Abstract
Modern analysis of food and feed is mostly focused on development of fast and reliable portable devices intended for field applications. In this review, electrochemical biosensors based on immunological reactions and aptamers are considered in the determination of mycotoxins as one of most common contaminants able to negatively affect human health. The characteristics of biosensors are considered from the point of view of general principles of bioreceptor implementation and signal transduction providing sub-nanomolar detection limits of mycotoxins. Moreover, the modern trends of bioreceptor selection and modification are discussed as well as future trends of biosensor development for mycotoxin determination are considered.
1. Introduction
Growing attention to the problems related to the contamination of foodstuffs and its negative effect on the health of the population call for new efforts in the area of fast and reliable detection of chemical hazards and toxic substances produced by microbiota. Although modern chromatographic and spectrometric instrumentation is able to detect and quantify all of the food contaminants, some complications related to the timeliness of the information required and to the adaptation of measurements for field conditions remain in the scope of intensive investigations. This is especially true for mycotoxins, toxic fungal secondary metabolites that are formed in microbiological contamination of cereals and some other agriculture products [1,2]. Concern about the mycotoxin contamination of food and feed was originally initiated by numerous turkey deaths in the 1960s caused by feed contaminated with Aspergillus flavus and aflatoxin B1 (AFB1) [3]. The United Nations Food and Agriculture Organization (FAO) and the World Health Organization (WHO) have confirmed high toxicological impact of mycotoxins on human and animal health. This resulted in a strict legislation of their residuals in a wide range of foodstuffs. The necessity for overall monitoring of mycotoxins and their metabolites is aggravated by the large scale of agriculture production and a large number of manufacturers, many of which are unable to maintain high standards of production and contamination prevention due to limited technical facilities and financial resources. In the period from 2002 to 2012, 15% of risk related to food contamination referred to mycotoxins against 20% attributed to persistent organic pollutants [4]. The globalization of world economics, with huge traffic flows, creates favorable conditions for dissemination of contaminated foodstuffs around the world [5]. Global warming stimulates the formation and dissemination of mycotoxins [6]. Besides, they are not easily eliminated in food processing because of their stability against heat, physical, and chemical treatments [7]. Feed contamination also poses hazards due to carry-over of mycotoxins to animal-derived products (milk, eggs, etc.) [8].
All of these reasons increase the burning necessity for portable instrumentation able to detect mycotoxins outside well-equipped chemical laboratories, i.e., directly in a field, barn, mill or brewery. Depending on the format and customer desire, such instrumentation can operate as warning devices for early detection of mycotoxins or as portable analytical systems able to determine in a semi-quantitative or quantitative way appropriate analytes and their metabolites in complex biological matrices.
Although many conventional analytical instruments can be adapted for sensor format, their sensitivity toward analyte present in very low quantities is mainly established by sample pre-treatment utilizing extraction or adsorption on mycotoxin residues on an appropriate support. This step, being labor- and time-consuming, is compensated for by the many advantages of chemical sensors, e.g., their simple design and fast measurement time. For this reason, preferences are given to the equipment utilizing new recognition principles, which are almost not used in a standard laboratory equipment. Biosensors are one of most successful examples of such an approach that offer unique opportunities for mycotoxin determination in “point-on-demand” format [9,10,11]. In them, biochemical components (proteins and nucleobases) are attached to the specific transducers converting a biochemical recognition event into an electric signal [12]. In the case of mycotoxins, specific reactions responsible for biorecognition involve inhibition of acetylcholinesterase [13] and formation of the complexes with specific antibodies [14] and aptamers [15]. Biosensors do not assume complicated sample treatment and a long measurement protocol. They are compatible with flow-through and microfluidics assay formats. Recently, some aspects of electrochemical biosensors for mycotoxin determination have been summarized in reviews [9,16,17,18]. In this review, application of electrochemical biosensors based on affine interactions is considered for sensitive mycotoxin analysis with particular emphasis on the sensor interface assembly and real sample assay problems.
2. Mycotoxins: General Characteristics and Conventional Analytical Techniques
2.1. Mycotoxins: General Description
Mycotoxins are secondary metabolites produced by some fungi (Aspergillus, Fusarium, and Penicillium spp.) and exerting effect on vertebrates [19]. Mycotoxicoses arise if environmental conditions combine high humidity and increased temperature and hence promote the molds’ growth. Mycotoxins can be accumulated in cereals and cereal based foods, fruits, wine, milk, coffee beans, cocoa bakery and meat products [20].
From a legislative point of view and acute toxicity, aflatoxins, ochratoxins, fumonisins, deoxynivalenol, T-2, HT-2, zearalenone, patulin and citrinin are most intensively investigated.
Aflatoxins (Figure 1) are produced by A. flavus and A. pseudotamarii (aflatoxins B1 (AFB1) and B2 (AFB2)) and A. parasiticus and A. nomius spp. (AFB1, AFB2, aflatoxin G1, and aflatoxin G2) and have drawn serious attention due to their high cytotoxicity and carcinogenicity.
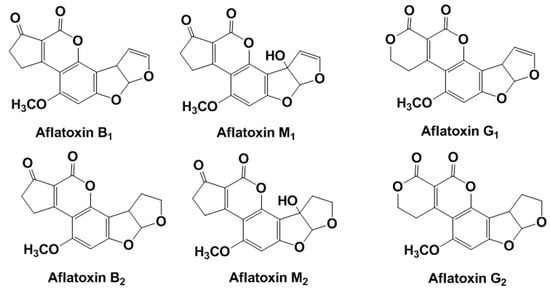
Figure 1.
Chemical structures of common aflatoxins.
