Pediatric Epilepsy Mechanisms: Expanding the Paradigm of Excitation/Inhibition Imbalance



Abstract
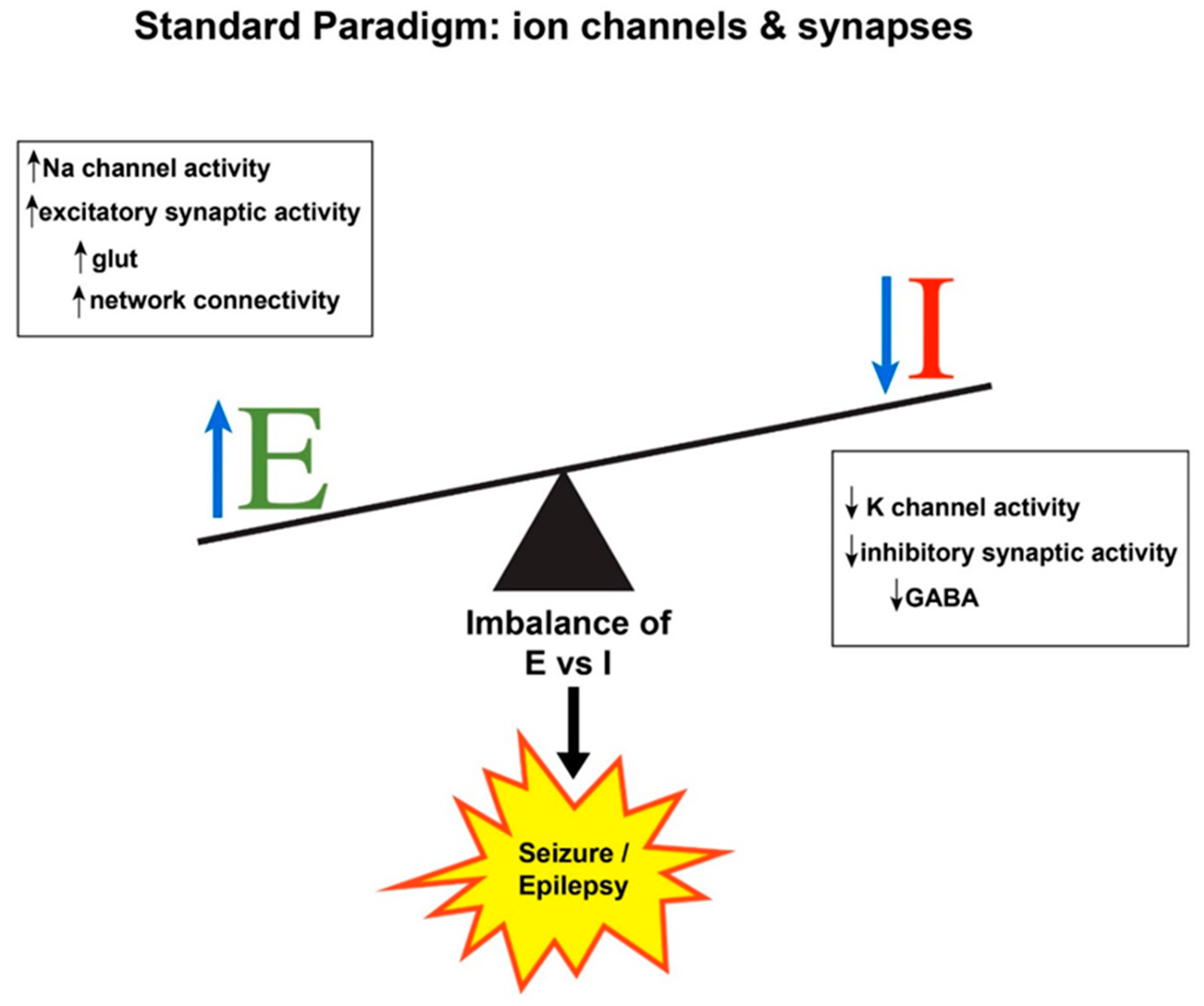
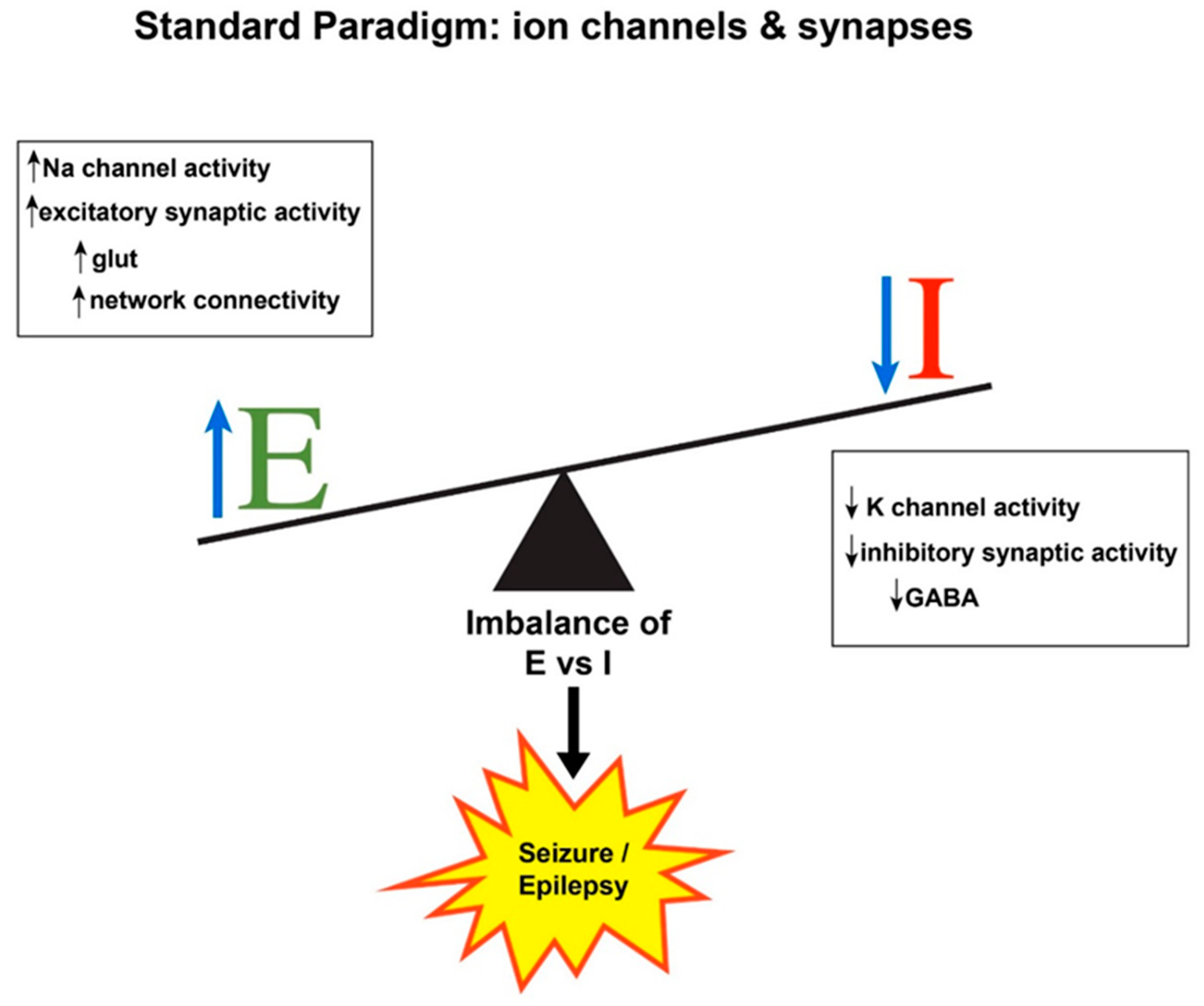
:1. Seizure Generation: Hyperexcitability and Hypersynchrony
2. Expanding the E/I Imbalance Paradigm
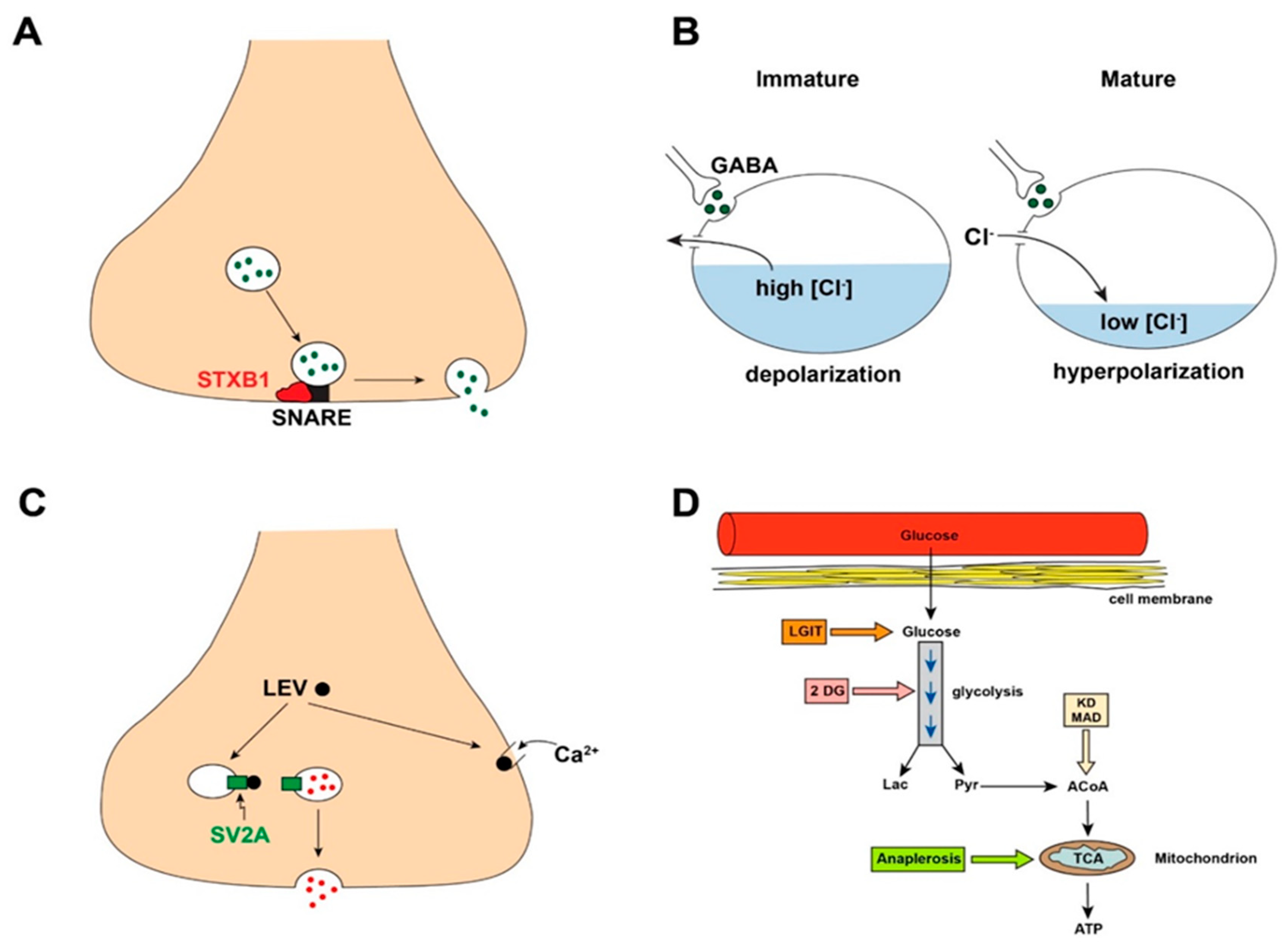
2.1. Newly Discovered Gene Mutations Responsible for Epilepsy Defy Traditional E/I Mechanisms
2.2. Neurotransmitters May Cause Paradoxical Physiological Actions
2.3. Antiseizure Drugs May Not Act via the Expected E/I Spectrum
2.4. Metabolic Regulation of Excitability and Epilepsy Requires Expansion of E/I Considerations
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stafstrom, C.E.; Rho, J.M. Neurophysiology of seizures and epilepsy. In Swaiman’s Pediatric Neurology: Principles and Practice, 6th ed.; Swaiman, K.F., Ashwal, S., Ferriero, D.M., Schor, N.F., Finkel, R.S., Gropman, A.L., Pearl, P.L., Shevell, M.I., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 506–512. [Google Scholar]
- Smith, E.H.; Schevon, C.A. Toward a mechanistic understanding of epileptic circuits. Curr. Neurol. Neurosci. Rep. 2016, 16, 97. [Google Scholar] [CrossRef] [PubMed]
- Jiruska, P.; de Curtis, M.; Jefferys, J.G. Modern concepts of focal epileptic networks. Int. Rev. Neurobiol. 2014, 114, 1–7. [Google Scholar] [PubMed]
- Uhlhaas, P.J.; Singer, W. Neural synchrony in brain disorders: Relevance for cognitive dysfunctions and pathophysiology. Neuron 2006, 52, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Trevelyan, A.J. Do cortical circuits need protecting from themselves? Trends Neurosci. 2016, 39, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, C.E. Recognizing seizures and epilepsy: Insights from pathophysiology. In Epilepsy; Miller, J.W., Goodkin, H.P., Eds.; John Wiley & Sons, Ltd.: West Sussex, UK, 2014; pp. 3–9. [Google Scholar]
- Bozzi, Y.; Provenzano, G.; Casarosa, S. Neurobiological bases of autism-epilepsy comorbidity: A focus on excitation/inhibition imbalance. Eur. J. Neurosci. 2018, 47, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Ajmone-Marsan, C. Cellular mechanisms in experimental epileptic seizures. Science 1964, 144, 193–194. [Google Scholar] [CrossRef] [PubMed]
