
Uncertainty of Size-Exclusion Chromatography Method in Quality Control of Bevacizumab Batches



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Size Exclusion Chromatography System
2.3. Analytical Method Validation
2.4. Statistical Model
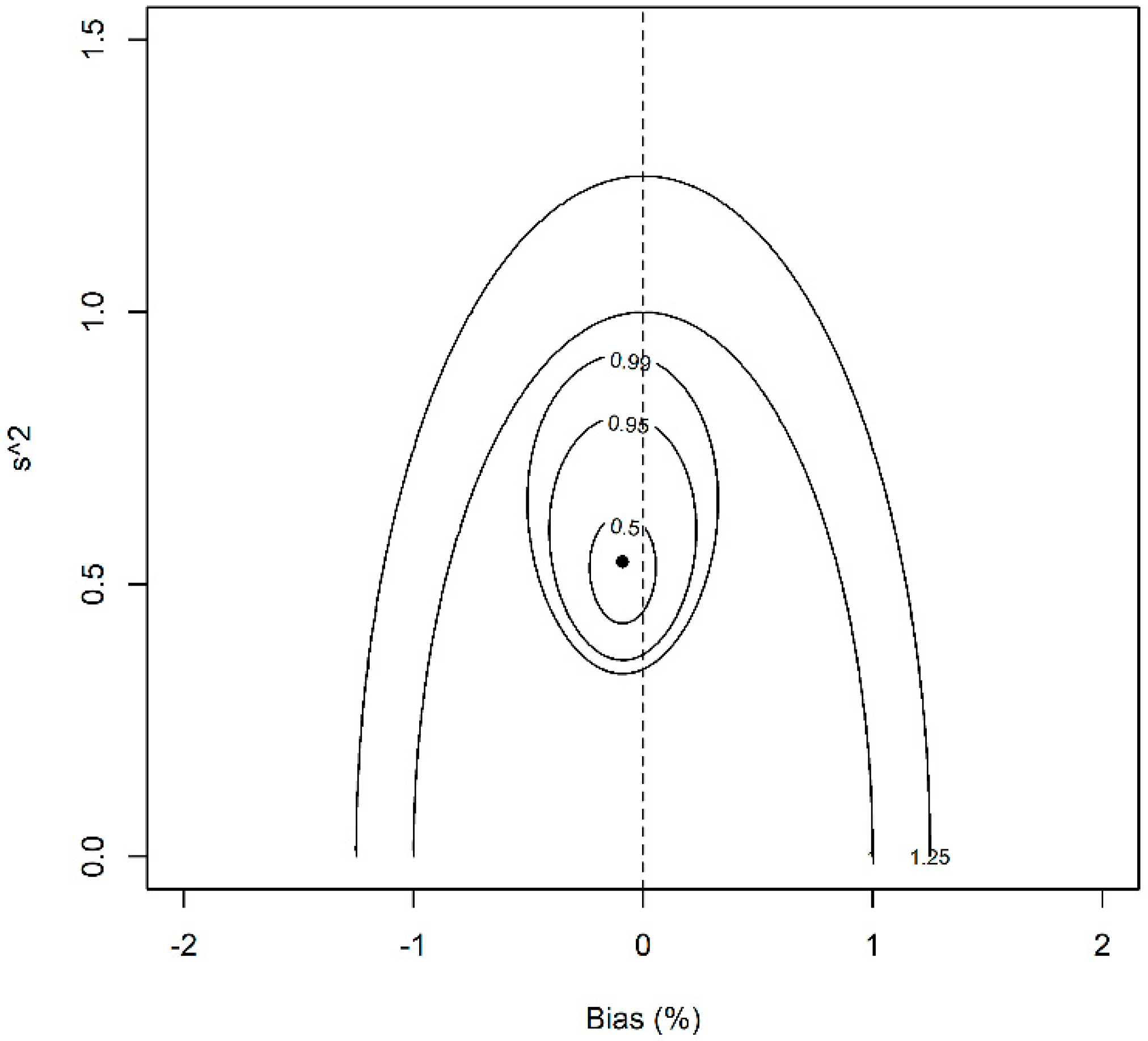
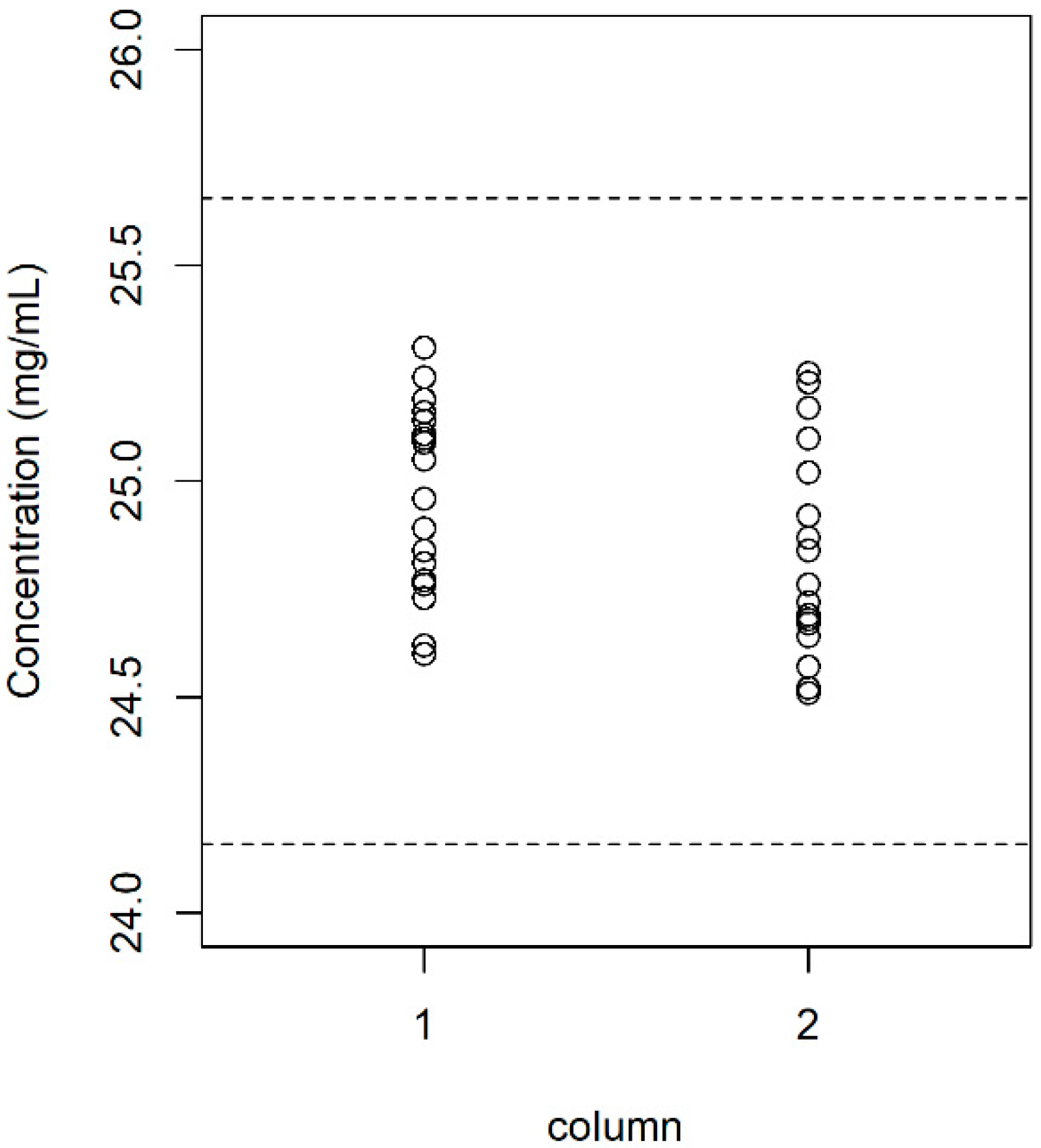
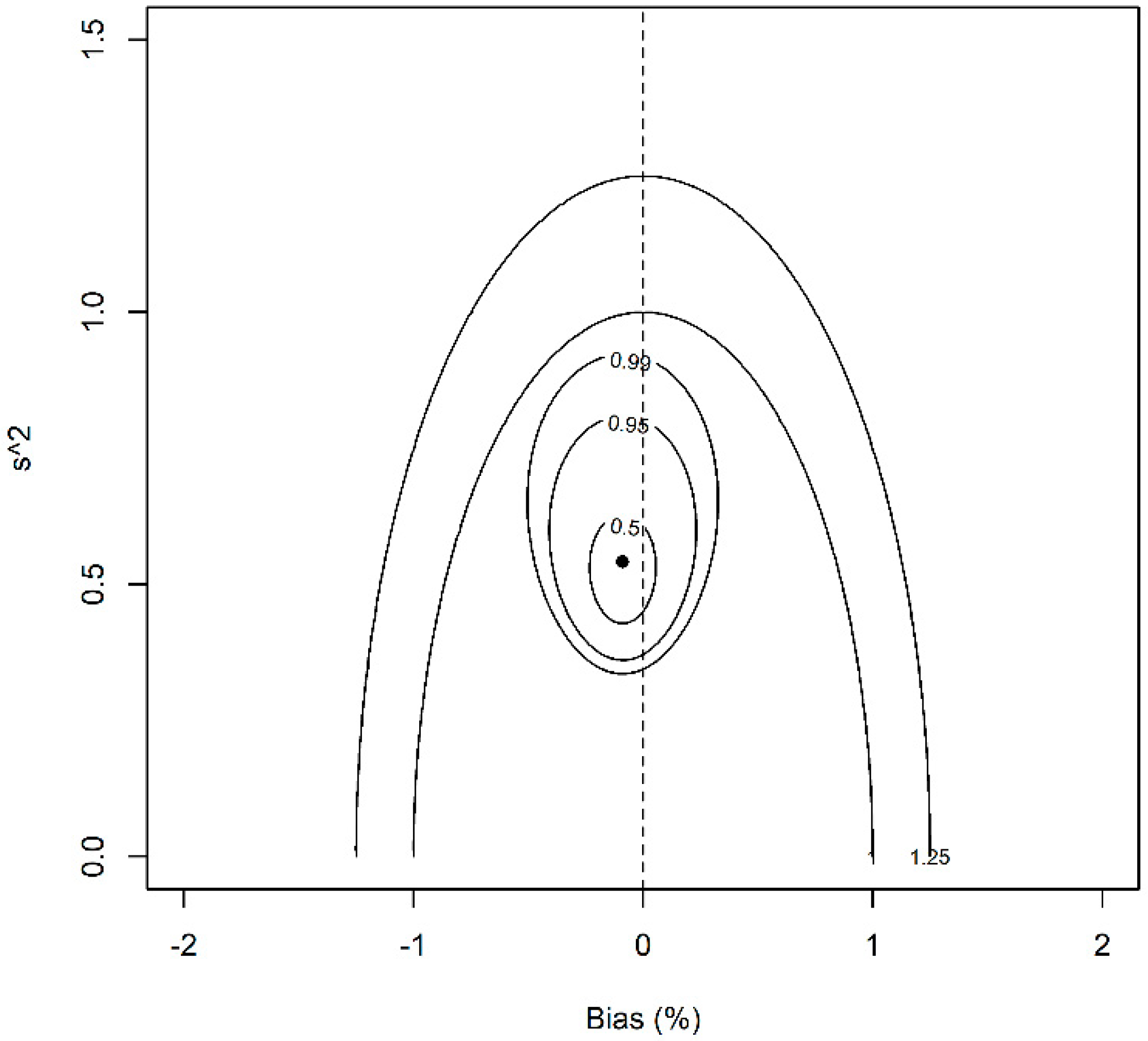
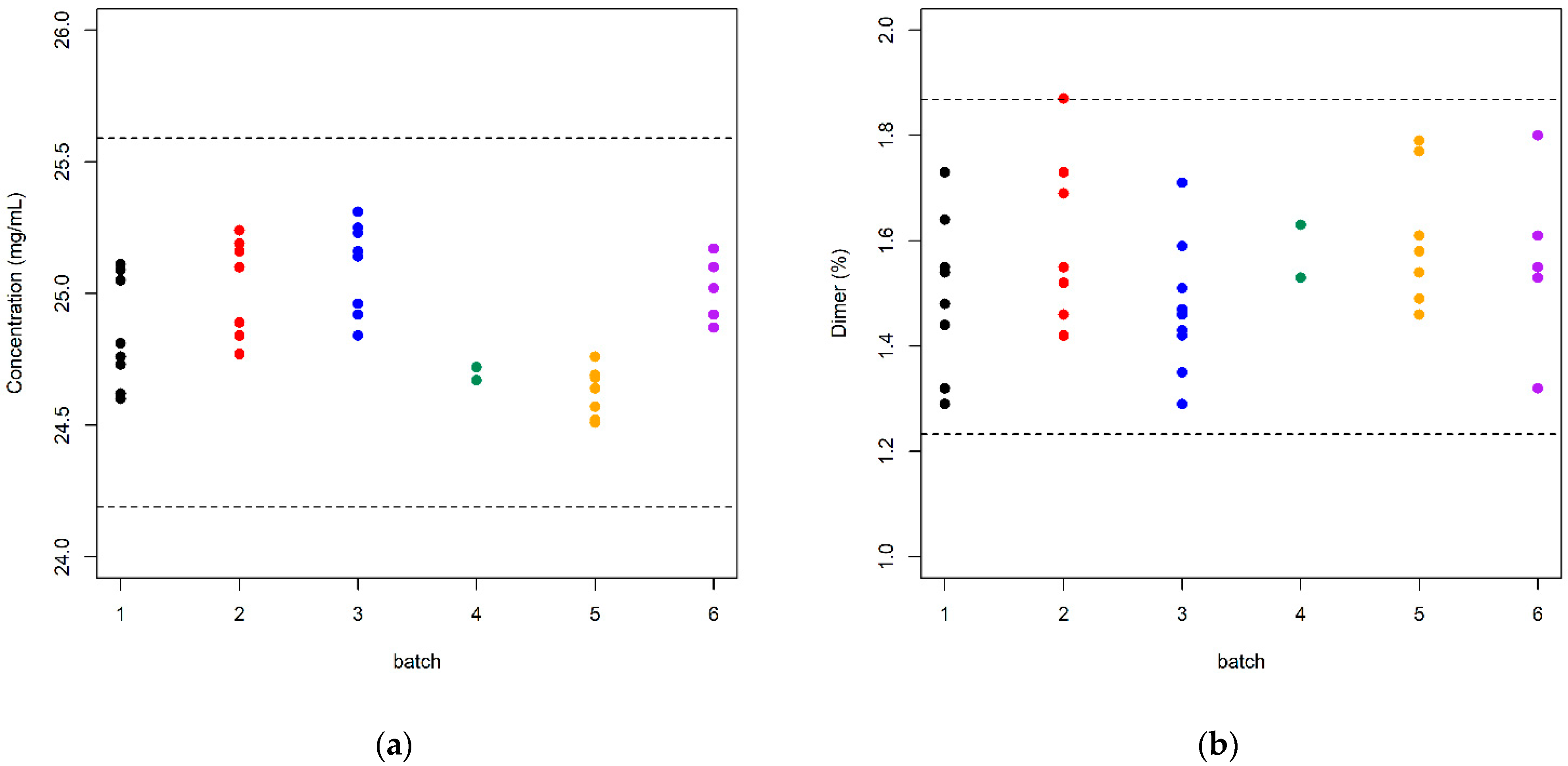
3. Results and Discussion
3.1. Analytical Method Validation
3.2. Data Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Regl, C.; Wohlschlager, T.; Esser-Skala, W.; Wagner, I.; Samonig, M.; Holzmann, J.; Huber, C.G. Dilute-and-shoot analysis of therapeutic monoclonal antibody variants in fermentation broth: A method capability study. mAbs 2019, 1, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int. J. Pharm. 1999, 185, 129–188. [Google Scholar] [CrossRef]