Letters ‘B’ and ‘G’ in the aflatoxin name indicate blue and green fluorescence produced by these compounds under ultraviolet (UV) irradiation. The indices ‘1’ and ‘2’ correspond to major and minor compounds produced, respectively. In accordance with the classification of the International Agency for Research on Cancer (IARC), aflatoxins belong to the first hazard class [21]. AFB1 can be metabolically converted to epoxide that also exerts carcinogenic and mutagenic effect on a human health. When ingested by cows, AFB1(2) is hydroxylated to AFM1(2), which are then secreted in the milk. They are quite stable in acidic conditions and high temperature typical for milk pasteurization and cheese maturation. The limits established in the European Union (EU) for AFB1 and total aflatoxin content in foods are 2 and 4 µg/kg, respectively. The maximum threshold limit for AFM1 in milk is set at 0.05 µg/kg and at half this value for baby food. The European Commission (EC) Regulations established the maximum acceptable level of AFB1 in cereals, peanuts and dried fruits for direct human consumption in 4 ng/g for total aflatoxins and 2 ng/g for the most toxic AFB1 [22].
Ochratoxin A (OTA) is produced mostly by A. ochraceus and Penicillium verrucosum sp. in maize, barley, wheat, oats, rye, hay and mixed feed and is mainly found after harvesting of insufficiently dried products (Figure 2).
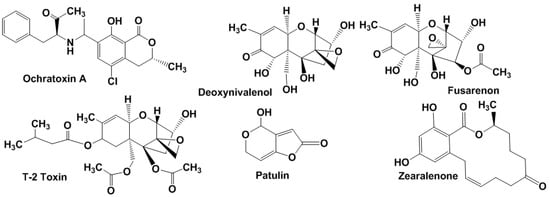
Figure 2.
Chemical structures of ochratoxin A, patulin, zearalenone and some trichothecene mycotoxins.
OTA-contaminated feeding stuffs reduce growth rates and productivity of cattle (especially pigs) and poultry. A maximal OTA limit of 5 μg/kg in cereals and 3 μg/kg in cereal products was established by the WHO.
Trichothecenes (Figure 2) are mainly produced by Fusarium spp. They affect animals and humans through contaminated grains (wheat, oats, barley, and rice) [23]. In accordance with the chemical structure and producing organisms, four groups of trichothecene mycotoxins are specified. Type A includes T-2 toxin, HT-2 toxin, neosolaniol and diacetoxyscirpenol, type B involves deoxynivalenol (DON), nivalenol, 3-acetyldeoxynivalenol and fusarenon, crotocin and baccharin belong to type C and satratoxin G, H, roridin A and verrucarin A to type D. Clinical signs of trichothecene poisoning in animals involve feed refusal, vomiting, growth retardation, reproductive disorders, and blood disorders (haematoxicity). As in the case of aflatoxins, trichothecene metabolites are less toxic than their precursors [24].
Patulin (Figure 2) is one of the most disseminated mycotoxins found in agriculture products. It is produced by Penicillium, Aspergillus and Byssochlamys spp. but also found in final products especially in apples and apple-based products [25]. Patulin poses mutagenic, teratogenic and carcinogenic effects [26]. The WHO established its maximum permitted level of 50 μg/L in apple juice and cider, 25 ng/g in solid apple products and 10 ng/g in products for infants and young children in the EU.
Zearalenone (Figure 2) is a non-steroidal estrogen produced by Fusarium spp., which is commonly present in soil fungi. It is able to competitively bind to estrogen receptors [27]. Zearalenone contaminates mostly corn and, to a lesser extent, barley, oats, wheat, sorghum, and rice. It is quite stable during storage and food cooking. When accumulated in food and feed, it is associated with the reproductive problems of some animals and possibly humans. IARC allocated zearalenone together with other Fusarium toxins in the third group (not classifiable as to their carcinogenicity to humans). Tolerable daily intake for zearalenone of 0.2 μg/kg of body weight is established in European Community (EC), while the FAO/WHO Expert Committee on Food Additives recommended maximum tolerable intake of the level of 0.5 μg/kg per day [28].
2.2. Conventional Analytical Methods for Determining Mycotoxins
Accurate and reliable analysis of mycotoxins is considered crucial for food safety and health when there is pollution. All of the conventional instrumentation used for this purpose assume three basic steps of analysis, i.e., sampling and extraction of analytes, purification and clean-up of the extracts and identification and quantification of certain species [29]. Sampling should take into account high heterogeneity of contamination by mycotoxins. Thus, DON is mostly concentrated in the pericarp of grain [30]. This makes conventional approaches insufficient for mycotoxin assay and increases the amount of sample for the following extraction of the analytes. In most cases, the sample is ground, homogenized in extraction solvent and then filtered. The organic solvent is selected to accept as many mycotoxins as possible. Recently, several new extraction methods have been adapted for mycotoxin assay, e.g., supercritical fluid extraction, accelerated solvent extraction and microwave-assisted extraction. They decrease the amounts of the extraction solvent required. Prior to analysis, the extract is cleaned to remove co-extracted interferences. This can be done by solid-phase extraction, immunoaffinity column chromatography or by treatment of the extracts with reagents providing chemical destruction or separation of co-extracted species in compact sediment [20].
2.2.1. Chromatography Techniques
Routine determination of mycotoxins is commonly performed by various chromatography techniques including thin-layer chromatography (TLC), high-performance liquid chromatography (HPLC), and the gas chromatography (GC). Among them, HPLC is most frequently used for quantification of mycotoxins whereas TLC and GC can be applied for the detection of appropriate pollution although their sensitivity is often insufficient for detection of mycotoxins at their permissible levels [31]. Both normal and reverse-phase HPLC are also applied for separation and extract purification. The HPLC coupled with various detectors (ultraviolet-visible (UV-VIS), fluorescence and mass sensitive detectors) offers the highest sensitivity and selectivity even at very small sample/extract portions. Regarding fluorescence detection, appropriate protocols are mostly applied for sensitive determination of mycotoxins with natural fluorescence, e.g., aflatoxins, ochratoxin A, and citrinin [32]. Other mycotoxins should be preliminary derivatized, e.g., by o-phthalylaldehyde or 9-(fluorenylmethyl)chloroformate [33].
