
Idiopathic Pulmonary Arterial Hypertension in Paediatrics Represents Still a Serious Challenge: A Case Series Study
 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Echocardiography
2.2. Right Heart Catheterisation
3. Statistics
4. Results
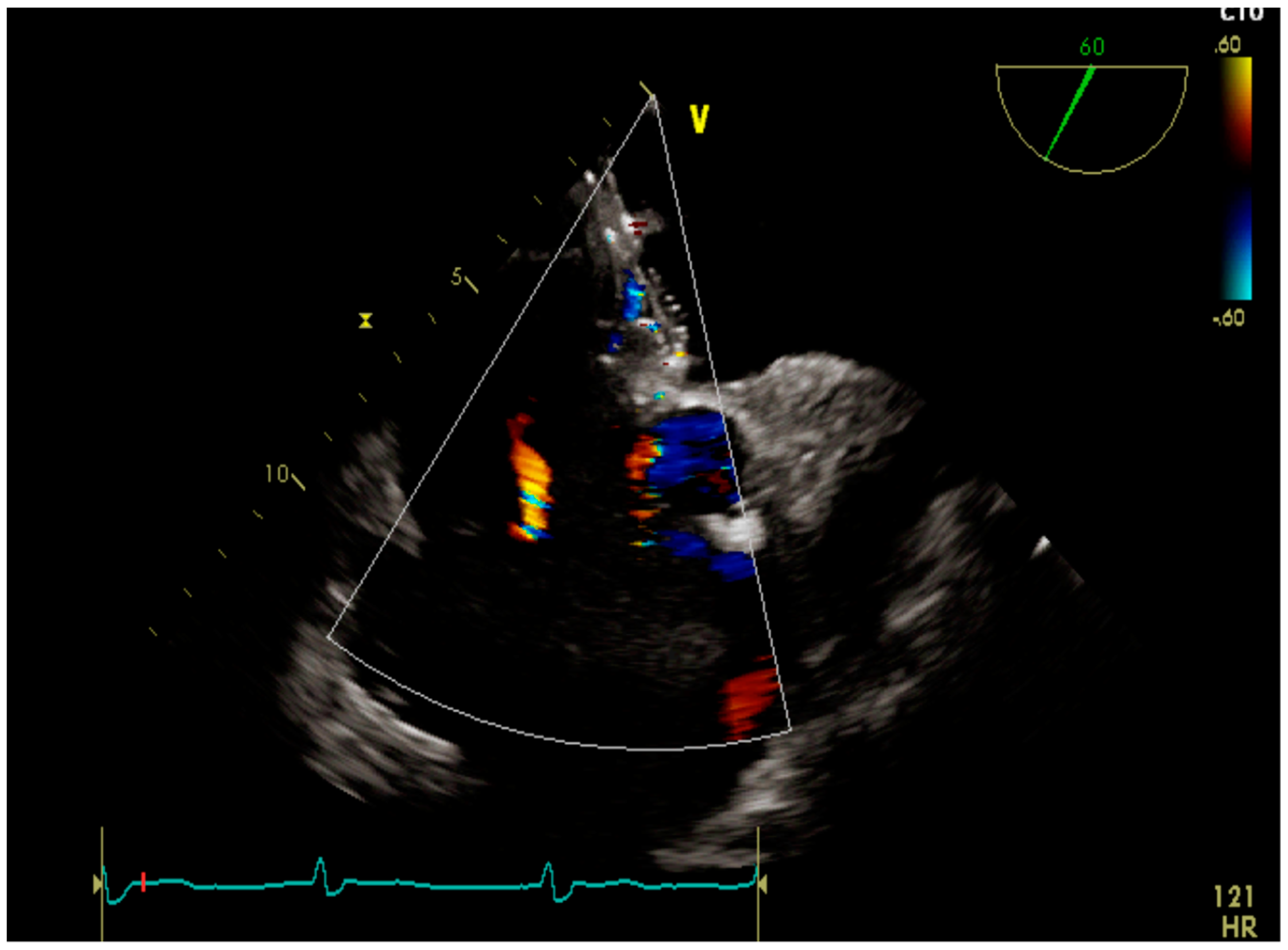
4.1. Case 1
4.2. Case 2
4.3. Case 3
5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hopper, R.K.; Abman, S.H.; Ivy, D.D. Persistent challenges in pediatric pulmonary hypertension. Chest 2016, 150, 226–236. [Google Scholar] [CrossRef]
- Calcaterra, G.; Bassareo, P.P.; Barilla, F.; Martino, F.; Fanos, V.; Fedele, F.; Romeo, F. Pulmonary hypertension in pediatrics. A feasible approach to bridge the gap between real world and guidelines. J. Matern. Fetal Neonatal Med. 2021, 34, 3820–3826. [Google Scholar] [CrossRef]
- Douwes, J.M.; van Loon, R.L.; Hoendermis, E.S.; Vonk-Noordegraaf, A.; Roofthooft, M.T.; Talsma, M.D.; Hillege, H.L.; Berger, R.M. Acute pulmonary vasodilator response in paediatric and adult pulmonary arterial hypertension: Occurrence and prognostic value when comparing three response criteria. Eur. Heart J. 2011, 32, 3137–3146. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [PubMed]
- Calcaterra, G.; Fanos, V.; Bassareo, P.P. Still puzzling about a clear definition of pulmonary arterial hypertension in newborns. Eur. Respir. J. 2019, 53, 1900005. [Google Scholar] [CrossRef] [PubMed]
- Yamaki, S.; Wagenvoort, C.A. Comparison of primary plexogenic arteriopathy in adults and children. A morphometric study in 40 patients. Br. Heart J. 1985, 54, 428–434. [Google Scholar] [CrossRef]
- Rosenzweig, E.B.; Abman, S.H.; Adatia, I.; Beghetti, M.; Bonnet, D.; Haworth, S.; Ivy, D.D.; Berger, R.M.F. Paediatric pulmonary arterial hypertension: Updates on definition, classification, diagnostics and management. Eur. Respir. J. 2019, 53, 1801916. [Google Scholar] [CrossRef]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 233–270. [Google Scholar] [CrossRef]
- Chokkalingam Mani, B.; Chaudhari, S.S. Right Heart Cardiac Catheterization; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557404/ (accessed on 3 March 2023).
- Krishnan, A.; Markham, R.; Savage, M.; Wong, Y.W.; Walters, D. Right Heart Catheterisation: How to do it. Heart Lung Circ. 2019, 28, e71–e78. [Google Scholar] [CrossRef]
- Bangalore, S.; Bhatt, D.L. Images in cardiovascular medicine. Right heart catheterization, coronary angiography, and percutaneous coronary intervention. Circulation 2011, 124, e428–e433. [Google Scholar] [CrossRef]
- Saji, T. Update on pediatric pulmonary arterial hypertension. Differences and similarities to adult disease. Circ. J. 2013, 77, 2639–2650. [Google Scholar] [CrossRef]
