

Rapid Determination of Five Residual Solvents in Ursodeoxycholic Acid Raw Materials

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Apparatus and Operations
2.3. Measurement Procedures
2.4. Preparation of Reference Solution and Sample Solution
2.4.1. Preparation of Residual Solvent Localization Solution
2.4.2. Preparation of Reference Solution
2.4.3. Preparation of Sample Solution
2.5. Method Validation
3. Results and Discussion
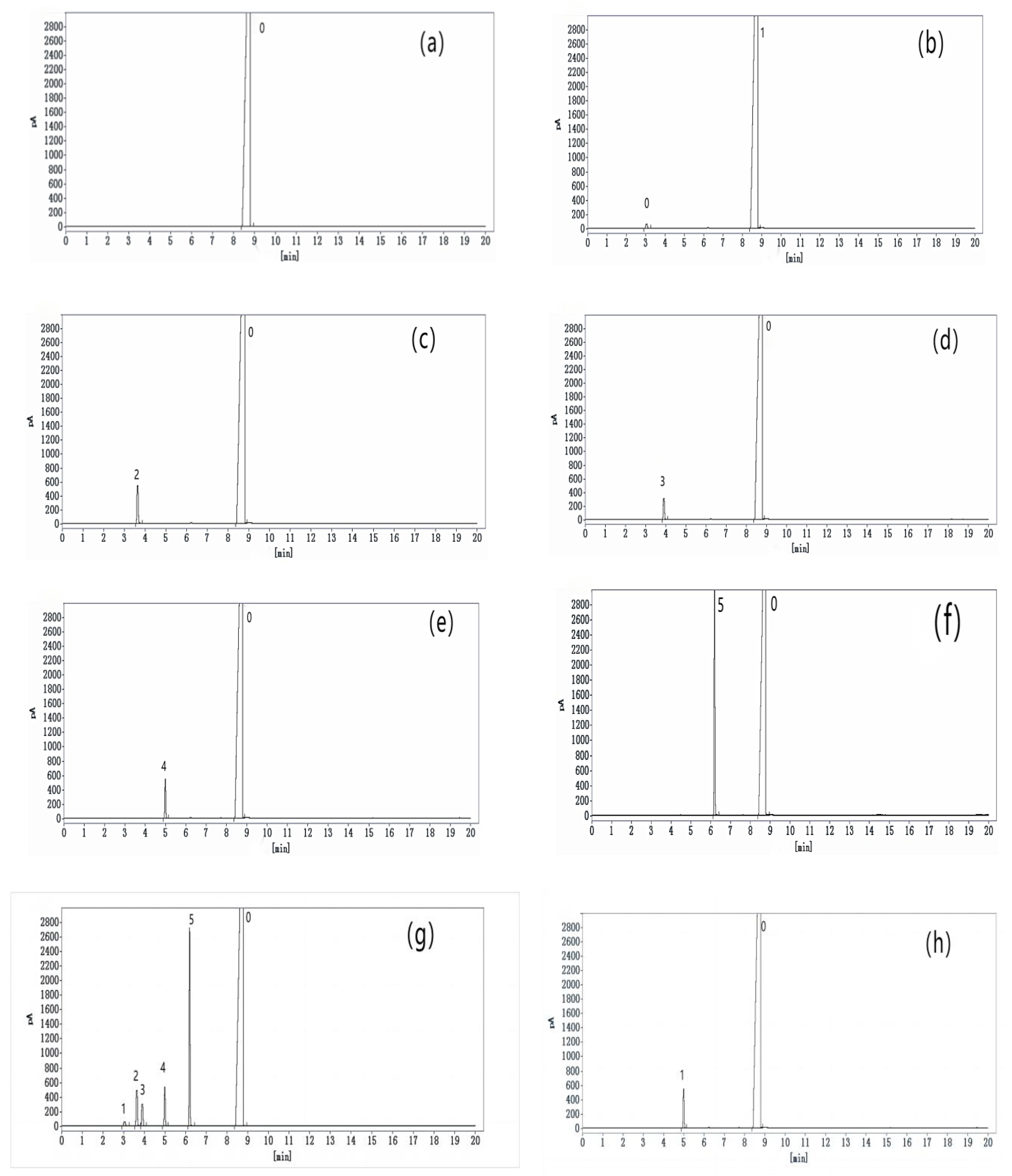
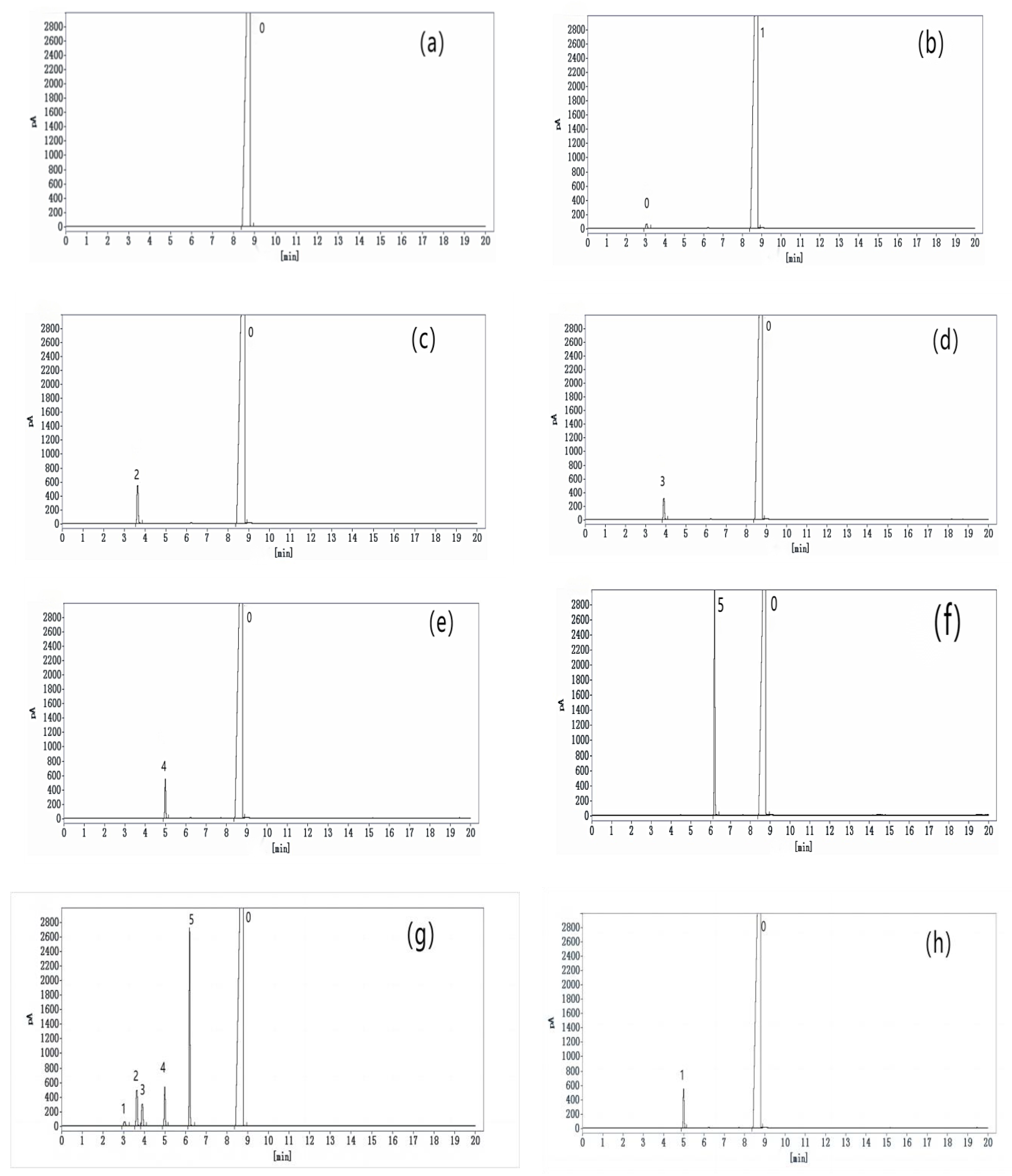
3.1. Optimization of Chromatographic Conditions
3.1.1. The Effect of Equilibrium Temperature
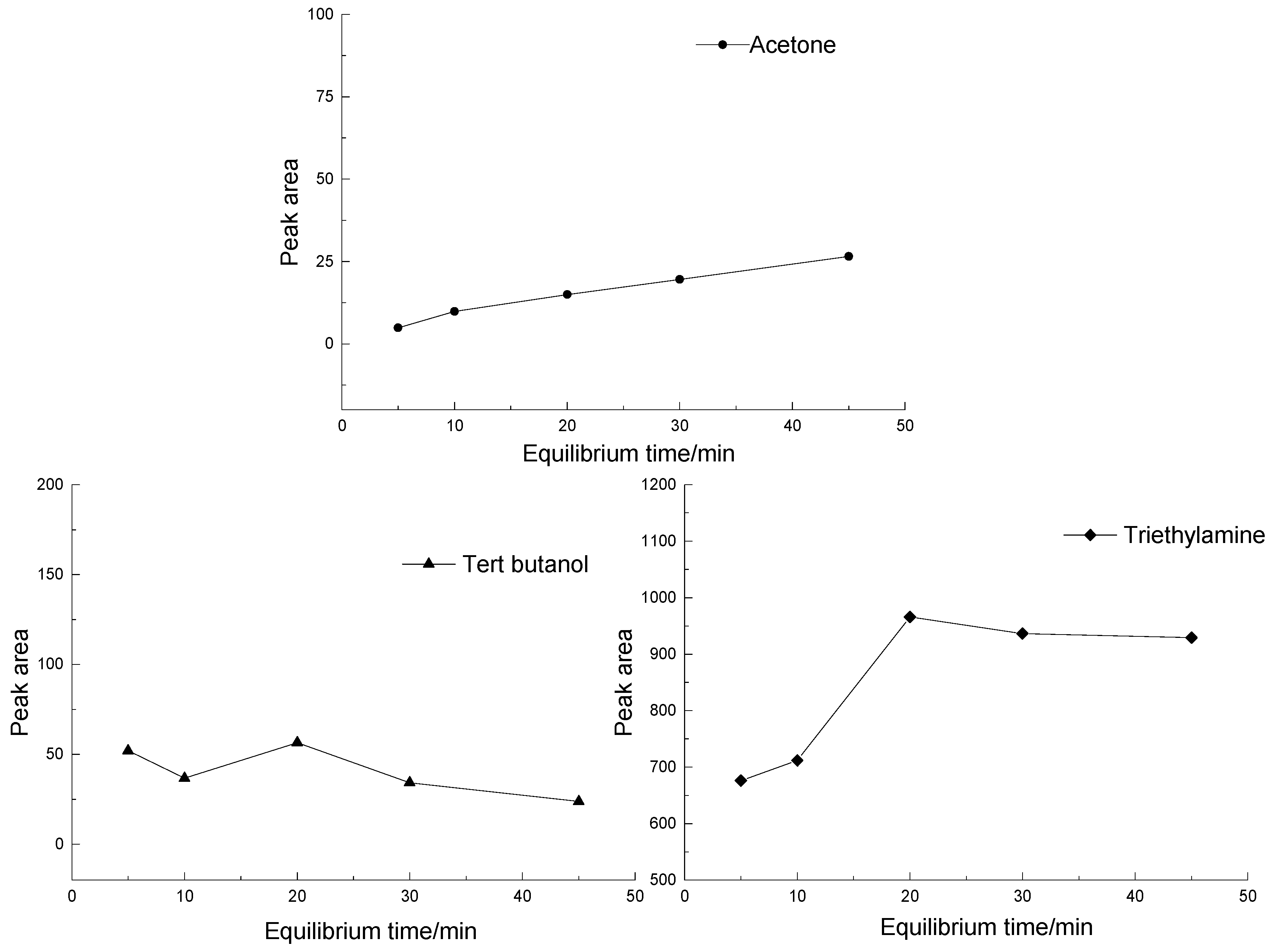
3.1.2. The Effect of Equilibrium Time
3.2. Method Validation
3.2.1. System Suitability
3.2.2. Repeatability Test
3.2.3. Linearity
3.2.4. LOD and LOQ
3.2.5. Accuracy
3.2.6. Robustness
3.3. Simultaneous Determination of Ursodeoxycholic Acid Samples
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gu, H.Z. The clinical application of ursodeoxycholic acid. Inn. Mong. Tradit. Chin. Med. 2012, 2, 52–53. [Google Scholar]
- Qi, Z.; Xu, M.L.; Zheng, Y.Y. Clinical use and research progress of ursodeoxycholic acid. J. Clin. Ration. Drug Use 2016, 9, 84–85. [Google Scholar]