HPLC hyphenated to tandem mass spectroscopy (HPLC-MS/MS) or sequential MS (MSn) detection with atmospheric pressure chemical ionization or electrospray ionization has become the method of choice for mycotoxin analysis due its advantages, i.e., high sensitivity and accuracy [34]. The HPLC-MS instrumentation has been applied for simultaneous determination of a number of structurally relevant mycotoxins in a single chromatographic run (multi-analysis approach). Microfluidic chips coupled with HPLC-MS allowed for the reduction of sample consumption and dead volume of the column/detector and increasing sensitivity in on-line sample pre-concentration [35]. Meanwhile, chromatographic techniques also have several drawbacks related to matrix effects. They can be overcome by matrix-matched calibration or application of internal standards. Lengthy sample pre-treatment, expensive instruments and trained personnel limit the application of chromatography for fast on-site analysis performed by importers, traders, and food and feed companies etc.
2.2.2. Immunological Methods
Immunological methods are based on specific interactions between protecting proteins produced by immune system (antibodies (Ab), or γ-globulins) and mycotoxin (antigen, Ag) [36]. The production of Ab by β-T-lymphocytes is activated by the intrusion of biological and chemical hazards. The reaction (1) results in the formation of the Ag-Ab complex, which is then destroyed or released from the organism.
Abs are highly specific to the structure of different compounds, but selectivity of its interaction with relative compounds can vary depending on the source and measurement protocol. The use of immunoassay techniques for mycotoxin analysis is complicated by the small size of analyte molecules. In living beings, such small molecules called haptens cannot initiate immune response. First, they form conjugates with serum proteins [37]. A similar protocol is used for the accumulation of native (polyclonal) Abs required for immunological methods of hapten analysis.
In biosensor format, the monitoring of immune reaction (1) requires special measurement protocols considered below. They usually involve a number of steps of reagent addition separated with washing. Such a multistep procedure makes the immunoassay time- and labor-consuming [38].
The determination of haptens is performed by (A) direct and (B) indirect competitive analysis and (C) label-free assay. Appropriate reaction schemes are presented in Figure 3.
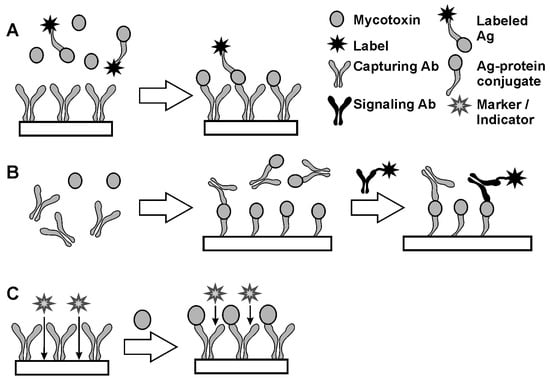
Figure 3.
Direct (A), indirect (B) immunoassay (A) and label-free protocol (C) of hapten immunoassay.
In the competitive assay (Figure 3A), the analyte molecule is conjugated with a label, which presence in the complex with Ab is easily detected. In 1970-s, radioactive elements were often used as labels (radioimmunoassay). At the present time, colored particles, chromogenic species and enzymes are more frequently used. The enzyme linked immunosorbent assay (ELISA) is called the “gold standard” of immunoassay [39]. Labeled Ag molecules are mixed with the sample so that a certain ratio of native and labeled Ag molecules is achieved. After incubation of the mixture with the Ab attached to a solid support, the number of labels bonded to the surface via Ag–Ab interaction depends on the part of the Ag molecules labeled. Thus, increasing concentration of an analyte results in a decreasing signal related to the label. Competitive immunoassay is a universal method of analysis applicable for various species and Abs which is quite popular due to the one-step protocol and compatibility with flow-through and flow-injection mode. The application of enzymes in labels offer higher sensitivity of immunoassay due to catalytic enhancement of the response toward Ag–Ab interaction. Among many enzymes, horseradish peroxidase (HRP) has found a broad application due to its low cost, high stability and many reactions resulted in the formation of colored, luminescent or electrochemically active products [40,41,42]. Its activity within a complex with Ag can be sensitively determined by conventional spectrophotometry with 4-aminoantipirine and tetramethylbenzidine as substrates [43,44] or by voltammetry with hydroquinone, catechol or iodide as organic substrates [45,46,47,48].
Regarding haptens, ELISA and related techniques of immunoassay have limitations related to the synthesis of labeled derivatives caused by the small size of appropriate molecules and negative influence of labeling on the reactivity and cross-selectivity of Ab binding. For this reason, indirect competitive immunoassay (Figure 3B) has been proposed [49,50]. Contrary to direct competitive assay, this combined homogeneous and heterogeneous reactions separated by the removal of excessive reagent. First, a sample containing hapten molecules is mixed with a certain excess of Abs. After that, Abs remained free to interact with hapten conjugates immobilized on the solid support. Finally, unoccupied hapten binding sites react with specific Ab bearing labels. This part of the reaction is performed in heterogeneous conditions on the solid interface. An indirect competitive assay is more sensitive than direct competitive assay due to the lack of necessity to modify hapten molecules. Then, increased concentration of an analyte results in a higher label signal. From general consideration, measurement of minor shift of a high signal is always less accurate than that of increased signal from its zero level. Both for direct and indirect competitive immunoassay, the sensitivity of hapten determination depends strongly on the selection of appropriate conjugates with inert carriers synthesized from immunoreagents and inert carriers like ovalbumin, soybean trypsin inhibitor or dextran. Steric accessibility of the immunoreagents in such conjugates and on the solid support are altered by the introduction of specific linkers and surfactants affecting both the 3D structure of appropriate particles and hydrophobicity of the reaction interface.
