
Separation of Serum and Plasma Proteins for In-Depth Proteomic Analysis


Abstract
1. Introduction
2. Plasma vs. Serum
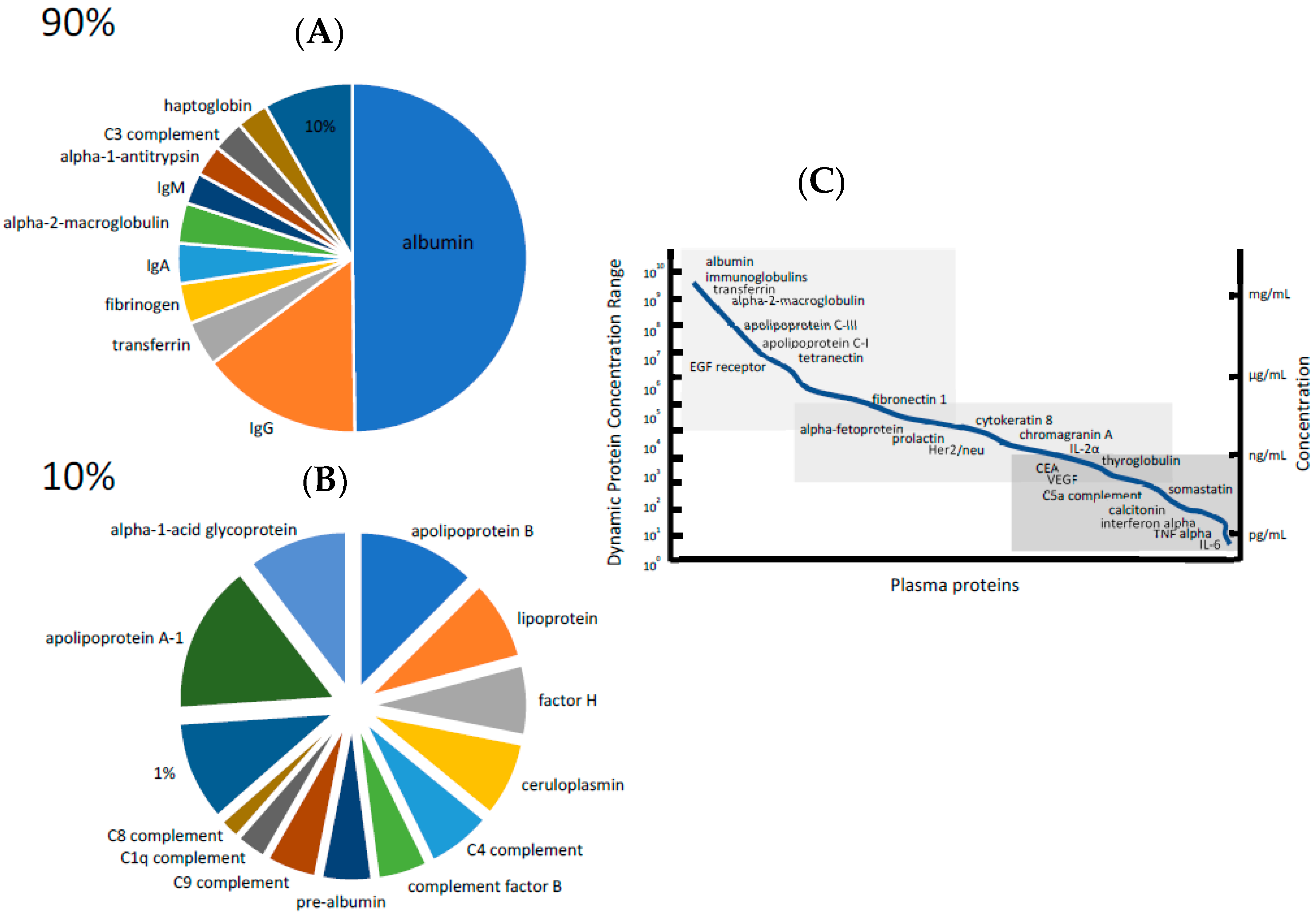
3. The Dynamic Range of Protein Concentration Problem
3.1. High-Abundance Protein Depletion
3.2. Low-Abundance Protein Enrichment
4. Electrophoresis for the Fractionation of Serum and Plasma Proteins
4.1. Two-Dimensional Gels Using Isoelectric Immobilized pH Gradients
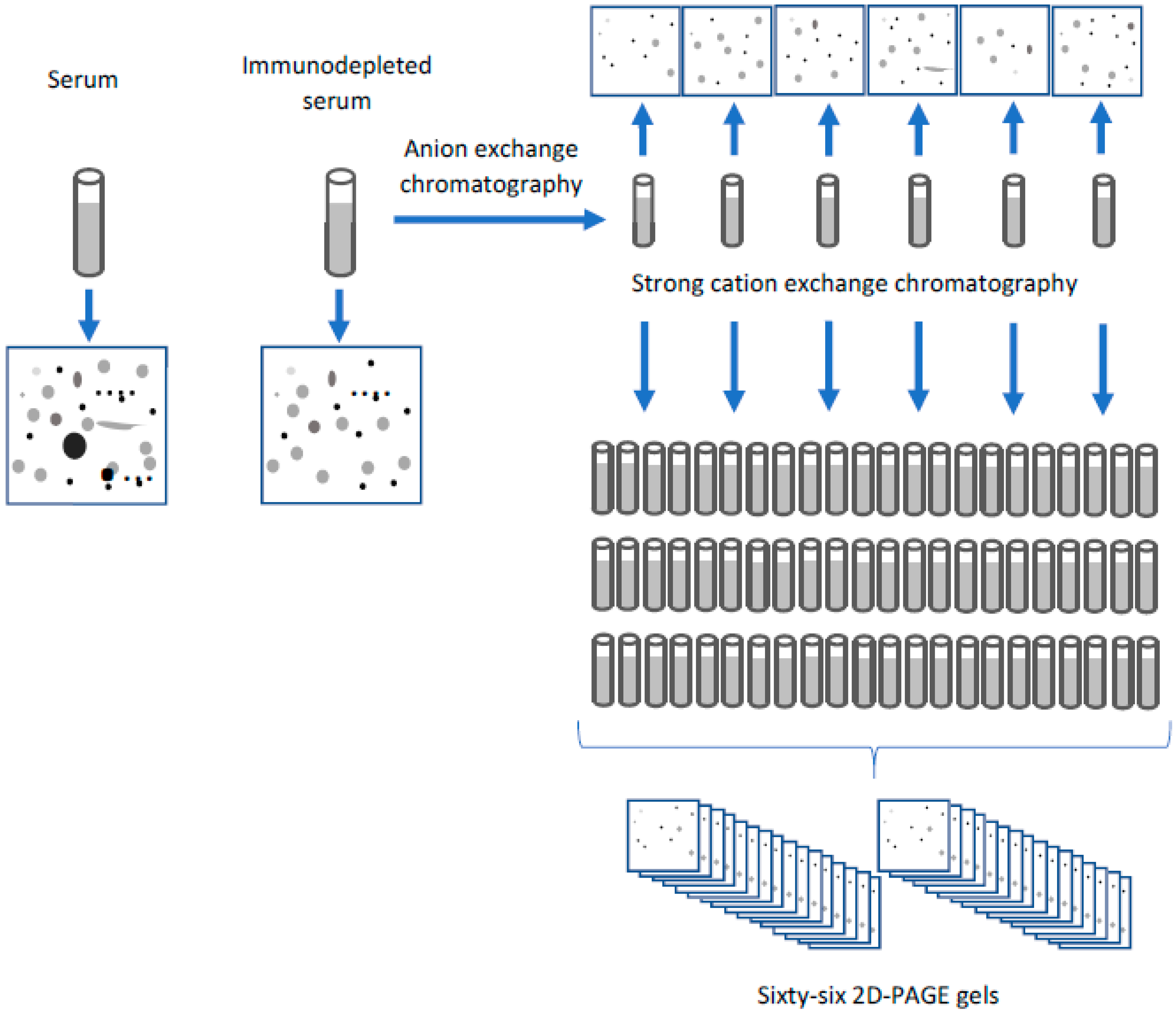
4.2. Combining Two-Dimensional Gels with Chromatography
5. Chromatography for the Fractionation of Serum and Plasma Proteins
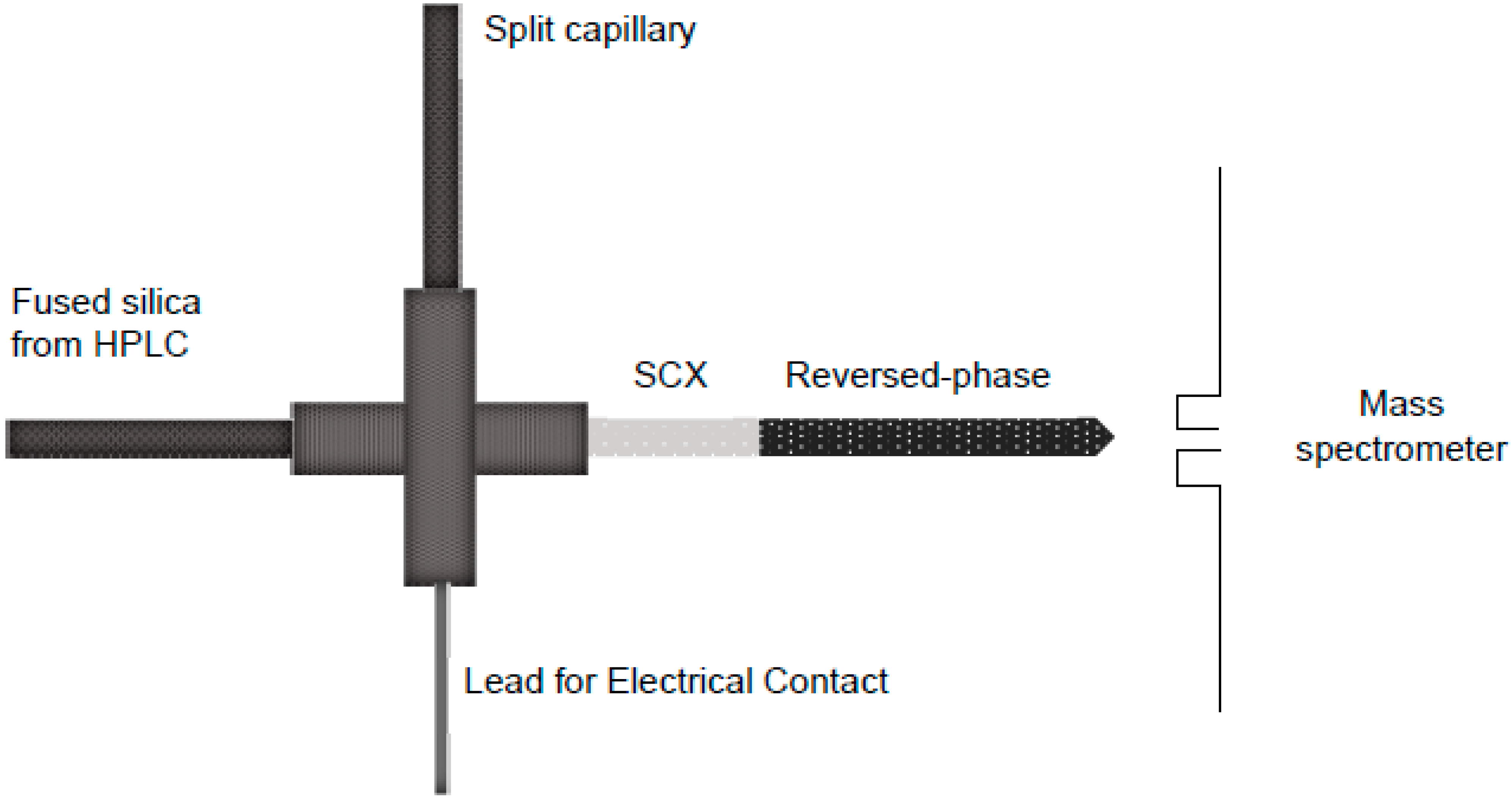
Multidimensional Protein Identification Technology
6. Other Separation Techniques for Characterizing the Serum/Plasma Proteome
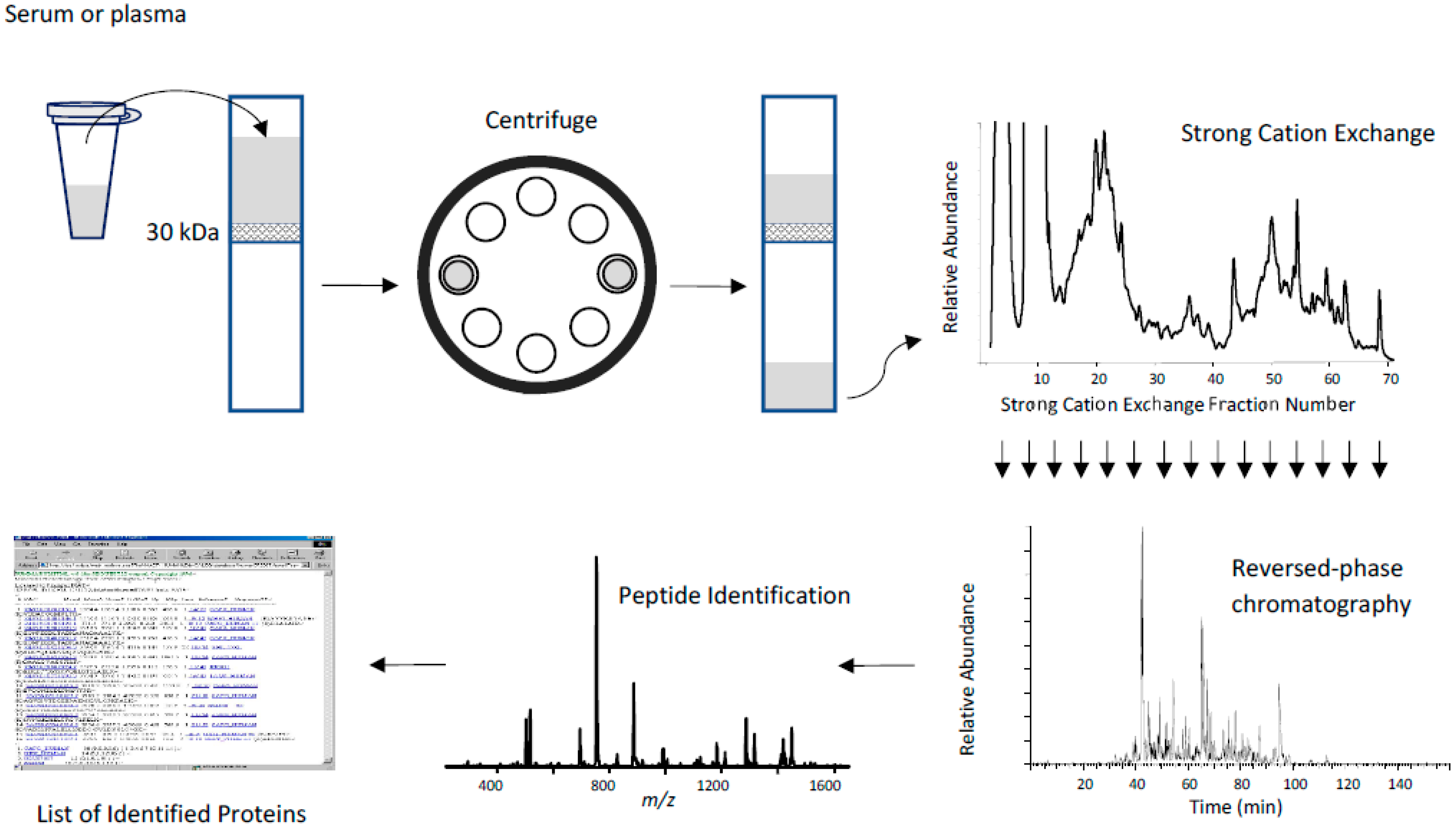
Low Molecular Weight Fractionation
7. Applications
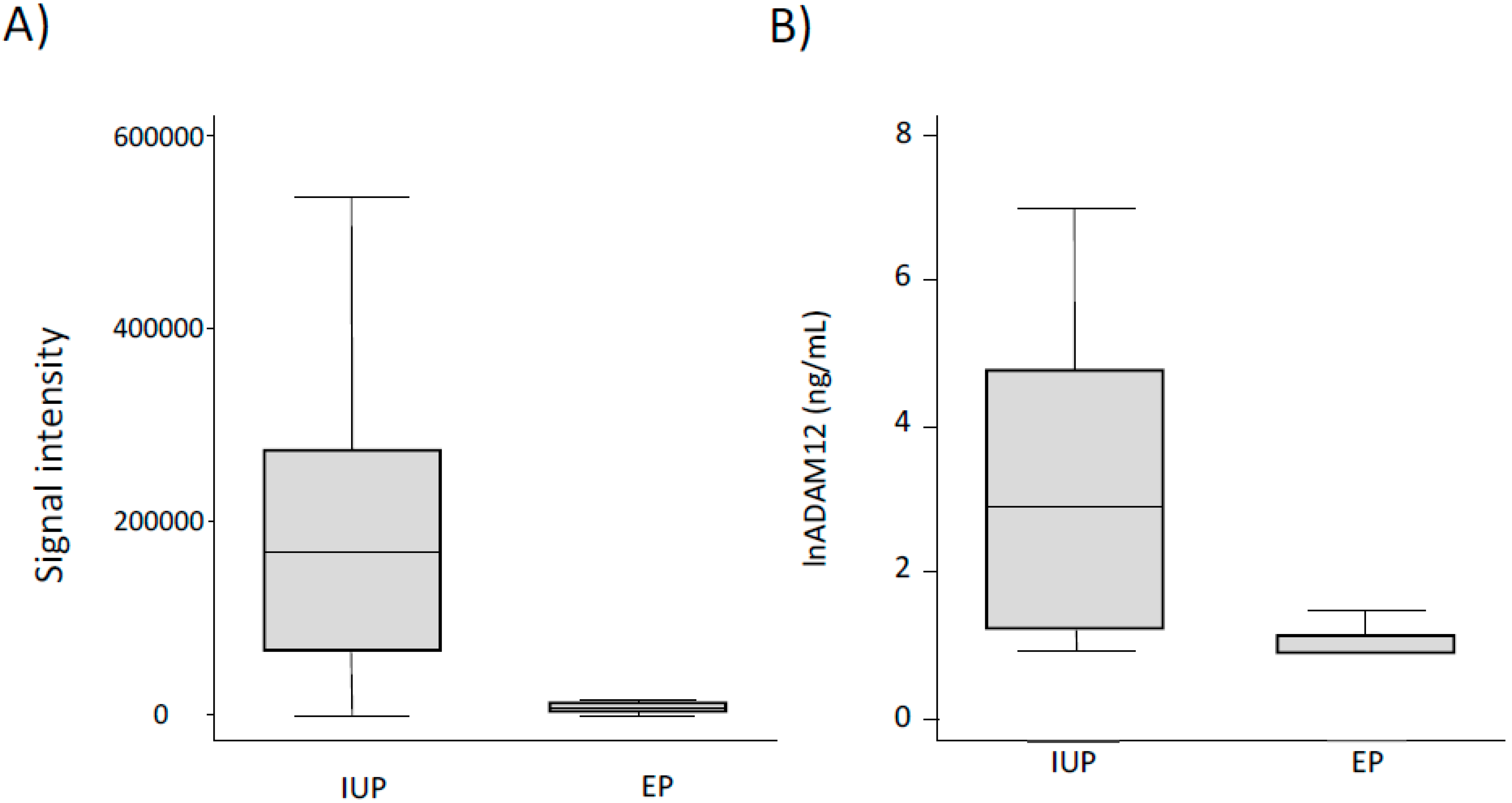
7.1. Biomarkers of Ectopic Pregnancy
7.2. Identification of a Novel Proatherogenic Peptide Hormone
8. Current Challenges in Serum and Plasma Proteomics
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shorthouse, J.; Wilson, S. Use of blood components in clinical practice. Nurs. Stand. 2019, 34, 76. [Google Scholar] [CrossRef] [PubMed]
- Blood Test. Available online: https://www.hopkinsmedicine.org/health/treatment-tests-and-therapies/blood-test (accessed on 4 January 2022).