- Adatia, I.; Kothari, S.S.; Feinstein, J.A. Pulmonary hypertension associated with congenital heart disease: Pulmonary vascular disease: The global perspective. Chest 2010, 137, 52S–61S. [Google Scholar] [CrossRef]
- Levine, D.J. Diagnosis and management of pulmonary arterial hypertension: Implications for respiratory care. Respir. Care 2006, 51, 368–381. [Google Scholar] [PubMed]
- Valencia, G.A.; Krishnan, U. Idiopathic Pulmonary Arterial Hypertension in Children: A Review. Pulm. Ther. 2017, 3, 67–92. [Google Scholar] [CrossRef]
- Berger, R.M.; Beghetti, M.; Humpl, T.; Raskob, G.E.; Ivy, D.D.; Jing, Z.C.; Bonnet, D.; Schulze-Neick, I.; Barst, R.J. Clinical features of paediatric pulmonary hypertension: A registry study. Lancet 2012, 379, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Abman, S.H.; Mullen, M.P.; Sleeper, L.A.; Austin, E.D.; Rosenzweig, E.B.; Kinsella, J.P.; Ivy, D.; Hopper, R.K.; Raj, J.U.; Fineman, J.; et al. Characterisation of paediatric pulmonary hypertensive vascular disease from the PPHNet Registry. Eur. Respir. J. 2021, 59, 2003337. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; McGoon, M.D.; Elliott, C.G.; Foreman, A.J.; Miller, D.P.; Ivy, D.D. Survival in childhood pulmonary arterial hypertension: Insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Circulation 2012, 125, 113–122. [Google Scholar] [CrossRef]
- Ploegstra, M.J.; Ivy, D.D.; Wheeler, J.G.; Brand, M.; Beghetti, M.; Rosenzweig, E.B.; Humpl, T.; Iriart, X.; Rouzic, E.M.; Bonnet, D.; et al. Growth in children with pulmonary arterial hypertension: A longitudinal retrospective multiregistry study. Lancet Respir. Med. 2016, 4, 281–290. [Google Scholar] [CrossRef]
- D’Alto, M.; Di Maio, M.; Romeo, E.; Argiento, P.; Blasi, E.; Di Vilio, A.; Rea, G.; D’Andrea, A.; Golino, P.; Naeije, R. Echocardiographic probability of pulmonary hypertension: A validation study. Eur. Respir. J. 2022, 60, 2102548. [Google Scholar] [CrossRef]
- Zuckerman, W.A.; Turner, M.E.; Kerstein, J.; Torres, A.; Vincent, J.A.; Krishnan, U.; Kerstein, D.; Rosenzweig, E.B. Safety of cardiac catheterization at a center specializing in the care of patients with pulmonary arterial hypertension. Pulm. Circ. 2013, 3, 831–839. [Google Scholar] [CrossRef]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar] [CrossRef] [PubMed]
- Beghetti, M.; Schulze-Neick, I.; Berger, R.M.; Ivy, D.D.; Bonnet, D.; Weintraub, R.G.; Saji, T.; Yung, D.; Mallory, G.B.; Geiger, R.; et al. Haemodynamic characterisation and heart catheterisation complications in children with pulmonary hypertension: Insights from the Global TOPP Registry (tracking outcomes and practice in paediatric pulmonary hypertension). Int. J. Cardiol. 2016, 203, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, J.; Petersen, C.L.; Kjaer, A.; Schaadt, B.K.; Oh, J.K.; Hassager, C. Evaluation of right ventricular volume and function by 2D and 3D echocardiography compared to MRI. Eur. J. Echocardiogr. 2006, 7, 430–438. [Google Scholar] [CrossRef]