Label-free techniques do not assume synthesis of auxiliary reagents. In them, the target interactions and formation of Ag–Ab complexes affect transfer of diffusionally free probes that are then involved in electrochemical or chemiluminescent reactions on the solid support (Figure 3C) [51,52]. Being simple and fast, such approaches are most encouraged in the development of tests and immunosensors intended to use outside chemical laboratory and with non-qualified labor stuff. However, label-free immunoassay should take into account non-specific adsorption of interferences from the samples tested on the electrode that are the reason for false positives and overestimations of the analyte content. Sensitivity toward by-processes that take place on the same support is considered as a weak point of such techniques.
Immunological methods are preferably employed for the first-level screening and survey studies on mycotoxin contamination [53]. ELISA methods for mycotoxin assay have been commercially available for aflatoxins, fumonisins and trichothecenes since the 1970s. The assay is mostly performed in a 96-well plate allowing simultaneous testing of up to 45 samples in duplicate within 1–2 h. The use of kinetic regime instead of equilibrium assessment reduces measurement time to several minutes. ELISA test kits are available in a number of formats, including direct and indirect competitive assay [54].
2.3. Other Sensors for Mycotoxin Determination
Although the topic of the present review is focused on electrochemical biosensors, other principles of mycotoxin determination should be mentioned, although some of them are rather far from the portable design of electrochemical devices. Appropriate devices cover the last five years.
Among optic sensors, a monolithically integrated optoelectronic biosensor has been proposed for OTA determination in beer [55]. A broad-band Mach–Zehnder interferometer was produced on s silicon chip (37 mm2) with one arm coated with immobilized Ab. Competitive immunoassay made it possible to detect down to 2.0 ng/mL with recoveries ranging from 90.6% to 116%. Similar design was used in polarization reflection ellipsometry based on silicon wafers consisted of Si3N4–SiO2 sandwich [56]. Immobilization of monoclonal Ab against AFB1 allowed detection of 0.01 ng/ml of the analyte by shift of refractive index. Total internal reflectance ellipsometry has been applied for detection of OTA bonded to specific aptamers immobilized on gold-coated glass slices [57]. A similar technique was exploited here for detection of AFB1 and AFM1 with specific aptamers immobilized on the surface of golden films [58]. The method proposed was limited in the detection of mycotoxins with 0.01 ng/mL level and was successfully applied for investigation of binding kinetics and calculation of binding constants.
A miniaturized optical immunosensor based on White Light Reflectance Spectroscopy (WLRS) has been described for sensitive detection of AFM1 in milk [59]. Silicon chip with a SiO2 layer on top was covered with AFM1 - bovine serum albumin (BSA) conjugate. Indirect competitive immunoassay allowed detecting 6 pg/mL of the analyte within 25 min.
Among mass-sensitive sensors, quartz crystal microbalance with dissipation monitoring (QCM-D) can be mentioned [60]. In the sandwich immunoassay, 0.2–40 ng/mL of OTA were measured using Au nanoparticles as labels of secondary Abs for signal amplification.
3. Biosensors for Mycotoxin Determination
3.1. Immunosensors–General Principles and Ab Immobilization
Immunosensors are intended to immunological determination of the analyte in a simpler and faster way against conventional techniques assuming the use of reagent solutions and time-consuming steps like washing etc. From this point of view, the immunosensors do not differ dramatically from the ELISA and related methods by sensitivity, and selectivity mainly depended on the Ag–Ab interactions. In many cases, the difference between immunotest and immunosensor assumes the reactions between analytes, auxiliary reagents and Ab molecules to be performed on the transducer (electrode) interface or on plastic support closely attached to such a transducer [61]. This allows avoiding any losses in the redox active species involved in the electron transfer between the label (indicator) and electrode. The design of immunosensors involves nevertheless some additional steps necessary to improve the operational characteristics of immunosensors, i.e., signal time, signal drift during the storage, deviation of the signal etc. In many cases, this can be optimized at the step of reactant immobilization [62].
The immobilization is considered as indispensable part of biosensor development. Biochemical reagents (Abs, conjugates with inert proteins etc.) are attached to the support/electrode interface to prevent their leaching during the incubation with the sample and washing. Among various protocols, the following have become most popular in immunosensor development:
- Physical immobilization by adsorption on appropriate support or entrapment in polymeric matrices [63];
- Covalent immobilization by cross-linking with high-molecular carriers (proteins, carbonaceous nanoparticles, Au films and nanoparticles) [64,65,66,67];
- Affinity immobilization via avidin-biotin and protein A/G binding [68].
Physical immobilization does not require formation of covalent bonds between the reactants. Instead, multi-point weak interactions take place, which are governed by electrostatic, hydrophobic, donor–acceptor interactions, H-bonding etc. In many cases, Ab is immobilized on polyelectrolytes [69] or inert biopolymer that offers a sufficient number of functional groups for such interactions. In any case, own surface charge of the Ab molecules is important and its changes with the pH and ionic strength of the working solution. Physical immobilization via adsorption is a thermodynamically reversible process. Increase in the electrolyte concentration, temperature increase and pH-caused surface recharging can result in desorption of the Ab molecules when the biosensor is in contact with solution in non-optimal conditions. To avoid such circumstances, immobilized biomolecules can be covered by another additional layer that mechanically prevents desorption. Similar effect can be reached by electropolymerization of specific monomers [70] or polycondensation of orthosilicates (sol-gel immobilization [71]) performed in the presence of the Ab molecules. Appropriate reaction schemes are presented in Figure 4.