- Wan, J.F.; Chu, S.F.; Zhou, X.; Li, Y.T.; He, W.B.; Tan, F.; Luo, P.; Ai, Q.D.; Wang, Q.; Chen, N.N. Ursodeoxycholic acid protects interstitial Cajal-like cells in the gallbladder from undergoing apoptosis by inhibiting TNF-α expression. Acta Pharmacol. Sin. 2018, 39, 1493–1500. [Google Scholar] [CrossRef]
- Gazda, J.; Drazilova, S.; Gazda, M.; Martin Janicko, M.; Koky, T.; Macej, M.; Carbone, M.; Jarcuska, P. Treatment response to ursodeoxycholic acid in primary biliary cholangitis: A systematic review and meta-analysis. Dig. Liver Dis. 2023, 55, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- National Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China: Part II; China Medical Science and Technology Press: Beijing, China, 2020.
- United States Pharmacopoeia Commission. Ursodeoxycholic Acid; USP43; US Pharmacopoeia: New York, NY, USA, 2020. [Google Scholar]
- European Pharmacopoeia Commission. Ursodeoxycholic Acid; EP10.0; European Pharmacopoeia: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Dawkins, M.S. From an animal’s point of view: Motivation, fitness, and animal welfare. Behav. Brain Sci. 1990, 13, 1–9. [Google Scholar] [CrossRef]