- Chace, H.D.; Kalas, T.A.; Naylor, E.W. Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns. Clin. Chem. 2003, 49, 1797. [Google Scholar] [CrossRef] [PubMed]
- Basic Metabolic Panel (BMP). Available online: https://medlineplus.gov/lab-tests/basic-metabolic-panel-bmp/ (accessed on 5 January 2022).
- Complete Blood Count (CBC). Available online: https://www.mayoclinic.org/tests-procedures/complete-blood-count/about/pac-20384919 (accessed on 5 January 2022).
- Tirumalai, R.S.; Chan, K.C.; Prieto, D.A.; Issaq, H.J.; Conrads, T.P.; Veenstra, T.D. Characterization of the low molecular weight serum proteome. Mol. Cell. Proteom. 2003, 2, 1096. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Polanski, M.; Pieper, R.; Gatlin, T.; Tirumalai, R.S.; Conrads, T.P.; Veenstra, T.D.; Adkins, J.N.; Pounds, J.G.; Fagan, R.; et al. The human plasma proteome: A nonredundant list developed by combination of four separate sources. Mol. Cell. Proteom. 2004, 3, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Voytik, E.; Treit, P.V.; Doll, S.; Kleinhempel, A.; Niu, L.; Müller, J.B.; Buchholtz, M.L.; Bader, J.M.; Teupser, D.; et al. Plasma proteome profiling to detect and avoid sample-related biases in biomarker studies. EMBO Mol. Med. 2019, 11, e10427. [Google Scholar] [CrossRef] [PubMed]
- Almeida, N.; Rodriguez, J.; Pla Parada, I.; Perez-Riverol, Y.; Woldmar, N.; Kim, Y.; Oskolas, H.; Betancourt, L.; Valdés, J.G.; Sahlin, K.B.; et al. Mapping the melanoma plasma proteome (MPP) using single-shot proteomics interfaced with the WiMT database. Cancers 2021, 13, 6224. [Google Scholar] [CrossRef]