- Schipper, S.; Aalbers, M.W.; Rijkers, K.; Swijsen, A.; Rigo, J.M.; Hoogland, G.; Vles, J.S. Tonic GABAA receptors as potential target for the treatment of temporal lobe epilepsy. Mol. Neurobiol. 2016, 53, 5252–5265. [Google Scholar] [CrossRef]
- Engel, J., Jr. Excitation and inhibition in epilepsy. Can. J. Neurol. Sci. 1996, 23, 167–174. [Google Scholar] [CrossRef]
- Cossart, R.; Bernard, C.; Ben-Ari, Y. Multiple facets of GABAergic neurons and synapses: Multiple fates of GABA signalling in epilepsies. Trends Neurosci. 2005, 28, 108–115. [Google Scholar] [CrossRef]
- Fritschy, J.-M. Epilepsy, E/I balance and GABA-A receptor plasticity. Front. Mol. Neurosci. 2008, 1, 5. [Google Scholar] [CrossRef]
- Kaila, K.; Ruusuvuori, E.; Seja, P.; Voipio, J.; Puskarjov, M. GABA actions and ionic plasticity in epilepsy. Curr. Opin. Neurobiol. 2014, 26, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Knoflach, F.; Hernandez, M.-C.; Bertrand, D. GABAA receptor-mediated neurotransmission: Not so simple after all. Biochem. Pharmacol. 2016, 115, 10–17. [Google Scholar] [PubMed]
- Maheshwari, A.; Noebels, J.L. Monogenic models of absence epilepsy: Windows into the complex balance between inhibition and excitation in thalamocortical microcircuits. Prog. Brain Res. 2014, 213, 223–252. [Google Scholar] [PubMed]
- Mody, I. Aspects of the homeostatic plasticity of GABAA receptor-mediated inhibition. J. Physiol. 2005, 562, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Staley, K. Molecular mechanisms of epilepsy. Nat. Neurosci. 2015, 18, 367–372. [Google Scholar] [CrossRef]
- Ran, X.; Li, J.; Shao, Q.; Lin, Z.; Wu, J. EpilepsyGene: A genetic resource for genes and mutations related to epilepsy. Nucleic Acids Res. 2015, 43, D893–D899. [Google Scholar] [CrossRef]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Niday, Z.; Tzingounis, A.V. Potassium channel gain of function in epilepsy: An unresolved paradox. Neuroscientist 2018, 24, 368–380. [Google Scholar] [CrossRef]
- Barcia, G.; Fleming, M.R.; Deligniere, A.; Gazula, V.R.; Brown, M.R.; Langouet, M.; Chen, H.; Kronengold, J.; Abhyankar, A.; Cilio, R.; et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Genet. 2018, 44, 1255–1259. [Google Scholar] [CrossRef]