- Kim, O.Y.; Lee, S.Y.; Lee, D.Y.; Hur, S.J. Developing a procedure to extract chenodeoxycholic acid and synthesize ursodeoxycholic acid from pig by-products. Heliyon 2023, 9, e18313. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.F.; Yin, W.C.; Wang, F.D.; Peng, D.M. Improvement of the synthesis process of ursodeoxycholic acid. Chin. J. Pharm. Ind. 2015, 46, 1058–1059. [Google Scholar]
- Chen, W.; Hu, D.; Feng, Z.; Liu, Z. An effective synthesis of ursodeoxycholic acid from dehydroepiandrosterone. Steroids 2021, 172, 108870. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.X.; Dong, D.D.; Wang, Z.F.; Wan, J.F.; Cao, X.J. Synthesis of ursodeoxycholic acid by electrochemical stereoselective reduction of 7-ketolithocholic acid in aprotic solvents. Sci. Rep. 2021, 1, 16273. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.W.; Fu, J.J.; Li, J.J.; Tang, Y.; Shao, Z.W.; Zhou, D.Y.; Song, L. Efficient encapsulation of curcumin into spent brewer’s yeast using a pH-driven method. Food Chem. 2022, 394, 133537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H. Construction and Application of a Recombinant Yeast Strain for Efficient Synthesis of Ursodeoxycholic Acid. Master’s Thesis, Tianjin University of Science and Technology, Tianjin, China, 2022. [Google Scholar]