- Hassan, L.A.; Shatat, S.M.; Eltanany, B.M.; Al-Ghobashy, M.A.; Abbas, S.S. Stability and biosimilarity assessment of infliximab using an orthogonal testing protocol and statistically-guided interpretation of peptide mapping. Anal. Methods 2019, 12, 3198–3211. [Google Scholar] [CrossRef]
- Lee, J.J.; Yang, J.; Lee, C.; Moon, Y.; Ahn, S.; Yang, J. Demonstration of functional similarity of a biosimilar adalimumab SB5 to Humira®. Biologicals 2019, 58, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Shaltout, E.L.; Al-Ghobashy, M.A.; Fathalla, F.A.; Salem, M.Y. Chromatographic and electrophoretic assessment of Filgrastim biosimilars in pharmaceutical formulations. J. Pharm. Biomed. Anal. 2014, 97, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Shatat, S.M.; Eltanany, B.M.; Mohamed, A.A.; Al-Ghobashy, M.A.; Fathalla, F.A.; Abbas, S.S. Coupling of on-column trypsin digestion-peptide mapping and principal component analysis for stability and biosimilarity assessment of recombinant human growth hormone. J. Chromatogr. B 2018, 1072, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Zhang, E.; Xu, Y.; Gao, W.; Wang, L.; Xie, M.H.; Qin, P.; Lu, L.; Li, S.; Shen, P.; et al. Demonstrating analytical similarity of trastuzumab biosimilar HLX02 to Herceptin® with a panel of sensitive and orthogonal methods inluding a novel FcyRIIIa affinity chromatography technology. Biodrugs 2020, 34, 363–379. [Google Scholar] [CrossRef] [Green Version]
- Parr, M.K.; Montacir, O.; Montacir, H. Physicochemical characterization of biopharmaceuticals. J. Pharm. Biomed. Anal. 2016, 130, 366–389. [Google Scholar] [CrossRef]
- Bobály, B.; Lauber, M.; Beck, A.; Guillarme, D.; Fekete, S. Utility of a high coverage phenyl-bonding and wide-pore superficially porous