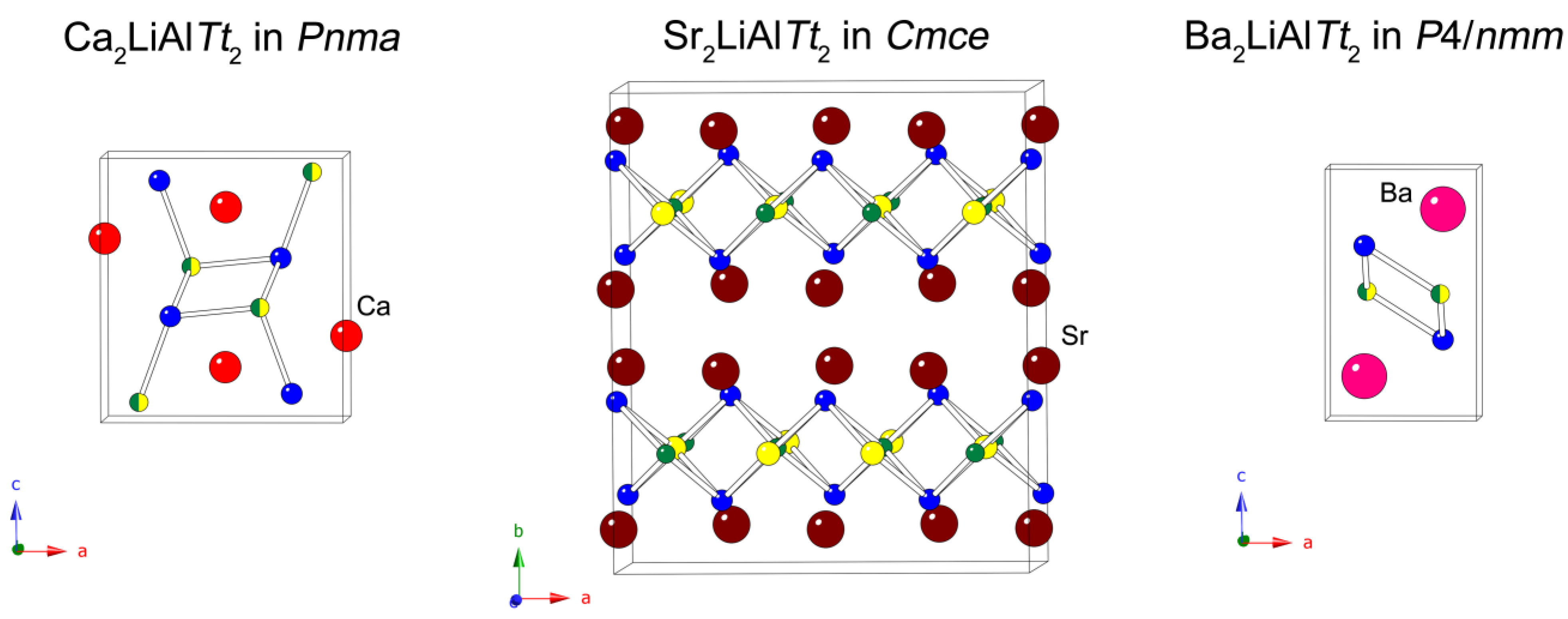
Tt = Si, Ge) and our starting points were the equiatomic nominal compositions. The syntheses quickly yielded several new compounds in each respective system. Below, we detail the synthetic work that led to the identification of the title compounds, as well as describe their crystal structures. At the outset, we draw attention to the fact that the structures, despite their same general chemical formula, are different (
Figure 1). Each compound’s differing anion (silicon or germanium) impacts only the size of the unit cell, with the silicides having smaller unit cell volumes than the germanides. The nature of the alkaline earth metal appears to influence which structure forms, with the calcium compounds being analogs of the TiNiSi/SrMgSi family (orthorhombic
Pnma space group, no. 62), the strontium compounds being a part of the Na
2LiAlP
2 structural family (orthorhombic
Cmce space group, no. 64), and the barium compounds crystallizing with the PbFCl structure type in the tetragonal
P4/
nmm space group (no. 129). To this end, attempts at the synthesis of Mg
2LiAlSi
2 and Mg
2LiAlGe
2 failed (
Vide infra).