- Lygirou, V.; Makridakis, M.; Vlahou, A. Biological sample collection for clinical proteomics: Existing SOPs. Methods Mol. Biol. 2015, 1243, 3–27. [Google Scholar]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef]
- Maloney, J.P.; Silliman, C.C.; Ambruso, D.R.; Wang, J.; Tuder, R.M.; Voelkel, N.F. In vitro release of vascular endothelial growth factor during platelet aggregation. Am. J. Physiol. 1998, 275, H105401061. [Google Scholar] [CrossRef]
- Omenn, G.S. Plasma proteomics, the human proteome project, and cancer-associated alternative splice variant proteins. Biochim. Biophys. Acta 2014, 1844, 866–873. [Google Scholar] [CrossRef][Green Version]
- Das, L.; Murthy, V.; Varma, A.K. Comprehensive analysis of low molecular weight serum proteome enrichment for mass spectrometric studies. ACS Omega 2020, 5, 28877–28888. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Shi, T.; Fillmore, T.L.; Schepmoes, A.A.; Brewer, H.; Gao, Y.; Song, E.; Wang, H.; Rodland, K.D.; Qian, W.J.; et al. Deep-dive targeted quantification for ultrasensitive analysis of proteins in nondepleted human blood plasma/serum and tissues. Anal. Chem. 2017, 89, 9139–9146. [Google Scholar] [CrossRef] [PubMed]