particle for the analysis of monoclonal antibodies and related products. J. Chromatogr. A 2018, 1549, 63–76. [Google Scholar] [CrossRef]
- Bobály, B.; D’Atri, V.; Lauber, M.; Beck, A.; Guillarme, D.; Fekete, S. Characterizing various monoclonal antibodies with milder reverse phase chromatography conditions. J. Chromatogr. B 2018, 1096, 1–10. [Google Scholar] [CrossRef]
- Fekete, S.; Beck, A.; Veuthey, J.L.; Guillarme, D. Theory and practice of size exclusion chromatography for the analysis of protein aggregates. J. Pharm. Biomed. Anal. 2014, 101, 43–55. [Google Scholar] [CrossRef]
- Goyon, A.; Guillarme, D.; Fekete, S. The importance of system band broadening in modern size exclusion chromatography. J. Pharm. Biomed. Anal. 2017, 135, 50–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakawa, T.; Ejima, D.; Li, T.; Philo, J.S. The critical role of mobile phase composition in size exclusion chromatography of protein pharmaceuticals. J. Pharm. Sci. 2010, 99, 1674–1692. [Google Scholar] [CrossRef] [PubMed]
- Oliva, A.; Llabres, M. Validation of a size-exclusion chromatography method for Bevacizumab quantitation in pharmaceutical preparations: Application in a biosimilar study. Separations 2019, 6, 43. [Google Scholar] [CrossRef] [Green Version]
- Goyon, A.; D’Atri, V.; Colas, O.; Fekete, S.; Beck, A.; Guillarme, D. Characterization of 30 therapeutic antibodies and related products by size exclusion chromatography: Feasibility assessment for future mass spectrometry hyphenation. J. Chromatogr. B 2017, 1065, 35–43. [Google Scholar] [CrossRef] [PubMed]
- FDA Guidance for Industry: Q6B Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products; Center for Drug Evaluation and Research Food and Drug Administration: Rockville, MD, USA, 1999.