Several europium and ytterbium compounds were also synthesized. The characterization work is still ongoing and the results will be detailed in a later publication. We will just note that the europium compounds appear to mirror the crystal chemistry of the barium compounds, but the ytterbium compounds have differing structures and compositions. For that reason, the synthesized rare earth metal compounds are not included here.
2. Results
Crystal data for
A2LiAl
Tt2 (
A = Ca, Sr, Ba;
Tt = Si, Ge) can be found in
Table 1,
Table 2 and
Table 3, respectively. We should note that our initial results from the system Ba–Li–Al–Ge, based on single-crystal X-ray diffraction work, showed the co-existence of Li and Al atoms on a single crystallographic site, which piqued our attention. Later on, the same result was replicated for the system Ba–Li–Al–Si. The Ca-compounds, although with a differing crystal structure, also showed Li and Al atoms on a single crystallographic site (
Table 4). The refined chemical formulae in all four cases were
ALi
xAl
1–xTt (
A = Ca, Ba;
Tt = Si, Ge) with
x ≈ ½; however, subsequent synthetic work showed that changes in the nominal Li:Al ratio do not contribute to changes in the unit cell volume and there were no peak shifts when experimental and simulated powder X-ray diffraction patterns were compared. For the Sr-based compounds, Li and Al were found to occupy individual sites and were not disordered. Therefore, in this article, we labeled all six new alumo-silicides and alumo-germanides with the base formula
A2LiAl
Tt2 (
A = Ca, Sr, Ba;
Tt = Si, Ge), neglecting the arguably small possibility of trivial deviations from the stated stoichiometry.
As seen from
Table 1,
Table 2 and
Table 3 (as well as from
Table 4, where the atomic coordinates of each of the six structures are given), there are three basic structures adopted by each of the Ca, Sr, and Ba compounds, respectively. All three types of structures (
Figure 1) share a common feature—tetrahedral (or rather distorted tetrahedral) coordination of Al and Li atoms by the atoms of the
Tt element. In the case of the Ca and Ba compounds, there is no long-range ordering and the refined models have Li and Al atoms occupying the same crystallographic positions, while in the case of the Sr compounds, there is no randomization and the derived crystal structures of Sr
2LiAlSi
2 and Sr
2LiAlGe
2 are devoid of disorder. Apparently, the packing of the Al
Tt4 and Li
Tt4 fragments together with the increasing size of the alkaline earth metal atom (in the expected order Ca < Sr < Ba) governs the formation of each structure, as it will be discussed in the next section. In that regard, we will mention that we attempted to synthesize Mg
2LiAlSi
2 and Mg
2LiAlGe
2 using the same synthesis procedure (elements in the targeted stoichiometric ratio loaded into niobium tube, arc welded shut, enclosed in a silica tube under vacuum, and heated in tube furnaces according to the same heating profile as for the Ca, Sr, and Ba counterparts). The resultant products were not the quaternaries Mg
2LiAlSi
2 and Mg
2LiAlGe
2. Instead, three different crystals could be identified with a naked eye: (1) blue, shiny gem-like crystals; (2) atmosphere-reactive silver crystals; and (3) dark, hard, brittle crystals. The blue crystals were determined by single-crystal X-ray diffraction to be cubic Mg
2Si or Mg
2Ge [
25]. The other crystals could not be identified reliably. We posit that Mg is either too small to form structures comparable to the Ca, Sr, and Ba compounds detailed in this paper, or may form covalent bonds with Al and the
Tt element. Reactions with the heavier congener of Si and Ge, Sn, were not explored as a part of this study due to time limitations.
In summary, the reaction method detailed in this paper, with or without significant deviations, may produce more A2LiAlTt2 (A = Mg, Yb; Tt = Si, Ge, Sn) compounds, but further inquiries must be made.