- Fredolini, C.; Pathak, K.V.; Paris, L.; Chapple, K.M.; Tsantilas, K.A.; Rosenow, M.; Tegeler, T.J.; Garcia-Mansfield, K.; Tamburro, D.; Zhou, W.; et al. Shotgun proteomics coupled to nanoparticle-based biomarker enrichment reveals a novel panel of extracellular matrix proteins as candidate serum protein biomarkers for early-stage breast cancer detection. Breast Cancer Res. 2020, 22, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Echan, L.A.; Tang, H.Y.; Ali-Khan, N.; Lee, K.; Speicher, D.W. Depletion of multiple high-abundance proteins improves protein profiling capacities of human serum and plasma. Proteomics 2005, 5, 3292–3303. [Google Scholar] [CrossRef] [PubMed]
- Hinerfeld, D.; Innamorati, D.; Pirro, J.; Tam, S.W. Serum/plasma depletion with chicken immunoglobulin Y antibodies for proteomic analysis from multiple Mammalian species. J. Biomol. Tech. 2004, 15, 184–190. [Google Scholar]
- Chromy, B.A.; Gonzales, A.D.; Perkins, J.; Choi, M.W.; Corzett, M.H.; Chang, B.C.; Corzett, C.H.; McCutchen-Maloney, S.L. Proteomic analysis of human serum by two-dimensional differential gel electrophoresis after depletion of high-abundant proteins. J. Proteome Res. 2004, 3, 1120–1127. [Google Scholar] [CrossRef]
- Björhall, K.; Miliotis, T.; Davidsson, P. Comparison of different depletion strategies for improved resolution in proteomic analysis of human serum samples. Proteomics 2005, 5, 307–317. [Google Scholar] [CrossRef]
- Multiple Affinity Removal System. Available online: https://www.agilent.com/en/product/agilent-multiple-affinity-removal-spin-columns-cartridges (accessed on 27 February 2022).