- Yang, B.Y.; You, Z.N.; Xue, J.T.; Pan, J.; Li, C.X.; Xu, J.H. Clean enzymatic production of ursodeoxycholic acid enabled by a newly identified NADH- dependent 7β-hydroxysteroid dehydrogenase. Mol. Catal. 2023, 537, 112946. [Google Scholar] [CrossRef]
- Zhang, C.L.; Li, C.S.; Yu, X.L. Progress in Chinese patents for the effective ingredient ursodeoxycholic acid in bear bile. Chin. J. Tradit. Chin. Med. 2012, 37, 2851–2854. [Google Scholar]
- Luo, L.L.; Yu, J.; Wei, H.Q.; Hou, W.B.; Li, W.L. Research progress in the synthesis of ursodeoxycholic acid. Synth. Chem. 2021, 29, 986–996. [Google Scholar]
- Li, J.; Li, X.; Wang, F.; Zhao, T.; Wang, C.H. Determination of three residual organic solvents in ursodeoxycholic acid by headspace gas chromatography. Prog. Mod. Biomed. Sci. 2015, 31, 6156–6159. [Google Scholar]
- Zhou, H.J. International Technical Requirements for Drug Registration Q3C; People’s Health Publishing House: Beijing, China, 2000; pp. 1–8. [Google Scholar]
- State Food and Drug Administration. Technical Guidelines for the Study of Residual Solvents in Chemical Drugs; State Food and Drug Administration: Silver Spring, MD, USA, 2005; pp. 6–7.