3. Discussion
We begin the discussion with the structure of Ca
2LiAlSi
2 and Ca
2LiAlGe
2, which crystallize with the orthorhombic space group
Pnma (no. 62). They are isotypic with TiNiSi structure type (Pearson symbol
oP12) [
25]; the structure has three unique positions in the asymmetric unit. As such, and as described previously, Li and Al atoms are considered to be statistically disordered.
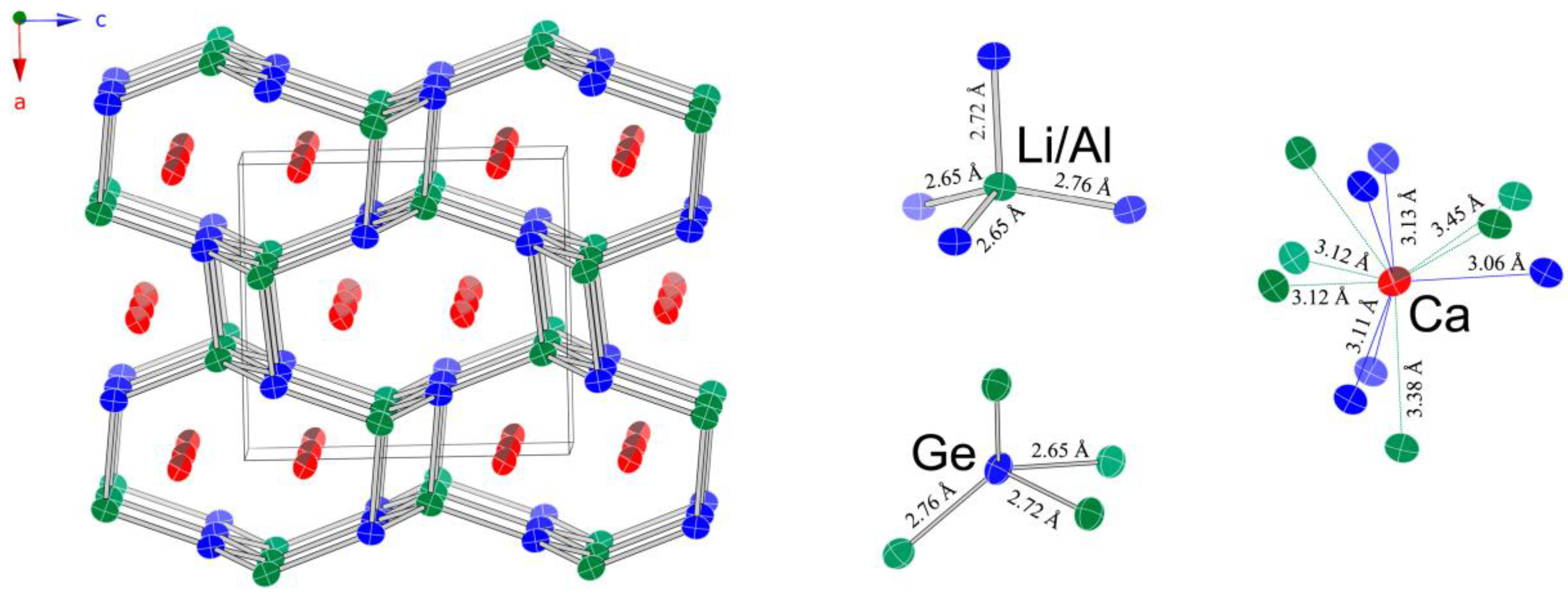
A schematic representation of the structure is given in
Figure 2. The chosen view shows that the structure can be viewed as a 3D network made up of edge- and corner-shared tetrahedra (Al
Tt4 and Li
Tt4), with the Ca atoms taking the space within the created channels. The tetrahedral units are slightly distorted from the ideal geometry and have two shorter and two longer Li/Al–
Tt bonds. The respective distances are tabulated in
Table 5.