- Dardé, V.M.; Barderas, M.G.; Vivanco, F. Depletion of high-abundance proteins in plasma by immunoaffinity subtraction for two-dimensional difference gel electrophoresis analysis. Methods Mol. Biol. 2007, 357, 351–364. [Google Scholar]
- Johansen, E.; Schilling, B.; Lerch, M.; Niles, R.K.; Liu, H.; Li, B.; Allen, S.; Hall, S.C.; Witkowska, H.E.; Regnier, F.E.; et al. A lectin HPLC method to enrich selectively-glycosylated peptides from complex biological samples. J. Vis. Exp. 2009, 1, 1398–1401. [Google Scholar] [CrossRef]
- Zhou, M.; Lucas, D.A.; Chan, K.C.; Issaq, H.J.; Petricoin, E.F., 3rd; Liotta, L.A.; Veenstra, T.D.; Conrads, T.P. An investigation into the human serum “interactome”. Electrophoresis 2004, 25, 1289–1298. [Google Scholar] [CrossRef]
- Chan, K.C.; Lucas, D.A.; Hise, D.; Schaefer, C.F.; Xiao, Z.; Janini, G.M.; Buetow, K.H.; Issaq, H.J.; Veenstra, T.D.; Conrads, T.P. Analysis of the human serum proteome. Clin. Proteom. 2004, 1, 101–225. [Google Scholar] [CrossRef]
- Finn, T.E.; Nunez, A.C.; Sunde, M.; Easterbrook-Smith, S.B. Serum albumin prevents protein aggregation and amyloid formation and retains chaperone-like activity in the presence of physiological ligands. J. Biol. Chem. 2012, 287, 21530–21540. [Google Scholar] [CrossRef] [PubMed]
- Petricoin, E.F.; Belluco, C.; Araujo, R.P.; Liotta, L.A. The blood peptidome: A higher dimension of information content for cancer biomarker discovery. Nat. Rev. Cancer 2006, 6, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.I.; Ross, S.; Lowenthal, M.S.; Fusaro, V.; Fishman, D.A.; Petricoin, E.F., 3rd; Liotta, L.A. Biomarker amplification by serum carrier protein binding. Dis. Markers 2003, 19, 1–10. [Google Scholar] [CrossRef]
- Lowenthal, M.S.; Mehta, A.I.; Frogale, K.; Bandle, R.W.; Araujo, R.P.; Hood, B.L.; Veenstra, T.D.; Conrads, T.P.; Goldsmith, P.; Fishman, D.; et al. Analysis of albumin-associated peptides and proteins from ovarian cancer patients. Clin. Chem. 2005, 51, 1933–1945. [Google Scholar] [CrossRef]
- Jonkers, J.; Meuwissen, R.; van der Gulden, H.; Petersen, H.; van der Valk, M.; Berns, A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 2001, 29, 418–425. [Google Scholar] [CrossRef]
- Moss, E.L.; Hollingworth, J.; Reynolds, T.M. The role of CS125 in clinical practice. J. Clin. Pathol. 2005, 58, 308–312. [Google Scholar] [CrossRef]
- Boschetti, E.; Zilberstein, G.; Righetti, P.G. Combinatorial peptides: A library that continuously probes low-abundance proteins. Electrophoresis 2022, 43, 355–369. [Google Scholar] [CrossRef]
- ProteoMiner Protein Enrichment Kits. Available online: https://www.bio-rad.com/en-us/product/proteominer-protein-enrichment-kits?ID=1dd94f06-7658-4ab4-b844-e29a1342a214 (accessed on 21 January 2022).