{kind=link}
{kind=link}
| Equilibrium Temperature | Test Samples | ||||
|---|---|---|---|---|---|
| Unknown Peak (2.758 min) | Acetone | Tert-Butanol | Triethylamine | ||
| 60 °C | Rt (min) | 2.758 | 3.646 | 3.948 | 6.218 |
| Resolution | —— | 5.709 | 2.191 | 22.044 | |
| Peak area | 45.542 | 411.721 | 2.783 | 12.881 | |
| Tailing factor | 1.533 | 0.934 | 1.056 | 1.718 | |
| Number of theoretical plates | 4269 | 10,628 | 13,918 | 119,444 | |
| 80 °C | Rt (min) | 2.758 | 3.645 | 3.949 | 6.212 |
| Resolution | —— | 5.702 | 2.215 | 22.277 | |
| Peak area | 39.932 | 641.844 | 3.963 | 26.420 | |
| Tailing factor | 1.479 | 1.092 | 0.923 | 1.685 | |
| Number of theoretical plates | 4335 | 10,403 | 14,349 | 123,073 | |
| 100 °C | Rt (min) | 2.740 | 3.633 | 3.935 | 6.193 |
| Resolution | —— | 6.345 | 2.283 | 22.551 | |
| Peak area | 3.255 | 712.048 | 9.883 | 36.692 | |
| Tailing factor | 1.349 | 1.145 | 0.985 | 1.500 | |
| Number of theoretical plates | 5708 | 11,287 | 15,126 | 118,512 | |
| Residue Solvent | Regression Equations | R | Linearity Range (mg/mL) |
|---|---|---|---|
| Methanol | Y = 981.35X + 10.04 | 0.9993 | 0.06–0.3 |
| Acetone | Y = 4517.4X + 78.569 | 0.9991 | 0.1–0.5 |
| Tert-butanol | Y = 2756.5X + 45.711 | 0.9990 | 0.1–0.5 |
| Ethyl acetate | Y = 3507.5X + 56.446 | 0.9990 | 0.1–0.5 |
| Triethylamine | Y = 14,349X + 252.25 | 0.9990 | 0.1–0.5 |
| Residue Solvents | Theoretical Concentration (mg/mL) | Actual Concentration (mg/mL) | Recovery (%) | Average Recovery (%) | RSD (%) |
|---|---|---|---|---|---|
| Methanol | 0.18 | 0.1808 | 100.4 | 100.6 | 3.45 |
| 0.18 | 0.1803 | 100.2 | |||
| 0.18 | 0.1773 | 98.5 | |||
| 0.24 | 0.2336 | 97.3 | |||
| 0.24 | 0.2410 | 100.4 | |||
| 0.24 | 0.2409 | 100.4 | |||
| 0.3 | 0.2974 | 99.1 | |||
| 0.3 | 0.2983 | 99.4 | |||
| 0.3 | 0.3282 | 109.4 | |||
| Acetone | 0.3 | 0.3004 | 100.1 | 98.2 | 2.60 |
| 0.3 | 0.2990 | 99.7 | |||
| 0.3 | 0.2894 | 96.5 | |||
| 0.4 | 0.3779 | 94.5 | |||
| 0.4 | 0.3962 | 99.1 | |||
| 0.4 | 0.3890 | 97.2 | |||
| 0.5 | 0.4746 | 94.9 | |||
| 0.5 | 0.4966 | 99.3 | |||
| 0.5 | 0.5107 | 101.8 | |||
| Tert-butanol | 0.3 | 0.3205 | 106.8 | 106.0 | 3.28 |
| 0.3 | 0.3185 | 106.2 | |||
| 0.3 | 0.3096 | 103.2 | |||
| 0.4 | 0.4074 | 101.9 | |||
| 0.4 | 0.4251 | 106.3 | |||
| 0.4 | 0.4270 | 106.7 | |||
| 0.5 | 0.5163 | 103.3 | |||