- Ramsdell, J.S. Neurological disease rises from ocean to bring model for human epilepsy to life. Toxins 2010, 2, 1646–1675. [Google Scholar] [CrossRef]
- Buckmaster, P.S.; Wen, X.; Toyoda, I.; Gulland, F.M.; Van Bonn, W. Hippocampal neuropathology of domoic acid-induced epilepsy in California sea lions (Zalophus californianus). J. Comp. Neurol. 2014, 522, 1691–1706. [Google Scholar] [CrossRef] [PubMed]
- Reid, C.A.; Rollo, B.; Petrou, S.; Berkovic, S.F. Can mutation-mediated effects occurring early in development cause long-term seizure susceptibility in genetic generalized epilepsies? Epilepsia 2018, 59, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.B.; Valakh, V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 2015, 87, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Jiruska, P.; de Curtis, M.; Jefferys, J.G.; Schevon, C.A.; Schiff, S.J.; Schindler, K. Synchronization and desynchronization in epilepsy: Controversies and hypotheses. J. Physiol. 2013, 591, 787–797. [Google Scholar] [CrossRef] [PubMed]
- McTague, A.; Howell, K.B.; Cross, J.H.; Kurian, M.A.; Scheffer, I.E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016, 15, 304–316. [Google Scholar] [CrossRef]
- Stamberger, H.; Weckhuysen, S.; DeJonghe, P. STXBP1 as a therapeutic target for epileptic encephalopathy. Expert Opin. Ther. Targets 2017, 21, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Suri, M.; Evers, J.M.G.; Laskowski, R.A.; O’Brien, S.; Baker, K.; Clayton-Smith, J.; Dabir, T.; Josifova, D.; Joss, S.; Kerr, B.; et al. Protein structure and phenotypic analysis of pathogenic and population missense variants in STXBP1. Mol. Genet. Genom. Med. 2017, 5, 495–507. [Google Scholar] [CrossRef]
- Toonen, R.F.; Wierda, K.; Sons, M.S.; de Wit, H.; Cornelisse, L.N.; Brussaard, A.; Plomp, J.J.; Verhage, M. Munc18-1 expression levels control synapse recovery by regulating readily releasable pool size. Proc. Natl. Acad. Sci. USA 2006, 103, 18332–18337. [Google Scholar] [CrossRef]
- Kovacevic, J.; Maroteaux, G.; Schut, D.; Loos, M.; Dubey, M.; Pitsch, J.; Remmelink, E.; Koopmans, B.; Crowley, J.; Cornelisse, L.N.; et al. Protein instability, haploinsufficiency, and cortical hyper-excitability underlie STXBP1 encephalopathy. Brain 2018, 141, 1350–1374. [Google Scholar] [CrossRef]