- D’Amato, A.; Bachi, A.; Fasoli, E.; Boschetti, E.; Peltre, G.; Sénéchal, H.; Righetti, P.G. In-depth exploration of cow’s whey proteome via combinatorial peptide ligand libraries. J. Proteome Res. 2009, 8, 3925–3936. [Google Scholar] [CrossRef]
- Martos, G.; López-Fandiño, R.; Molina, E. Immunoreactivity of hen egg allergens: Influence on in vitro gastrointestinal digestion of the presence of other egg white proteins and of egg yolk. Food Chem. 2013, 136, 775–781. [Google Scholar] [CrossRef]
- Boschetti, E.; Bindschedler, L.V.; Tang, C.; Fasoli, E.; Righetti, P.G. Combinatorial peptide ligand libraries and plant proteomics: A winning strategy at a price. J. Chromatogr. A. 2009, 1216, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Fekkar, A.; Pionneau, C.; Brossas, J.Y.; Marinach-Patrice, C.; Snounou, G.; Brock, M.; Mazier, D. DIGE enables the detection of a putative serum biomarker of fungal origin in a mouse model of invasive aspergillosis. J. Proteome 2012, 75, 2536–2549. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, P.H. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [CrossRef]
- Wasinger, V.C.; Cordwell, S.J.; Cerpa-Poljak, A.; Yan, J.X.; Gooley, A.A.; Wilkins, M.R.; Duncan, M.W.; Harris, R.; Williams, K.L.; Humphery-Smith, I. Progress with gene-product mapping of the Mollicutes: Mycoplasma genitalium. Electrophoresis 1995, 16, 1090–1094. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.L., Jr. High resolution two-dimensional polyacrylamide electrophoresis of human serum proteins. Am. J. Clin. Pathol. 1972, 57, 173–185. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Anderson, L.; Anderson, N.G. High resolution two-dimensional electrophoresis of human plasma proteins. Proc. Natl. Acad. Sci. USA 1977, 74, 5421–5425. [Google Scholar] [CrossRef] [PubMed]
- Görg, A.; Drews, O.; Lück, C.; Weiland, F.; Weiss, W. 2-DE with IPGs. Electrophoresis 2009, 30, S122–S132. [Google Scholar] [CrossRef] [PubMed]
- Vasudev, N.S.; Ferguson, R.E.; Cairns, D.A.; Stanley, A.J.; Selby, P.J.; Banks, R.E. Serum biomarker discovery in renal cancer using 2-DE and prefractionation by immunodepletion and isoelectric focusing; increasing coverage or more of the same? Proteomics 2008, 8, 5074–5085. [Google Scholar] [CrossRef]
- Kim, H.J.; Kang, H.J.; Lee, H.; Lee, S.T.; Yu, M.H.; Kim, H.; Lee, C. Identification of S100A8 and S100A9 as serological markers for colorectal cancer. J. Proteome Res. 2009, 8, 1368–1379. [Google Scholar] [CrossRef]
- Pieper, R.; Gatlin, C.L.; Makusky, A.J.; Russo, P.S.; Schatz, C.R.; Miller, S.S.; Su, Q.; McGrath, A.M.; Estock, M.A.; Parmar, P.P.; et al. The human serum proteome: Display of nearly 3700 chromatographically separated protein spots on two-dimensional electrophoresis gels and identification of 325 distinct proteins. Proteomics 2003, 3, 1345–1364. [Google Scholar] [CrossRef]
- Issaq, H.J.; Veenstra, T.D. The role of electrophoresis in disease biomarker discovery. Electrophoresis 2007, 28, 1980–1988. [Google Scholar] [CrossRef] [PubMed]
- Arentz, G.; Weiland, F.; Oehler, M.K.; Hoffmann, P. State of the art of 2D DIGE. Proteom. Clin. Appl. 2015, 9, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Lazensky, R.; Silva-Sanchez, C.; Kroll, K.J.; Chow, M.; Chen, S.; Tripp, K.; Walsh, M.T.; Denslow, N.D. Investigating an increase in Florida manatee mortalities using a proteomic approach. Sci. Rep. 2021, 11, 4282–4295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yu, W.; Cao, X.; Wang, Y.; Zhu, C.; Guan, J. Identification of serum biomarkers in patients with Alzheimer’s disease by 2D-DIGE proteomics. Gerontology 2022, 12, 1–13. [Google Scholar] [CrossRef]
- Molnár, I.; Horváth, C. Reverse-phase chromatography of polar biological substances: Separation of catechol compounds by high-performance liquid chromatography. Clin. Chem. 1976, 22, 1497–1502. [Google Scholar] [CrossRef]
- Atack, C.V.; Magnusson, T. Individual elution of noradrenaline (together with adrenaline), dopamine, 5-hydroxytryptamine and histamine form a single, strong cation exchange column, by means of mineral acid-organic solvent mixtures. J. Pharm. Pharmacol. 1970, 22, 625–627. [Google Scholar] [CrossRef]
- Wolters, D.A.; Washburn, M.P.; Yates, J.R., 3rd. An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 2001, 73, 5683–5690. [Google Scholar] [CrossRef]
- Perrot, M.; Sagliocco, F.; Mini, T.; Monribot, C.; Schneider, U.; Shevchenko, A.; Mann, M.; Jenö, P.; Boucherie, H. Two-dimensional gel protein database of Saccharomyces cerevisiae (update 1999). Electrophoresis 1999, 20, 2280–2298. [Google Scholar] [CrossRef]
- Adkins, J.N.; Varnum, S.M.; Auberry, K.J.; Moore, R.J.; Angell, N.H.; Smith, R.D.; Springer, D.L.; Pounds, J.G. Toward a human blood serum proteome: Analysis by multidimensional separation coupled with mass spectrometry. Mol. Cell. Proteom. 2002, 1, 947–955. [Google Scholar] [CrossRef]
- Barnhart, K.T. Clinical practice. Ectopic pregnancy. N. Engl. J. Med. 2009, 361, 379–387. [Google Scholar] [CrossRef]
- Seeber, B.E.; Barnhart, K.T. Suspected ectopic pregnancy. Obstet. Gynecol. 2006, 107, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Kirk, E.; Bottomley, C.; Bourne, T. Diagnosing ectopic pregnancy and current concepts in the management of pregnancy of unknown location. Hum. Reprod. Update 2014, 20, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Refaat, B.; Bahathiq, A.O. The performances of serum activins and follistatin in the diagnosis of ectopic pregnancy: A prospective case-control study. Clin. Chim. Acta. 2020, 500, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Tay, J.I.; Moore, J.; Walker, J.J. Ectopic pregnancy. BMJ 2000, 320, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Beer, L.A.; Tang, H.Y.; Sriswasdi, S.; Barnhart, K.T.; Speicher, D.W. Systematic discovery of ectopic pregnancy serum biomarkers using 3-D protein profiling coupled with label-free quantitation. J. Proteome Res. 2011, 10, 1126–1138. [Google Scholar] [CrossRef] [PubMed]