| 0.5 | 0.5284 | 105.7 | |||
| 0.5 | 0.5695 | 113.9 | |||
| Ethyl acetate | 0.3 | 0.3027 | 100.9 | 98.8 | 2.72 |
| 0.3 | 0.3005 | 100.2 | |||
| 0.3 | 0.2897 | 96.6 | |||
| 0.4 | 0.3804 | 95.1 | |||
| 0.4 | 0.3991 | 99.8 | |||
| 0.4 | 0.3946 | 98.6 | |||
| 0.5 | 0.4762 | 95.2 | |||
| 0.5 | 0.5002 | 100.0 | |||
| 0.5 | 0.5152 | 103.0 | |||
| Triethylamine | 0.3 | 0.2939 | 98.0 | 92.9 | 3.80 |
| 0.3 | 0.2922 | 97.4 | |||
| 0.3 | 0.2744 | 91.5 | |||
| 0.4 | 0.3527 | 88.2 | |||
| 0.4 | 0.3733 | 93.3 | |||
| 0.4 | 0.3572 | 89.3 | |||
| 0.5 | 0.4485 | 89.7 | |||
| 0.5 | 0.4755 | 95.1 | |||
| 0.5 | 0.4694 | 93.9 |
| Conditions | Contents (%) | |||
|---|---|---|---|---|
| Flow Rate of Carrier (mL/min) | Equilibrium Temperature (°C) | Acetone | Tert-Butanol | Triethylamine |
| 0.9 mL/min | 100 °C | 0.2068 | 0.0026 | 0.0056 |
| 1.0 mL/min | 100 °C | 0.2036 | 0.0024 | 0.0057 |
| 1.1 mL/min | 100 °C | 0.1999 | 0.0024 | 0.0043 |
| 1.0 mL/min | 90 °C | 0.1999 | 0.0024 | 0.0043 |
| Batch Number | Content (%) | ||||
|---|---|---|---|---|---|
| Methanol | Acetone | Tert-Butanol | Ethyl Acetate | Triethylamine | |
| 113UA2160817 | -- | 0.1898 | 0.0076 | -- | 0.0077 |
| 113UA2160906 | -- | 0.1803 | 0.0072 | -- | 0.0063 |
| 113UA2160917 | -- | 0.1937 | 0.0074 | -- | 0.0034 |
| 113UA2160106 | -- | 0.1933 | 0.0077 | -- | 0.0039 |
| Average content (%) | -- | 0.1893 | 0.0075 | -- | 0.0053 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, R.; He, J.; Shi, Y.; Gao, X.; Wang, F.; Liu, S. Rapid Determination of Five Residual Solvents in Ursodeoxycholic Acid Raw Materials. Separations 2023, 10, 573. https://doi.org/10.3390/separations10110573
Zhou R, He J, Shi Y, Gao X, Wang F, Liu S. Rapid Determination of Five Residual Solvents in Ursodeoxycholic Acid Raw Materials. Separations. 2023; 10(11):573. https://doi.org/10.3390/separations10110573
Chicago/Turabian StyleZhou, Ran, Jingyu He, Yi Shi, Xiaoying Gao, Fei Wang, and Sijie Liu. 2023. "Rapid Determination of Five Residual Solvents in Ursodeoxycholic Acid Raw Materials" Separations 10, no. 11: 573. https://doi.org/10.3390/separations10110573
APA StyleZhou, R., He, J., Shi, Y., Gao, X., Wang, F., & Liu, S. (2023). Rapid Determination of Five Residual Solvents in Ursodeoxycholic Acid Raw Materials. Separations, 10(11), 573. https://doi.org/10.3390/separations10110573