- Marsh, E.D.; Nasrallah, M.P.; Walsh, C.; Murray, K.A.; Nicole Sunnen, C.; McCoy, A.; Golden, J.A. Developmental interneuron subtype deficits after targeted loss of Arx. BMC Neurosci. 2016, 17, 35. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Excitatory actions of GABA during development: The nature of the nurture. Nat. Rev. Neurosci. 2002, 3, 728–739. [Google Scholar] [PubMed]
- Luhmann, H.J.; Kirischuk, S.; Sinning, A.; Kilb, W. Early GABAergic circuitry in the cerebral cortex. Curr. Opin. Neurobiol. 2014, 26, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Dzhala, V.I.; Talos, D.M.; Sdrulla, D.A.; Brumback, A.C.; Mathews, G.C.; Benke, T.A.; Delpire, E.; Jensen, F.E.; Staley, K.J. NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 2005, 11, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Glykys, J.; Dzhala, V.; Egawa, K.; Balena, T.; Saponijan, Y.; Kuchibhotla, K.V.; Bacskai, B.J.; Kahle, K.T.; Zeuthen, T.; Staley, K.J. Local impermeant anions establish the neuronal chloride concentration. Science 2014, 343, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Staley, K. Wrong-way chloride transport: Is it a treatable cause of some intractable seizures? Epilepsy Curr. 2006, 6, 124–127. [Google Scholar] [PubMed]
- Huberfeld, G.; Wittner, L.; Clemenceau, S.; Baulac, M.; Kaila, K.; Miles, R.; Rivera, C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 2007, 27, 9866–9873. [Google Scholar] [CrossRef]
- Dzhala, V.; Valeeva, G.; Glykys, J.; Khazipov, R.; Staley, K. Traumatic alterations in GABA signaling disrupt hippocampal network activity in the developing brain. J. Neurosci. 2012, 32, 4017–4031. [Google Scholar] [CrossRef] [PubMed]
- Brumback, A.C.; Staley, K.J. Thermodynamic regulation of NKCC1-mediated Cl- cotransport underlies plasticity of GABA(A) signaling in neonatal neurons. J. Neurosci. 2008, 28, 1301–1312. [Google Scholar] [CrossRef]
- Khazipov, R.; Valeeva, G.; Khalilov, I. Depolarizing GABA and developmental epilepsies. CNS Neurosci. Ther. 2015, 21, 83–91. [Google Scholar] [CrossRef]
- Deshpande, L.S.; DeLorenzo, R.J. Mechanisms of levetiracetam in the control of status epilepticus and epilepsy. Front. Neurol. 2014, 5, 11. [Google Scholar] [CrossRef]