- Gilpin, B.J.; Loechel, F.; Mattei, M.G.; Engvall, E.; Albrechtsen, R.; Wewer, U.M. A novel, secreted form of human ADAM 12 (meltrin alpha) provokes myogenesis in vivo. J. Biol. Chem. 1998, 273, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Rausch, M.E.; Beer, L.; Sammel, M.D.; Takacs, P.; Chung, K.; Shaunik, A.; Speicher, D.; Barnhart, K.T. A disintegrin and metalloprotease protein-12 as a novel marker for the diagnosis of ectopic pregnancy. Fertil. Steril. 2011, 95, 1373–1378. [Google Scholar] [CrossRef]
- Parnham, A.J.; Tarbit, I.F. “Delfia” and “Amerlite”: Two sensitive nonisotopic immunoassay systems for assay of thyrotropin compared. Clin. Chem. 1987, 33, 1421–1424. [Google Scholar] [CrossRef]
- Masaki, T.; Kodera, Y.; Terasaki, M.; Fujimoto, K.; Hirano, T.; Shichiri, M. GIP_HUMAN[22-51] is a new proatherogenic peptide identified by native plasma peptidomics. Sci. Rep. 2021, 11, 14470–14483. [Google Scholar] [CrossRef]
- Zhou, C.X.; Xie, S.C.; Li, M.Y.; Huang, C.Q.; Zhou, H.Y.; Cong, H.; Zhu, X.Q.; Cong, W. Analysis of the serum peptidome associated with Toxoplasma gondii infection. J. Proteom. 2020, 222, 103805–103813. [Google Scholar] [CrossRef]
- Padoan, A. The impact of pre-analytical conditions on human serum peptidome profiling. Proteom. Clin. Appl. 2018, 12, e1700183–e1700184. [Google Scholar] [CrossRef] [PubMed]
- Bhawal, R.; Oberg, A.L.; Zhang, S.; Kohli, M. Challenges and opportunities in clinical applications of blood-based proteomics in cancer. Cancers 2020, 12, 2428. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Picotti, P.; Huttenhain, R.; Heinzelmann-Schwarz, V.; Jovanovic, M.; Aebersold, R.; Vitek, O. Protein significance analysis in selected reaction monitoring (SRM) measurements. Mol. Cell Proteom. 2012, 11, M111.014662. [Google Scholar] [CrossRef] [PubMed]
- Skates, S.J.; Gillette, M.A.; LaBaer, J.; Carr, S.A.; Anderson, L.; Liebler, D.C.; Ransohoff, D.; Rifai, N.; Kondratovich, M.; Tezak, Z.; et al. Statistical design for biospecimen cohort size in proteomics-based biomarker discovery and verification studies. J. Proteome Res. 2013, 12, 5383–5394. [Google Scholar] [CrossRef]
- Muntel, J.; Kirkpatrick, J.; Bruderer, R.; Huang, T.; Vitek, O.; Ori, A.; Reiter, L. Comparison of protein quantification in a complex background by dia and tmt workflows with fixed instrument time. J. Proteome Res. 2019, 18, 1340–1351. [Google Scholar] [CrossRef]
- Cho, K.C.; Oh, S.; Wang, Y.; Rosenthal, L.S.; Na, C.H.; Zhang, H. Evaluation of the sensitivity and reproducibility of targeted proteomic analysis using data-independent acquisition for serum and cerebrospinal fluid proteins. J. Proteome Res. 2021, 20, 4284–4291. [Google Scholar] [CrossRef]
- Wang, H.; Dey, K.K.; Chen, P.C.; Li, Y.; Niu, M.; Cho, J.H.; Wang, X.; Bai, B.; Jiao, Y.; Chepyala, S.R.; et al. Integrated analysis of ultra-deep proteomes in cortex, cerebrospinal fluid and serum reveals a mitochondrial signature in Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 43–62. [Google Scholar] [CrossRef]
- Goldman, A.R.; Beer, L.A.; Tang, H.Y.; Hembach, P.; Zayas-Bazan, D.; Speicher, D.W. Proteome analysis using gel-LC-MS/MS. Curr. Protoc. Protein Sci. 2019, 96, e93. [Google Scholar] [CrossRef]
- Borras, E.; Canto, E.; Choi, M.; Maria Villar, L.; Alvarez-Cermeno, J.C.; Chiva, C.; Montalban, X.; Vitek, O.; Comabella, M.; Sabido, E. Protein-based classifier to predict conversion from clinically isolated syndrome to multiple sclerosis. Mol. Cell Proteom. 2016, 15, 318–328. [Google Scholar] [CrossRef]
- Yates, J.R.; Eng, J.K.; Clauser, K.R.; Burlingame, A.L. Search of sequence databases with ininterpreted high-energy collision-induced dissociation spectra of peptides. J. Am. Soc. Mass Spectrom. 1996, 7, 1089–1098. [Google Scholar] [CrossRef]
- Chalkley, R.J.; Baker, P.R.; Huang, L.; Hansen, K.C.; Allen, N.P.; Rexach, M.; Burlingame, A.L. Comprehensive analysis of a multidimensional liquid chromatography mass spectrometry dataset acquired on a quadrupole selecting, quadrupole collision cell, time-of-flight mass spectrometer: New developments in Protein Prospector allow for reliable and comprehensive automatic analysis of large datasets. Mol. Cell. Proteom. 2005, 4, 1194–1204. [Google Scholar]
- Willems, P.; Fels, U.; Staes, A.; Gevaert, K.; Van Damme, P. Use of hybrid data-dependent and -independent acquisition spectral libraries empowers dual-proteome profiling. J. Proteome Res. 2021, 20, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Horvatovich, P.; Qiao, L. Fragment mass spectrum prediction facilitates site localization of phosphorylation. J. Proteome Res. 2021, 20, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Shu, Q.; Li, M.; Shu, L.; An, Z.; Wang, J.; Lv, H.; Yang, M.; Cai, T.; Hu, T.; Fu, Y.; et al. Large-scale identification of N-linked intact glycopeptides in human serum using HILIC enrichment and spectral library search. Mol. Cell. Proteom. 2020, 19, 672–689. [Google Scholar] [CrossRef] [PubMed]