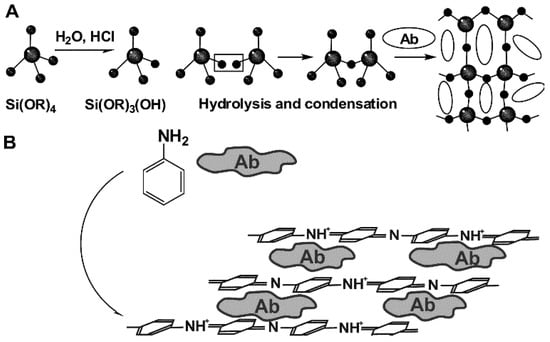
Figure 4.
Physical Ab immobilization by sol-gel immobilization (A) and entrapment in electropolymerized film (B).
Covalent immobilization involves the formation of covalent bonds between functional groups of a carrier and Ab molecules. Although the number of appropriate reagents is rather high, two protocols have found a broad application, i.e., carbodiimide binding and cross-linking with glutaraldehyde (Figure 5).
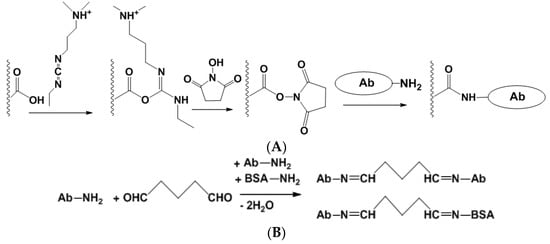
Figure 5.
Carbodiimide binding (A) and cross-linking with glutaraldehyde (B) for the covalent immobilization of Ab.
Affinity immobilization is based on natural receptor-ligand interaction [72,73]. Avidin (streptavidin)-biotin binding is mostly used due to simple implementation of biotin residue in biopolymer structure. Its dissociation constant (KD~10−14–10−15 M for avidin) is sufficient for the reliable immobilization of biomolecules. Each protein part (avidin or streptavidin) can bind up to four amines (biotin). This makes it possible to assemble multicomponent associates, e.g., conjugates of Ab with enzymes in ELISA format of immunoassay. Besides biotin–avidin binding, affinity immobilization involves reaction between concanavalin A, a plant protein of the lectin family, and carbohydrate residues in glycoproteins [74]. Proteins A and G can also non-specifically bind immunoglobulins [75].
3.1.1. Antibodies for Mycotoxin Determination
Based on the production protocol, polyclonal, monoclonal and recombinant Ab molecules are specified. Polyclonal Abs are extracted from the blood of immunized animals, they consist of a number of structurally relative globulins with similar affinity toward Ag. They can show insufficient selectivity toward certain mycotoxins. Monoclonal Abs are produced from hybridomas by fusing myeloma or spleen cells from immunized mice. Monoclonal Abs are more uniform in their affinity in comparison with polyclonal Abs. In the synthesis of recombinant Abs, the functional Ab gene is transmitted into prokaryotic or eukaryotic organisms from positive hybridoma or spleen cells [76].
In the mycotoxin assay, the IgG type of Ab is most frequently used. Its structure is outlined in Figure 6.
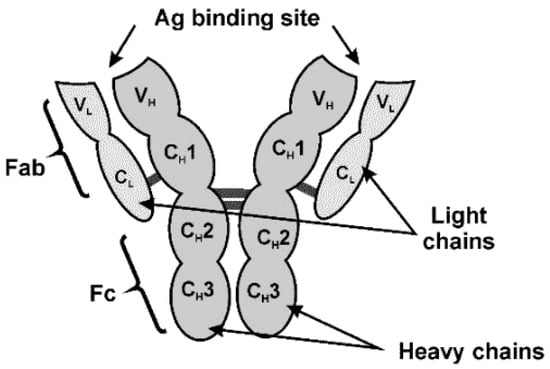
Figure 6.
IgG structure.
IgG consists of two pairs of identical light (L) and heavy (H) chains folded into globular domains (VL, CH1, CH2 and CH3). Specificity of the Ag binding is achieved by variation of specific binding sites at the N-terminal part of the sequences. The opposite C-terminal end of Ab is constant. In modern immunosensors, Abs described are often modified to reach higher efficiency of immobilization and better performance of appropriate biosensor. For this reason, fragments of IgG molecule are obtained [77] and are used in the assembly of immunosensors. Reduced Ab parts contain the tips of Y-shape molecule (Fab) or single-chain variable fragments (scFv). Being much smaller than native Ab, such derivatives minimize steric limitations of Ag–Ab interactions [78]. Examples of mycotoxin detection with scFv fragments of Abs are presented in Table 1.

Table 1.
Single-chain variable fragment (scFv) antibodies of Abs for mycotoxin determination (IC50 – concentration corresponded to 50% change of the signal. ELISA – enzyme linked immune sorbent assay, K-affinity constant, KD-dissociation constant, LOD—limit of detection).
3.1.2. Electrochemical Immunosensors
Electrochemical detection offers unique opportunities to measure the signal on Ab-Ag interaction in colored, turbid and viscous media using minimal sample volume and conventional instrumentation. Although there are many electrochemical techniques, most of the biosensors based on affine interactions utilize detection mode, i.e., voltammetric and impedimetric devices.