- Yan, H.D.; Ishihara, K.; Seki, T.; Hanaya, R.; Kurisu, K.; Arita, K.; Serikawa, T.; Sasa, M. Inhibitory effects of levetiracetam on the high-voltage-activated L-type Ca2+ channels in hippocampal CA3 neurons of spontaneously epileptic rat (SER). Brain Res. Bull. 2013, 90, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Klitgaard, H.; Matagne, A.; Gobert, J.; Wülfert, E. Evidence for a unique profile of levetiracetam in rodent models of seizures and epilepsy. Eur. J. Pharmacol. 1998, 353, 191–206. [Google Scholar] [CrossRef]
- Surges, R.; Volynski, K.E.; Walker, M.C. Is levetiracetam different from other antiepileptic drugs? Levetiracetam and its cellular mechanism of action in epilepsy revisited. Ther. Adv. Neurol. Disord. 2008, 1, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Ceulemans, B.; Schoonjans, A.S.; Marchau, F.; Paelinck, B.P.; Lagae, L. Five-year extended follow-up status of 10 patients with Dravet syndrome treated with fenfluramine. Epilepsia 2016, 57, e129–e134. [Google Scholar] [CrossRef] [PubMed]
- Sourbron, J.; Smolders, I.; de Witte, P.; Lagae, L. Pharmacological analysis of the anti-epileptic mechanisms of fenfluramine in scn1a mutant zebrafish. Front. Pharmacol. 2017, 8, 191. [Google Scholar] [CrossRef]
- Patel, M. A metabolic paradigm for epilepsy. Epilepsy Curr. 2018, 18, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.M.; Stafstrom, C.E. The ketogenic diet: What has science taught us? Epilepsy Res. 2012, 100, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Simeone, T.A.; Simeone, K.A.; Stafstrom, C.E.; Rho, J.M. Do ketone bodies mediate the anti-seizure effects of the ketogenic diet? Neuropharmacology 2018, 133, 233–241. [Google Scholar] [CrossRef]
- Masino, S.A.; Kawamura, M., Jr.; Ruskin, D.N.; Geiger, J.D.; Boison, D. Purines and neuronal excitability: Links to the ketogenic diet. Epilepsy Res. 2012, 100, 229–238. [Google Scholar] [CrossRef]
- Levin, B.E. Glucosensing neurons do more than just sense glucose. Int. J. Obes. Relat. Metab. Disord. 2001, 25, S68–S72. [Google Scholar] [CrossRef]
- Ma, W.; Berg, J.; Yellen, G. Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J. Neurosci. 2007, 27, 3618–3625. [Google Scholar] [CrossRef] [PubMed]