- Koestenberger, M.; Ravekes, W.; Everett, A.D.; Stueger, H.P.; Heinzl, B.; Gamillscheg, A.; Cvirn, G.; Boysen, A.; Fandl, A.; Nagel, B. Right ventricular function in infants, children and adolescents: Reference values of the tricuspid annular plane systolic excursion (TAPSE) in 640 healthy patients and calculation of z score values. J. Am. Soc. Echocardiogr. 2009, 22, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Badagliacca, R.; Poscia, R.; Pezzuto, B.; Nocioni, M.; Mezzapesa, M.; Francone, M.; Giannetta, E.; Papa, S.; Gambardella, C.; Sciomer, S.; et al. Right ventricular remodeling in idiopathic pulmonary arterial hypertension: Adaptive versus maladaptive morphology. J. Heart Lung Transplant. 2015, 34, 395–403. [Google Scholar] [CrossRef]
- Sato, T.; Tsujino, I.; Ohira, H.; Oyama-Manabe, N.; Yamada, A.; Ito, Y.M.; Goto, C.; Watanabe, T.; Sakaue, S.; Nishimura, M. Validation study on the accuracy of echocardiographic measurements of right ventricular systolic function in pulmonary hypertension. J. Am. Soc. Echocardiogr. 2012, 25, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.M.; Dwyer, N.; Celermajer, D.; Kritharides, L.; Marwick, T.H. Follow-Up of Pulmonary Hypertension with Echocardiography. JACC Cardiovasc. Imaging 2016, 9, 733–746. [Google Scholar] [CrossRef] [PubMed]
- Lammers, A.E.; Apitz, C.; Zartner, P.; Hager, A.; Dubowy, K.O.; Hansmann, G. Diagnostics, monitoring and outpatient care in children with suspected pulmonary hypertension/paediatric pulmonary hypertensive vascular disease. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 2016, 102, ii1–ii13. [Google Scholar]
- Raymond, R.J.; Hinderliter, A.L.; Willis, P.W.; Ralph, D.; Caldwell, E.J.; Williams, W.; Ettinger, N.A.; Hill, N.S.; Summer, W.R.; de Boisblanc, B.; et al. Echocardiographic predictors of adverse outcomes in primary pulmonary hypertension. J. Am. Coll. Cardiol. 2002, 39, 1214–1219. [Google Scholar] [CrossRef]
- Khirfan, G.; Almoushref, A.; Naal, T.; Abuhalimeh, B.; Dweik, R.A.; Heresi, G.A.; Tonelli, A.R. Mixed Venous Oxygen Saturation is a Better Prognosticator than Cardiac Index in Pulmonary Arterial Hypertension. Chest 2020, 158, 2546–2555. [Google Scholar] [CrossRef]
- D’Alto, M.; Badagliacca, R.; Argiento, P.; Romeo, E.; Farro, A.; Papa, S.; Sarubbi, B.; Russo, M.G.; Vizza, C.D.; Golino, P.; et al. Risk Reduction and Right Heart Reverse Remodeling by Upfront Triple Combination Therapy in Pulmonary Arterial Hypertension. Chest 2020, 157, 376–383. [Google Scholar] [CrossRef]