Four-fold coordination environments exist around the Si and Ge atoms too, but in that case, the distortions from the ideal tetrahedral angle, in particular, are more severe. Si—Li/Al and Ge—Li/Al distances are about 10% longer than the sum of the single-bonded Pauling covalent radii (
rLi = 1.225 Å;
rAl = 1.248 Å;
rSi = 1.173 Å;
rGe = 1.242 Å) [
26]. These distances match the reported values in a number of alumo-silicides and alumo-germanides, such as Li
8Al
3Si
5 [
27,
28], LiAlSi [
28], CaAl
2Si
2 [
29,
30], SrAl
2Si
2 [
31,
32], BaAl
2Si
2 [
33],
LnAl
2Si
2 (
Ln = Y, Sm, Tb, Dy, Yb) [
34],
Ln2Al
3Si
2 and
Ln2AlSi
2 (
Ln = Y, Tb-Lu) [
35], Ba
7.5Al
13Si
29 [
36], LiAlGe [
37], CaAl
2Ge
2 [
38,
39],
LnAl
2Ge
2 (
Ln = Y, La, Nd, Gd, Tb, Lu) [
40], BaAl
2Ge
2 [
41],
A3Al
2Ge
2 (
A = Ca, Sr, Ba) [
42], and
LnAl
2X2 (
Ln = Eu, Yb;
X = Si, Ge) [
43], among others. Ca atoms have a total coordination number 11, with 6 nearest Li/Al and 5 nearest
Tt atoms. The shortest contact is
dCa—Si = 3.06 Å, which also fairly closely matches the sum of the single-bonded Pauling covalent radii of Si and Ca (
rCa = 1.736 Å).
In the fully ionic approximation, the structures of Ca
2LiAlSi
2 and Ca
2LiAlGe
2 can be rationalized as (Ca
2+)
2(Li
+)(Al
3+)(Si
4−)
2 and (Ca
2+)
2(Li
+)(Al
3+)(Ge
4−)
2, i.e., the compounds should be considered intrinsic semiconductors. Applying the Zintl concept [
1] also leads to the same conclusion—when treating Li as a cation and Al as a part of the polyanionic four-bonded substructure, it becomes possible to partition the valence electrons the following way: (Ca
2+)
2(Li
+)(4
b-Al
1−)(4
b-Si
0)(0
b-Si
4−) and (Ca
2+)
2(Li
+)(4
b-Al
1−)(4
b-Ge
0)(0
b-Ge
4−). To arrive at this formula breakdown, we are assuming covalent Si—Al and Ge—Al interactions; Si and Al are coordinated to Al atoms 50% of the time. In the remaining 50% of the time, Li takes the Al position and covalency of the Si—Li and Ge—Li is neglected, i.e., Si and Ge are treated as isolated, closed-shell anions.
Following periodic trends, the structure of Sr
2LiAlSi
2 and Sr
2LiAlGe
2 will be discussed next. These compounds are also orthorhombic, but with the base-centered space group
Cmce (no. 64). They are isotypic with Na
2LiAlP
2 (Pearson symbol
oS48) [
44]. The structure has six unique positions in the asymmetric unit—two for the Sr atom, two for the Si or Ge atoms, and one each for the Li and Al atoms,
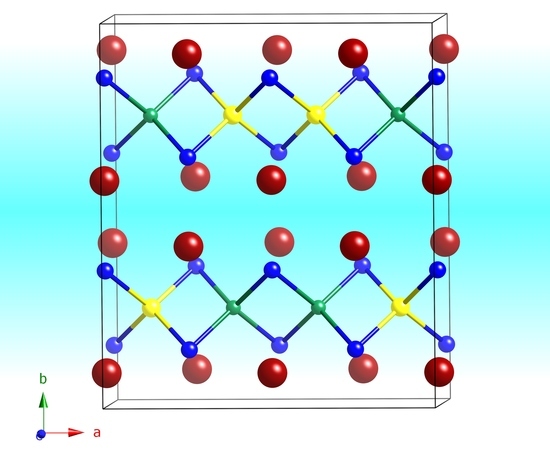
Table 4. As such, and as described previously, this is the only “true 2-1-1-2” structure, given that Li and Al atoms are ordered crystallographically here.