- Lutas, A.; Yellen, G. The ketogenic diet: Metabolic influences on brain excitability and epilepsy. Trends Neurosci. 2013, 36, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Sada, N.; Inoue, T. Electrical control in neurons by the ketogenic diet. Front. Cell. Neurosci. 2018, 12, 208. [Google Scholar] [CrossRef] [PubMed]
- Thio, L.; Wong, M.; Yamada, K. Ketone bodies do not directly alter excitatory or inhibitory hippocampal transmission. Neurology 2000, 54, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Simeone, K.A.; Simeone, T.A.; Pandya, J.D.; Wilke, J.C.; Ahn, Y.; Geddes, J.W.; Sullivan, P.G.; Rho, J.M. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann. Neurol. 2015, 78, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Augustin, K.; Boddum, K.; Williams, S.; Sun, M.; Terschak, J.A.; Hardege, J.D.; Chen, P.E.; Walker, M.C.; Williams, R.S. Seizure control by decanoic acid through direct AMPA receptor inhibition. Brain 2016, 139, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, C.E.; Ockuly, J.C.; Murphee, L.; Valley, M.T.; Roopra, A.; Sutula, T.P. Anticonvulsant and antiepileptic actions of 2-deoxy-D-glucose in epilepsy models. Ann. Neurol. 2009, 65, 435–447. [Google Scholar] [CrossRef]
- Hartman, A.L.; Rubenstein, J.E.; Kossoff, E.H. Intermittent fasting: A “new” historical strategy for controlling seizures? Epilepsy Res. 2013, 104, 275–279. [Google Scholar] [CrossRef]
- Pfeifer, H.H.; Thiele, E.A. Low-glycemic index treatment: A liberalized ketogenic diet for treatment of intractable epilepsy. Neurology 2005, 65, 1810–1812. [Google Scholar] [CrossRef]
- McDonald, T.; Puchowicz, M.; Borges, K. Impairments in oxidative glucose metabolism in epilepsy and metabolic treatments thereof. Front. Cell. Neurosci. 2018, 12, 274. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| GENE | GENE PRODUCT | ROLE | EPILEPSY SYNDROME |
|---|---|---|---|
| STXBP1 | Syntaxin binding protein 1 | Vesicle fusion with presynaptic membrane allowing neurotransmitter release | Ohtahara syndrome |
| ARX | Aristaless-related homeobox protein | Tangential migration of interneurons into the cortical plate | Multiple seizure types, infantile spasms |