- Schweppe, D.K.; Chavez, J.D.; Navare, A.T.; Wu, X.; Ruiz, B.; Eng, J.K.; Lam, H.; Bruce, J.E. Spectral library searching to identify cross-linked peptides. J. Proteome Res. 2016, 15, 1725–1731. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lin, L.; Qiao, L. Deep learning approaches for data-independent acquisition proteomics. Expert Rev. Proteom. 2021, 18, 1031–1043. [Google Scholar] [CrossRef]
- Wang, J.H.; Choong, W.K.; Chen, C.T.; Sung, T.Y. Calibr improves spectral library search for spectrum-centric analysis of data independent acquisition proteomics. Sci. Rep. 2022, 12, 2045–2057. [Google Scholar] [CrossRef]
- Lam, H.; Deutsch, E.W.; Eddes, J.S.; Eng, J.K.; King, N.; Stein, S.E.; Aebersold, R. Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 2007, 7, 655–667. [Google Scholar] [CrossRef]
- Shiferaw, G.A.; Vandermarliere, E.; Hulstaert, N.; Gabriels, R.; Martens, L.; Volders, P.J. COSS: A fast and user-friendly tool for spectral library searching. J. Proteome Res. 2020, 19, 2786–2793. [Google Scholar] [CrossRef]
- Dasari, S.; Chambers, M.C.; Martinez, M.A.; Carpenter, K.L.; Ham, A.J.; Vega-Montoto, L.J.; Tabb, D.L. Pepitome: Evaluating improved spectral library search for identification complementarity and quality assessment. J. Proteome Res. 2012, 11, 1686–1695. [Google Scholar] [CrossRef]
- Biskup, E.; Wils, R.S.; Hogdall, C.; Hogdall, E. Prospects of improving early ovarian cancer diagnosis using cervical cell swabs. Anticancer Res. 2022, 42, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kulczyńska-Przybik, A.; Dulewicz, M.; Słowik, A.; Borawska, R.; Kułakowska, A.; Kochanowicz, J.; Mroczko, B. The clinical significance of cerebrospinal fluid reticulon 4 (RTN4) levels in the differential diagnosis of neurodegenerative diseases. J. Clin. Med. 2021, 10, 5281. [Google Scholar] [CrossRef] [PubMed]
- Duca, R.B.; Massillo, C.; Dalton, G.N.; Farré, P.L.; Graña, K.D.; Gardner, K.; De Siervi, A. MiR-19b-3p and miR-101-3p as potential biomarkers for prostate cancer diagnosis and prognosis. Am. J. Cancer Res. 2021, 11, 2802–2820. [Google Scholar] [PubMed]
- Anderson, N.L. The clinical plasma proteome: A survey of clinical assays for proteins in plasma and serum. Clin. Chem. 2010, 56, 177–185. [Google Scholar] [CrossRef]
- Cao, Z.; Tang, H.Y.; Wang, H.; Liu, Q.; Speicher, D.W. Systematic comparison of fractionation methods for in-depth analysis of plasma proteomes. J. Proteome Res. 2012, 11, 3090–3100. [Google Scholar] [CrossRef]
- Addona, T.A.; Shi, X.; Keshishian, H.; Mani, D.R.; Burgess, M.; Gillette, M.A.; Clauser, K.R.; Shen, D.; Lewis, G.D.; Farrell, L.A.; et al. A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat. Biotechnol. 2011, 29, 635–643. [Google Scholar] [CrossRef]
- Keshishian, H.; Burgess, M.W.; Gillette, M.A.; Mertins, P.; Clauser, K.R.; Mani, D.R.; Kuhn, E.W.; Farrell, L.A.; Gerszten, R.E.; Carr, S.A. Multiplexed, quantitative workflow for sensitive biomarker discovery in plasma yields novel candidates for early myocardial injury. Mol. Cell. Proteom. 2015, 14, 2375–2393. [Google Scholar] [CrossRef]
- Ewing, M.A.; Glover, M.S.; Clemmer, D.E. Hybrid ion mobility and mass spectrometry as a separation tool. J. Chromatogr. A 2016, 1439, 3–25. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasma | Serum |
|---|---|
| Straw-colored | Clear, yellowish fluid |
| Composed of serum and clotting factors | Blood without clotting factors |
| Acquired by centrifuging blood to which an anticoagulant has been added | Acquire by centrifuging blood that has been allowed to clot for ~30 min |
| Easier and faster to prepare | Takes longer and is more difficult to prepare |
| Long shelf-life (i.e., up to ten years) | Shorter shelf life (i.e., a few months) |
| Product | Manufacturer | Capture Agent | Captured Proteins |
|---|---|---|---|
| Albumin Depletion Kit | ThermoFisher Scientific | Antibody | Albumin |
| High-Select HSA/Immunoglobulin Depletion Spin Columns | ThermoFisher Scientific | Antibodies | Albumin, IgG, IgM, IgE, IgD, and IgA |
| High-Select Top14 Abundant Protein Depletion Spin Columns | ThermoFisher Scientific | Antibodies | Albumin, IgG, IgA, IgM, IgD, IgE, Alpha-1-Acid glycoprotein, Alpha-1-Antitrypsin, Alpha-2-Macroglobulin, Apolipoproteins A-I, Fibrinogen, Haptoglobin, Transferrin |
| Albumin/IgG Removal Kit | ThermoFisher Scientific | Cibacron Blue Dye and Protein A | Albumin and IgG |
| Multiple Affinity Removal Column Human 14 | Agilent | Antibodies | albumin, IgG, antitrypsin, IgA, transferrin, haptoglobin, fibrinogen, alpha-2-macroglobulin, alpha1-acid glycoprotein, IgM, apolipoprotein AI, apolipoprotein AII, complement C3 and transthyretin |
| ProteoExtract Albumin/IgG Removal Kit | Millipore Sigma | albumin-specific affinity resin and protein A | Albumin and IgG |
| ProteoMiner Protein Enrichment | BioRad | Combinatorial library of hexapeptides | All proteins |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paul, J.; Veenstra, T.D. Separation of Serum and Plasma Proteins for In-Depth Proteomic Analysis. Separations 2022, 9, 89. https://doi.org/10.3390/separations9040089
Paul J, Veenstra TD. Separation of Serum and Plasma Proteins for In-Depth Proteomic Analysis. Separations. 2022; 9(4):89. https://doi.org/10.3390/separations9040089
Chicago/Turabian StylePaul, Joseph, and Timothy D. Veenstra. 2022. "Separation of Serum and Plasma Proteins for In-Depth Proteomic Analysis" Separations 9, no. 4: 89. https://doi.org/10.3390/separations9040089
APA StylePaul, J., & Veenstra, T. D. (2022). Separation of Serum and Plasma Proteins for In-Depth Proteomic Analysis. Separations, 9(4), 89. https://doi.org/10.3390/separations9040089