Voltammetric detection is carried out in an electrochemical cell consisting of a working electrode (biosensor), a reference and auxiliary (counter) electrode. In some assemblies like planar interdigitated electrodes, reference and auxiliary electrodes are combined together. A working solution should contain inert (supporting) electrolyte with a constant ionic strength. In the measurement, the working electrode is polarized against the reference electrode using an external source of power (voltammograph, potentiostat). In linear sweep voltammetry (Figure 7), working potential E is changing linearly between its limit values E1 and E2 and is defined prior to measurement with the program interface. In amperometric mode, the potential remains constant within the whole measurement period. The resulting current recorded on the working electrode depends on the rate of electrode reaction of oxidation (reduction) of species able to electron transfer and moved to the electrode surface preferably by diffusion. For constant current voltammetry and the rather high rate of electron exchange, the current corresponded to the conversion of a certain compound forms on the dependence of the current from the potential applied a peak with a typical S-shape (Figure 7).
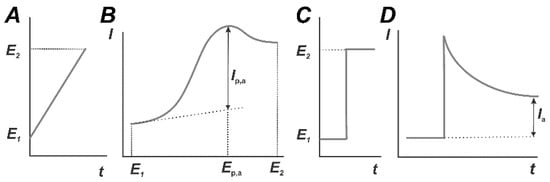
Figure 7.
Linear sweep (A,B) and potentiostatic (C,D) modes of constant current voltammetry. Temporal changes in the working potential (A,C) and voltammetric curves (B,D).
Here and below, oxidative reactions and oxidation currents are considered because they are mostly used in affine biosensors. The peak current Ip,a linearly depends on the volume concentration of the analyte and measurement conditions in accordance with the Randles–Ševcik Equation (2).
where S is the active electrode surface area, m2, c0 the concentration of the electroactive species in the bulk solution, mol m−3, D is its diffusion coefficient, m2 s−1, n is the number of electrons transferred and ν is the scan rate, V s−1. The Equation (2) is used also for the assessment of real electrode surface and for confirmation of the diffusional nature of the current recorded. The latter corresponds to the dependence of the peak current on the square root from the scan rate ν1/2, whereas the surface confined reaction showed linear dependence with the scan rate ν. The peak potential depends on the nature of the oxidized compounds and can be used for establishment of the reaction mechanism and selection of optimal working conditions. In potentiostatic (amperometric) mode, polarization potential is chosen near the peak potential in direct current voltammetry. Once the electrode is polarized, the current recorded is decreasing in accordance with the Cottrell Equation (3):
If the electrode is covered with surface film containing Ab, the current is rather quickly stabilized on the level directly proportional to the volume concentration of the reactant.
In addition to constant current techniques, other modulations of the working potential are used mostly to improve sensitivity of the method. Differential pulse voltammetry (DPV) and square wave voltammetry (SWV) are most common. The modulation of potential and response are illustrated in Figure 8. The bell-shape peak similar to first derivative of conventional I-E curves is recorded on both cases with the peak current and peak square proportional to the analyte concentration. The concentrations recorded with DPV and SWV are lower than those on direct current voltammetry by about one order of magnitude.
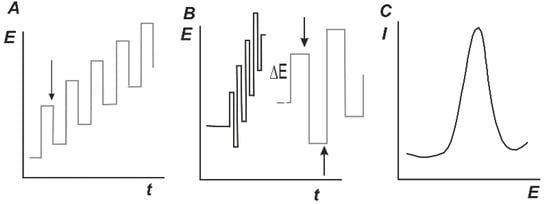
Figure 8.
Differential pulse voltammetry (DPV) (A) and square-wave voltammetry (SWV) (B) and signal shape (C). Arrows mark sampling points.
In electrochemical immunosensors, voltammetry is used for the detection of redox active labels and indicators and for electrochemical detection of enzymes applied in ELISA-based biosensors. Thus, the activity of HRP is measured using hydroquinone and that of alkaline phosphatase using 1-napthylphosphate as labels. In the latter case, α-naphtol is formed in enzymatic hydrolysis of the substrate. Its oxidation to naphtoquinone produces current measured as a signal of appropriate immunosensor.
Label-free detection in immunosensors is mostly based on the use of another electrochemical technique, electrochemical impedance spectroscopy (EIS). In so called Faradaic EIS, measurements are performed in the presence of redox probe, most frequently, the ferrocyanide pair [Fe(CN)6]3−/4−. The electrode is polarized at its formal electrode potential. After that, the sinusoidal potential of small amplitude is applied and appropriate current is recorded as a function of the frequency of potential changes. The results of the measurements are often presented in the Nyquist diagram in plots of imaginary and real components of the impedance. At high frequency, appropriate dependence has the shape of a semicircle the diameter of which gives the value of charge transfer resistance Rct. Target interaction of Ab with Ag increases the density of the surface layer and hence hinders the access of ferricyanide ions to the electrode. Besides, Ag binding can change electrostatic interactions with negatively charged redox probe and increase hydrophobicity of the interface. In both cases, the Ret value increases with the Ag concentration. EIS techniques are very sensitive to changes in the surface layer but especially for bulky analyte molecules.
Analytical characteristics of immunosensors for mycotoxin determination are summarized in Table 2 for the period from 2014 to 2018.
Table 2.
Analytical characteristics of immunosensors for mycotoxin determination (2014–2018).
4. Aptasensors
4.1. Aptamers Against Mycotoxins
Aptamers are synthetic DNA/RNA sequences obtained by combinatorial chemistry from the random library of oligonucleotides by affinity chromatography [120,121]. Aptamers usually contain up to 60 nucleotides and can recognize a wide variety of targets, from small ions to cells and Ab molecules. Contrary to DNA probes applied in DNA sensors intended for hybridization event detection, aptamers do not have direct analogs in nature. They are selected from random oligonucleotide libraries that can contain up to 1015 individual sequences. This is made by special protocols, among them the systematic evolution of ligands by exponential enrichment (SELEX) is mostly known [15]. Until 2007, aptamers mostly consisted of RNA, recently DNA aptamers have become dominant in analytical applications. They are stable to ward nuclease digestion, show excellent batch-to-batch reproducibility of the main properties, including analyte binding, and rather low cost in mass production.