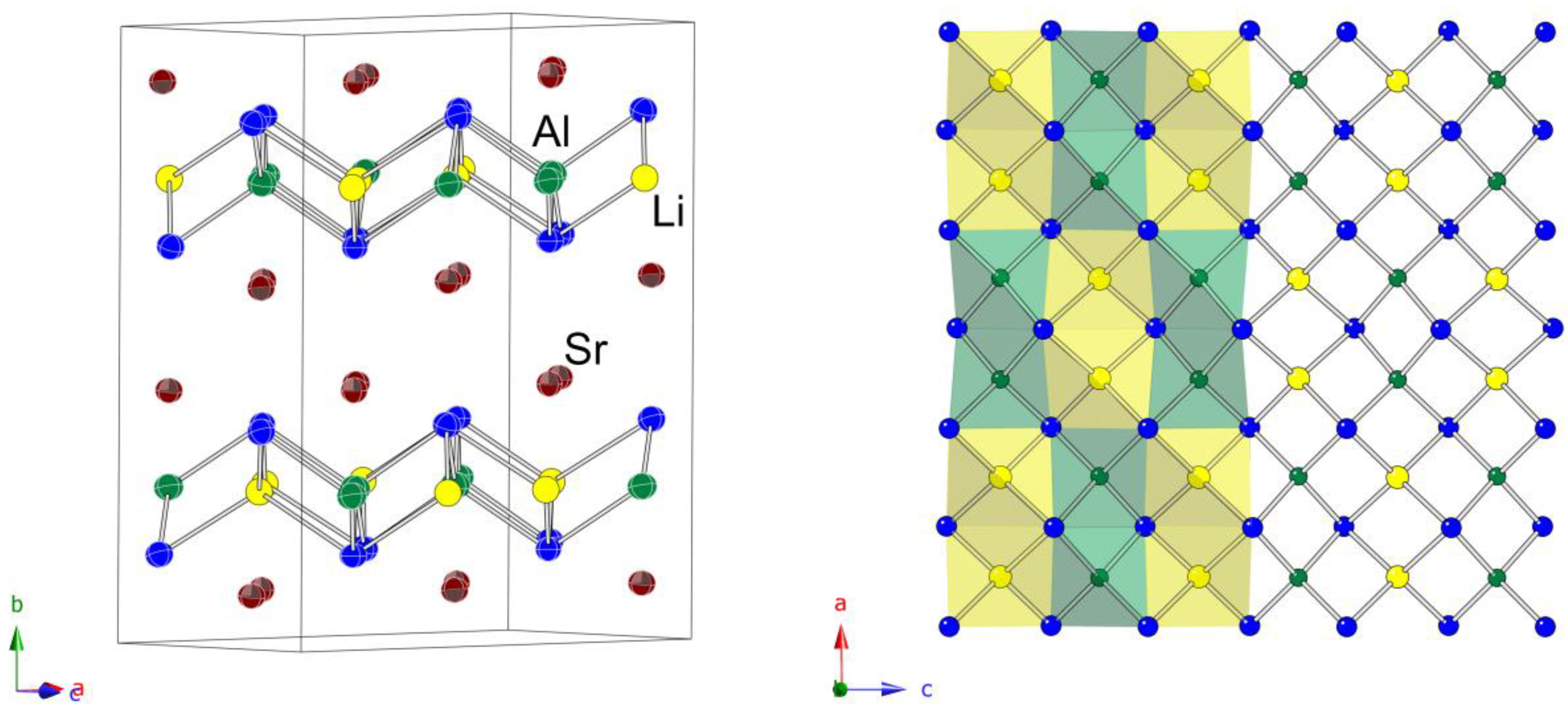
A schematic representation of the structure is given in
Figure 3. The chosen view shows that the structure can be viewed as slabs made up of all edge-shared Al
Tt4 and Li
Tt4 tetrahedra parallel to the
ac plane. The general topology of the 2D polyanionic substructure is reminiscent of the PbO-like layers that are frequently observed in the structures of many pnictides and chalcogenides [
45,
46,
47], and analogous to the layers seen in the tetragonal structures of Ba
2LiAlSi
2 and Ba
2LiAlGe
2, which will be discussed next. The difference is that here, given that there is no Li/Al disorder, the exact positions of Li and Al atoms are known and the ordering pattern is checkerboard-like, with a repeating sequence of Al
Tt4 × 2 by Li
Tt4 × 2 (
Figure 3, right panel).