| CDKL5 | Cyclin-dependent kinase-like 5 | Actin cytoskeleton, dendritic arborization, MeCP2 phosphorylation | Multiple seizure types, infantile spasms |
| PCDH19 | Protocadherin 19 | Neuron adhesion during migration | Female-restricted epilepsy +/− ID, multiple seizure types, infantile spasms |
| UBE3A | Ubiquitin protein ligase E3A | Targets proteins for intracellular degradation | Angelman syndrome |
| PTEN | Phosphatase and tensin homolog | Tumor and cell growth/migration suppression | Cowden syndrome, focal seizures |
| ASD | Mechanism | E/I Alteration |
|---|---|---|
| Phenobarbital | Enhances GABAA receptor function by increasing chloride channel open time | ↑ I |
| Phenytoin | Blocks Na channels | ↓ E |
| Carbamazepine, Oxcarbazepine | Blocks Na channels | ↓ E |
| Valproate | Multiple—enhances GABA action, blocks Na and Ca channels | ↓ E, ↑ I |
| Ethosuximide | Blocks T-type Ca channels | ↓ E |
| Benzodiazepines | Enhance GABAA receptor function by increasing frequency of chloride channel openings | ↑ I |
| Levetiracetam, Brivaracetam | Modulate synaptic vesicle protein SV2A | Unclear |
| Topiramate | Multiple—enhances GABA action, blocks AMPA receptors and Na channels | ↓ E, ↑ I |
| Vigabatrin | Inhibits GABA transaminase | ↑ I |
| Zonisamide | Multiple—blocks Na and Ca channels, alters neurotransmitter transport | ↓ E |
| Perampanel | Blocks AMPA receptors | ↓ E |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, L.-R.; Habela, C.W.; Stafstrom, C.E. Pediatric Epilepsy Mechanisms: Expanding the Paradigm of Excitation/Inhibition Imbalance. Children 2019, 6, 23. https://doi.org/10.3390/children6020023
Shao L-R, Habela CW, Stafstrom CE. Pediatric Epilepsy Mechanisms: Expanding the Paradigm of Excitation/Inhibition Imbalance. Children. 2019; 6(2):23. https://doi.org/10.3390/children6020023
Chicago/Turabian StyleShao, Li-Rong, Christa W. Habela, and Carl E. Stafstrom. 2019. "Pediatric Epilepsy Mechanisms: Expanding the Paradigm of Excitation/Inhibition Imbalance" Children 6, no. 2: 23. https://doi.org/10.3390/children6020023
APA StyleShao, L.-R., Habela, C. W., & Stafstrom, C. E. (2019). Pediatric Epilepsy Mechanisms: Expanding the Paradigm of Excitation/Inhibition Imbalance. Children, 6(2), 23. https://doi.org/10.3390/children6020023