- Lee, J.; Kang, H.A.; Bae, J.S.; Kim, K.D.; Lee, K.H.; Lim, K.J.; Choo, M.J.; Chang, S.J. Evaluation of analytical similarity between trastuzumab biosimilar CT-P6 and reference product using statistical analyses. mAbs 2018, 10, 547–571. [Google Scholar] [CrossRef] [Green Version]
- ICH Q2-(R1): Validation of Analytical Procedures: Text and methodology. ICH Guideline, November 2005.
- Prior, S.; Metcalfe, C.; Hufton, S.E.; Wadhwa, M.; Schneider, C.K.; Burns, C. Maintaining standards for biosimilar monoclonal antibodies. Nat. Biotechnol. 2021, 39, 276–280. [Google Scholar] [CrossRef]
- FDA. Guidance on Statistical Approaches to Evaluate Analytical Similarity; The United States Food and Drug Administrations: Silver Spring, MD, USA, 2017.
- Oliva, A.; Monzon, C.; Santovena, A.; Fariña, J.B.; Llabrés, M. Development of an ultra-high-performance liquid chromatography method for determining triamcinolone acetonide in hydrogels using the design of experiments/design space strategy in combination with process capability index. J. Sep. Sci. 2016, 39, 2689–2701. [Google Scholar] [CrossRef]
- Bates, D.M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67, 1. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org (accessed on 10 May 2021).
- Hoffman, D.; Kringle, R. Two-sided tolerance intervals for balanced and unbalanced random effects models. J. Biopharm. Stat. 2005, 15, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Box, G.P.; Tiao, G. Bayesian Inference in Statistical Analysis; Addison-Wesley Publishing Company Inc.: New York, NY, USA, 1973. [Google Scholar]
- European Medicines Agency Assessment Report on ABP 215 Biosimilar. Available online: https://www.ema.europa.eu/en/documents/assessment-report/mvasi-epar-public-assessment-report_en.pdf (accessed on 10 May 2021).
- Montes, R.O.; Burdick, R.K.; Leblond, D.J. Simple approach to calculate random effects model tolerance intervals to set release and shelf-life specification limits of pharmaceutical products. PDA J. Pharm. Sci. Tech. 2019, 73, 39–59. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Batch | Sample Size | Content (mg/mL) | Dimer (%) | ||
|---|---|---|---|---|---|
| Mean | SD | Mean | SD | ||
| 1 | 8 | 24.85 | 0.209 | 1.499 | 0.150 |
| 2 | 7 | 25.03 | 0.189 | 1.606 | 0.162 |
| 3 | 9 | 25.11 | 0.167 | 1.470 | 0.125 |
| 4 | 2 | 24.69 | 0.035 | 1.580 | 0.071 |
| 5 | 8 | 24.61 | 0.096 | 1.587 | 0.130 |
| 6 | 5 | 25.02 | 0.124 | 1.562 | 0.172 |
| Parameter | Content (mg/mL) | Dimer (%) |
|---|---|---|
| 24.89 (24.71, 25.06) | 1.543 (1.490, 1.600) | |
| 0.190 (0.093, 0.364) | 0.029 (0.00, 0.098) | |
| 0.162 (0.130, 0.210) | 0.142 (0.114, 0.181) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliva, A.; Llabrés, M. Uncertainty of Size-Exclusion Chromatography Method in Quality Control of Bevacizumab Batches. Separations 2021, 8, 133. https://doi.org/10.3390/separations8090133
Oliva A, Llabrés M. Uncertainty of Size-Exclusion Chromatography Method in Quality Control of Bevacizumab Batches. Separations. 2021; 8(9):133. https://doi.org/10.3390/separations8090133
Chicago/Turabian StyleOliva, Alexis, and Matías Llabrés. 2021. "Uncertainty of Size-Exclusion Chromatography Method in Quality Control of Bevacizumab Batches" Separations 8, no. 9: 133. https://doi.org/10.3390/separations8090133
APA StyleOliva, A., & Llabrés, M. (2021). Uncertainty of Size-Exclusion Chromatography Method in Quality Control of Bevacizumab Batches. Separations, 8(9), 133. https://doi.org/10.3390/separations8090133