The Al
Tt4 and Li
Tt4 tetrahedral units are ever so slightly distorted from the ideal geometry and have two shorter and two longer Li/Al–
Tt bonds. The refined bond lengths match the sum of the single-bonded Pauling covalent radii of the elements well. Sr atoms are arranged in double layers between the [LiAlSi
2] and [LiAlGe
2] slabs. They have a total coordination number of nine, with five nearest
Tt atoms, two nearest Al, and two neighboring Li atoms. All respective distances are tabulated in
Table 6.
The application of the Zintl concept here [
1] is more instructive than what we considered before on the examples of Ca
2LiAlSi
2 and Ca
2LiAlGe
2, given that no disorder is present and no assumptions need to be made. Hence, using the same scheme to partition the valence electrons, we can propose the following formulae breakdown: (Sr
2+)
2(Li
+)(4
b-Al
1−)(2
b-Si
2−)
2 and (Sr
2+)
2(Li
+)(4
b-Al
1−)(2
b-Ge
2−)
2. To arrive at these formulae, only Si—Al and Ge—Al interactions were treated as covalent. In a more extreme case, where Si—Li and Ge—Li bonds are also treated as covalent, all
Tt atoms will be four-bonded and will bear no formal charge. Li with four covalent bonds must be considered formally as 4
b-Li
3−, in analogy with the Zintl clathrate Ba
8Li
5Ge
41 [
11]. In this scenario, the valence electron count can be ascribed as follows: (Sr
2+)
2(4
b-Li
3−)(4
b-Al
1−)(4
b-Si
0)
2 and (Sr
2+)
2(4
b-Li
3−)(4
b-Al
1−)(4
b-Ge
0)
2.
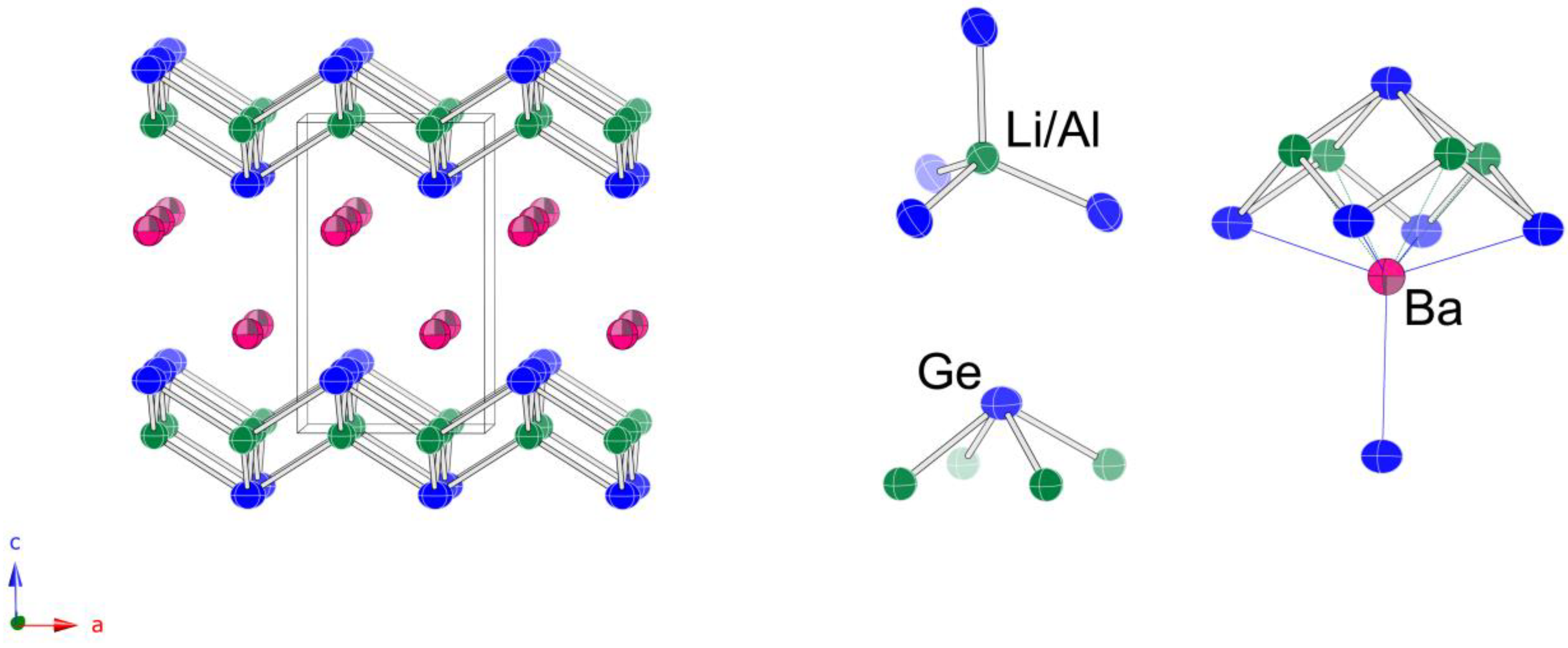
The last remaining structure to briefly discuss is that of Ba
2LiAlSi
2 and Ba
2LiAlGe
2. The latter two compounds crystallize with the tetragonal space group
P4/
nmm (no. 129). This is a structure type known as PbFCl (Pearson symbol
tP6) [
25], which is very common and well-known (
Figure 4). There are three unique positions in the asymmetric unit (