- Boudjemline, Y.; Patel, M.; Malekzadeh-Milani, S.; Szezepanski, I.; Lévy, M.; Bonnet, D. Patent ductus arteriosus stenting (transcatheter Potts shunt) for palliation of suprasystemic pulmonary arterial hypertension: A case series. Circ. Cardiovasc. Interv. 2013, 6, e18–e20. [Google Scholar] [CrossRef] [PubMed]
- Reichenberger, F.; Pepke-Zaba, J.; McNeil, K.; Parameshwar, J.; Shapiro, L.M. Atrial septostomy in the treatment of severe pulmonary arterial hypertension. Thorax 2003, 58, 797–800. [Google Scholar] [CrossRef] [PubMed]
- George, M.P.; Champion, H.C.; Pilewski, J.M. Lung transplantation for pulmonary hypertension. Pulm. Circ. 2011, 1, 182–191. [Google Scholar] [CrossRef]
- Moser, B.; Jaksch, P.; Taghavi, S.; Muraközy, G.; Lang, G.; Hager, H.; Krenn, C.; Roth, G.; Faybik, P.; Bacher, A.; et al. Lung transplantation for idiopathic pulmonary arterial hypertension on intraoperative and postoperatively prolonged extracorporeal membrane oxygenation provides optimally controlled reperfusion and excellent outcome. Eur. J. Cardiothorac. Surg. 2018, 53, 178–185. [Google Scholar] [CrossRef]
- Bassareo, P.P.; McMahon, C.J. Metabolomics: A New Tool in Our Understanding of Congenital Heart Disease. Children 2022, 9, 1803. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.C.; Chen, H.L.; Liu, Y.C.; Wu, Y.H.; Lo, S.H.; Hsu, J.H.; Yin, H.L.; Hsu, J.S.; Wu, B.N.; Dai, Z.K. Unique Pulmonary Hypertension in Young Children: A Case Series Study. Children 2022, 9, 1064. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bassareo, P.P.; Argiento, P.; McMahon, C.J.; Dunne, E.; Walsh, K.P.; Russo, M.G.; D’Alto, M. Idiopathic Pulmonary Arterial Hypertension in Paediatrics Represents Still a Serious Challenge: A Case Series Study. Children 2023, 10, 518. https://doi.org/10.3390/children10030518
Bassareo PP, Argiento P, McMahon CJ, Dunne E, Walsh KP, Russo MG, D’Alto M. Idiopathic Pulmonary Arterial Hypertension in Paediatrics Represents Still a Serious Challenge: A Case Series Study. Children. 2023; 10(3):518. https://doi.org/10.3390/children10030518
Chicago/Turabian StyleBassareo, Pier Paolo, Paola Argiento, Colin Joseph McMahon, Esme Dunne, Kevin Patrick Walsh, Maria Giovanna Russo, and Michele D’Alto. 2023. "Idiopathic Pulmonary Arterial Hypertension in Paediatrics Represents Still a Serious Challenge: A Case Series Study" Children 10, no. 3: 518. https://doi.org/10.3390/children10030518
APA StyleBassareo, P. P., Argiento, P., McMahon, C. J., Dunne, E., Walsh, K. P., Russo, M. G., & D’Alto, M. (2023). Idiopathic Pulmonary Arterial Hypertension in Paediatrics Represents Still a Serious Challenge: A Case Series Study. Children, 10(3), 518. https://doi.org/10.3390/children10030518