The interest in aptamers as biorecognition elements in food analysis, including mycotoxin detection, is related to their high sensitivity (dissociation constants in nanomolar range). Besides, they are easily modified to involve labels and functional groups required for aptamer immobilization and detection. In some cases, the aptamer molecule can contain a long linker consisted of homonucleic sequence (oligoT as example) or lineal hydrocarbon radical (C12-C16) with terminal amino group. They allow partially suppress possible steric limitations of the reaction with the analyte molecule that take place near the electrode surface.
Aptamers are often considered as an alternative to Ab due to the ability of selective binding of appropriate analytes via non-covalent interactions. Thus, the reaction with the aptamer does not affect chemical nature of an analyte and is reversible. For this reason, many approaches to the assembling of aptamer-based biosensors (aptasensors) and detection of target aptamer-analyte interactions are rather similar to those described above for immunosensors. Meanwhile, aptamers are more stable than Ab toward hydrolysis, they are rather easy modified for implementing in the biosensor assembly [122]. Besides, aptamers meet requirements of bioethics because their design and production do not need animals.
Aptamers for mycotoxin determination have been designed since 2008. To date, aptamers against AFB1, AFB2, AFM1, T-2 toxin and zearalenone have been described and characterized mostly using fluorescence spectroscopy. The examples are summarized in Table 3. The affinity of target interactions and LOD values were preferably estimated by surface plasmon resonance (SPR) technique [123] and fluorescence spectroscopy.
Table 3.
Aptamers selected against various mycotoxins for their determination (KD—dissociation constant of the aptamer-mycotoxin complex, LOD—limit of detection).
4.2. Aptasensors—Assembling and Signal Transduction
The reaction of aptamer with a target results in changing the environment resulted in optical or electrochemical response. In case of fluorescence aptasensors, changes in the intensity of emission can be observed both in homogeneous solutions and on the appropriate support [15,16]. Besides, the aptamers bearing labels can be folded in three-dimensional structures using specific interactions of partially hybridized items or auxiliary DNA/RNA sequences. In the case of electrochemical devices, the reaction takes place on the surface of the electrode and results in changes in the accessibility of redox labels or in permeability of the layer for small charge carriers used as a redox probe. Although the size of mycotoxin molecules is rather small, they also affect the hydrophobicity of the surface layer and to some extend hydrogen ion exchange. This contributes to the increased sensitivity of the analyte detection. Besides, mycotoxins can provoke folding of the linear aptamer structure to bulky 3D architectures, among which flat squares formed by four guanine residues (G4 quadruplexes) are most known (Figure 9).
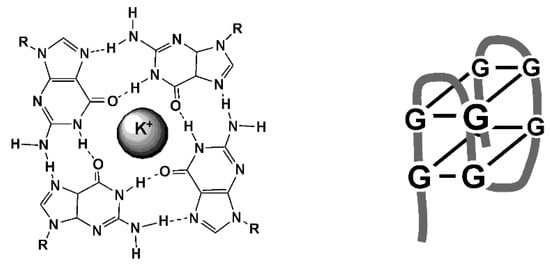
Figure 9.
Structure of G4 quadruplex and folded aptamer.
Such a transformation involves guanine reach fragments of the aptamer sequence and is stabilized by K+ ions pronounced for negatively charged ferricyanide ion most frequently applied for measurement of the signal with a label-free aptasensor. Changes in the hydrophilicity and charge separation between the aptamer and polar matrix are other reasons altering the current of ferricyanide redox probe or EIS parameters.
Among G4-quadruplexes, aptamers can contain fragments able to self-hybridize due to the existence of partially complementary sequences of nucleobases. Such aptamers form one or several loops and are able to reversible convert to linear pieces [139]. The appropriate formats of the signal transduction based on such pinhole aptamers are discussed below in the following sections.
As in the case of Abs, immobilization can affect their affinity and the long-term stability of bioreceptors in a real sample assay. Immobilization of aptamers in the aptasensor assembly is performed using the same protocols as the Ab immobilization. However, modification of the aptamer is mostly performed by terminal groups and directed to introduce thiol, amino, and carboxylic groups or biotin residues. This makes it possible to fix aptamers in the surface layer by one-point binding and to achieve site-specific immobilization of the receptor with maximal steric accessibility of the immobilized molecule toward analyte species. Thus, aptamers bearing terminal thiol groups spontaneously interact with Au nanoparticles that can be dispersed in appropriate support. Such a scheme provides both electric conductivity of the surface layer and reliable fixation of the aptamer on the electrode interface. In a similar manner, various labels can be introduced to the opposite terminus of the molecule. Double-labeled aptamers with amino (carboxylic) and thiol groups are used in so-called E-sensors (Figure 10) [140].
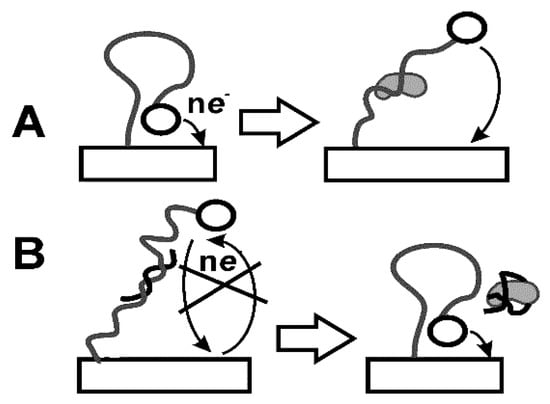
Figure 10.
E-sensor based on double-labeled loop aptamer (A) and application of partially complementary auxiliary DNA sequence (B).