Table 4), meaning that Li and Al atoms are, again, statistically disordered.
From the schematic representation of the structure in
Figure 4, it is evident that the overall arrangement of the atoms is nearly identical to that described above for the orthorhombic Sr
2LiAlSi
2 and Sr
2LiAlGe
2; the major difference is that the ordering of the tetrahedral Al
Tt4 and Li
Tt4 units is averaged in this symmetry. The Ba atoms take the space between the layers and have the exact same coordination environment (CN 9) as the Sr atoms in Sr
2LiAlSi
2 and Sr
2LiAlGe
2 (
vide supra). Another Si atom is 3.9 Å away, i.e., is the topmost Si atom in
Figure 4, for which a line denoting Si—Ba contact is not drawn.
Naturally, given that Ba is the largest element from group 2, all distances here are slightly longer compared to the Ca- and Sr-based compounds. Specifically, the average bond lengths of A–Al and A–Li (A = Ca, Sr, Ba) in Ca2LiAlTt2, Sr2LiAlTt2, and Ba2LiAlTt2 (Tt = Si, Ge) increase as the differing cation increases in size. In other words, the average A–Al and A–Li contacts in Ca2LiAlSi2 are shorter than those in Sr2LiAlSi2, which are in turn shorter than those in Ba2LiAlSi2. The same principle applies to the analogous germanides. For example, for the silicides, the average Ca–Al/Li distance is 3.14 Å, whereas the average Ba–Al/Li is 3.36 Å (recall that both structures have Al and Li atoms disordered).
Of note is also the fact that the fourfold symmetry in Ba
2LiAl
Tt2 makes the tetrahedral units much closer to the ideal geometry, as they have four equal Li/Al–
Tt bonds. Ge coordination environment is square pyramidal. All respective distances are tabulated in
Table 7.
Just like with the cases of Ca2LiAlTt2 and Sr2LiAlTt2, in the fully ionic approximation, the structures of Ba2LiAlSi2 and Ba2LiAlGe2 can be rationalized as (Ba2+)2(Li+)(Al3+)(Si4−)2 and (Ba2+)2(Li+)(Al3+)(Ge4−)2.
4. Materials and Methods
4.1. Synthesis
Objective: To synthesize and establish the structure of a series of novel quaternary alkaline earth metal and rare earth metal silicides and germanides, with the base formula A2LiAlTt2 (A = Mg, Ca, Sr, Ba, Eu, Yb; Tt = Si, Ge). For this purpose, given that Li and most of the alkaline earth metals are reactive towards air and moisture, all synthetic and post-synthetic manipulations were performed inside an argon-filled glove box or under vacuum.
All metals were used as received from Alfa or Aldrich (>99.9% wt.), except for Ba and Li, whose surfaces had to be carefully cleaned with a blade each time the metal rods were cut. In a typical experiment, a mixture of elements with the desired stoichiometric ratio (total weight ca. 500 mg) was loaded into a Nb tube, which was then sealed by arc welding under an Ar atmosphere. The welded Nb tube was subsequently enclosed in a fused silica tube, which was flame-sealed under vacuum (below discharge).
To synthesize A2LiAlSi2 and A2LiAlGe2, the tubes with the reactant mixtures inside were heated in tube furnaces to 1223 K (rate of 300 K/h) and equilibrated for 8 h, followed by a cooling to 573 K at a rate of 200 K/h. The tubes were rotated in the furnaces at temperatures > 973 K. At the completion of their heat treatment, all tubes were quenched in cold water. The products of this method were not phase-pure.
Note: Many of the compounds detailed in this paper react rapidly with air and/or moisture. Corresponding with periodic trends, the germanides are more reactive than the silicide compounds. Moreover, the compounds’ reactivity can be represented as Ca2LiAlTt2 < Sr2LiAlTt2 < Ba2LiAlTt2. A2LiAlGe2 (A = Ca, Sr, Ba) samples reacted with the ambient air and turned opaquely yellow within a few minutes. The A2LiAlSi2 samples experienced no visible color changes, but were not air stable beyond one hour, even when covered under an oil film. In direct contact with deionized water, both A2LiAlSi2 and A2LiAlGe2 ignited and volatilized. Due to these compounds’ extreme sensitivity to the ambient atmosphere, further characterization besides the crystallographic work could not be effectively completed.
4.2. Crystallographic Studies
Powder X-ray diffraction (PXRD) measurements were carried out at room temperature on a Rigaku Miniflex diffractometer (filtered Cu Kα radiation, λ = 1.5418 Å). Small portions of the obtained samples were ground into powder using agate mortars and pestles inside the argon-filled glovebox. The specimens for PXRD were prepared by covering the polycrystalline material with a light film of oil and were transferred to another glovebox, which housed the diffractometer. The data were collected as quickly as possible with a typical θ–θ scan of 0.05°/step and data acquisition rate of 2 s/step. The collected powder X-ray diffraction patterns were used for phase identification of the reaction products only.
The PXRD measurements before and after exposure to air over confirmed the previously stated instability in air.
Single-crystal X-ray diffraction data for the six compounds were collected at 200 K using a Bruker SMART CCD-based diffractometer (three-circle goniometer; monochromated Mo
Kα radiation (
λ = 0.710 73 Å)). Firstly, several crystals from each batch were selected and checked before the best ones were chosen for intensity data collection. The experiments were managed in batch runs consisting of an appropriate number of frames to ensure 98+% coverage in angular range in 2θ of up to ca. 60° (frame width was typically 0.5–0.6° in ω and θ with a data acquisition rate of 6–8 s/frame). Data processing was performed with the Bruker supplied software. SADABS was used for semi-empirical absorption correction based on equivalents [
48]. The structure factors were sorted and merged by the subprogram XPREP in the SHELXTL software package [
49], which was also employed in the space group determination. The structures were solved by direct methods and refined to convergence by full-matrix least-squares methods on
F2. Selected details of the data collection and relevant crystallographic parameters are given in
Table 1,
Table 2,
Table 3 and
Table 4. Refined distances are tabulated in
Table 5,
Table 6 and
Table 7.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}