Prior to contact with the analyte, both labels are positioned near each other so that electron exchange between the label and electrode is quite effective. The reaction with the analyte stimulated conformation change. The distance between the redox label and electrode increases with the analyte concentration and appropriate current decays. Opposite effect can be reached in so-called displacement aptasensors. In them, auxiliary DNA sequence with two terminal labels is used. The first one is utilized for immobilization of the molecule to the electrode and second one produces signal measured in direct current (DC) or DPV mode. The primary sequence of auxiliary DNA is complementary to the part of the aptamer molecule. Prior to contact with the analyte, both DNA and aptamer are attached to the electrode due to the formation of rather rigid double-stranded product of hybridization. After addition of the analyte, aptamer forms a specific complex and is removed from the surface. The DNA molecule, being free, forms loops so that the redox label approach the electrode surface. Resulting signal increases with the analyte concentration. The E-sensors described can be easily regenerated after the measurement. For this purpose, they are heated to destroy double-stranded pieces and treated with aptamer molecules. Ferrocene and methylene blue are mostly used in E-sensors as redox labels [141]. They can also be used together for higher robustness of the response. Thus, the signal of first label attached to the loop will decrease whereas second one sterically hindered prior to analyte binding will increase the signal with the analyte concentration [142].
Physical immobilization of aptamers has received certain attention in the last years. Aptamers can be adsorbed on the carbonaceous materials via π–π stacking interactions and via formation of double-stranded DNA pieces with partially complementary DNA probes immobilized on the surface of electrode or nanoparticles used as immobilization supports [143,144]. The electrostatic immobilization of negatively charged aptamer molecules can be accelerated by positively charged supports like poly(L-lysine) or poly(ethylene imine) and their specific adsorption on materials with a high affinity toward phosphate groups like zirconia oxide.
The performance of electrochemical aptasensors applied for mycotoxin determination is summarized in Table 4 for the period from 2014 to 2018. Earlier works have been discussed in reviews [11,16,145,146].
Table 4.
Electrochemical aptasensors for mycotoxin determination (2014–2018).
As can be seen, modification of electrodes can alter the detectable concentrations of mycotoxins by several orders of magnitude. There is no direct connection between the affinity characteristics obtained in homogeneous conditions (KD constants) and analytical parameters of appropriate biosensors. In both cases, the reason can be related to the contribution of heterogeneous factors, namely, transfer of the reactants and analyte on the electrode interface. In comparison with immunosensors considered above, EIS became the most popular protocol of signal recording. This might be not only due to higher efficiency of this detection system but also due to the higher cost of aptamer modification against labeling Ab for immunosensors. To some extent, the availability of reactants is achieved by commercial products, including already labeled aptamers ready for implementation in the biosensor assembly. Their combination with screen-printed electrodes mares the cost of single measurement just compatible with routine ELISA techniques.
Other modifiers used for aptasensor assembly play the role of aptamer support and mediators of electron transfer. Reduced graphene oxide, carbon nanotubes and Au nanoparticles are most popular due to the rather simple preparation for deposition on the bare electrode, chemical and electrochemical stability and compatibility with aptamers and auxiliary reagents. It can be expected, that the list of such reagents will be expected by broad application of metal–organic framework (MOF) materials and nanocomposites obtained in situ on the electrode surface.
In some aptasensors, biochemical amplification previously developed for DNA sensors devoted to hybridization detection was used like the telomerase/EXO III amplification cycle [149]. This approach increases the time and cost of measurement but decreases the concentrations detected by more than one order of magnitude against conventional analogs. They might be quite applicable for infant food analysis or for screening ultra-low levels of contamination if they required.
Ultra-low levels of mycotoxins to be detected dictate the choice of enzyme label. An appropriate reaction scheme [162] is similar to the electrochemical ELISA technique but mostly uses alkaline phosphatase which has sensitive detection schemes based on α-naphtol oxidation (Figure 11).
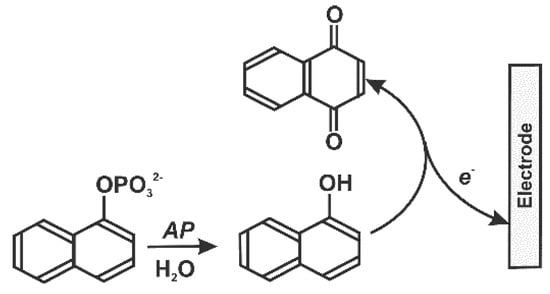
Figure 11.
Voltammetric determination of alkaline phosphatase (AP) activity with α-naphtyl phosphate as substrate.
Regarding real sample assay, most works show results obtained with spiked samples containing a certain amount of mycotoxin without reference to gas-liquid chromatography (GLC) or another standard method of analysis. In the case of juices from fruits and extracts from nuts and vegetables, sample treatment assumes filtering from solid impurities and pH correction. In some cases, extraction by methanol followed by solvent evaporation and/or re-extraction of sediment in aqueous ethanol is proposed. Such a protocol is adapted from conventional protocols elaborated for HPLC assay. Its application counterbalances advantages of biosensors like fast response and minimal sample treatment. To some extent, the negative effect of the sample matrix related to non-specific adsorption of proteins and other interferences is avoided by the deposition of components covering the free surface of the electrode in the stage of biosensor manufacture. The use of mercaptans for golden electrodes or BSA for glassy carbon minimize positive false results yielding from adsorption of native sample components on the electrode.
5. Conclusions
Various affine biosensors based on antibodies and aptamers have been developed for reliable and sensitive detection of mycotoxins. Most of them are directed to the analysis of agriculture samples, food and feed in order to detect nanogram levels of hazardous species able to negatively affect human health. To be competitive with conventional analytical instrumentation, biosensors should offer fast and reliable detection in field conditions with minimal sample treatment and auxiliary reagents required. Regarding the characteristics of the detection system, electrochemical transducers allow for quantifying the changes on the electrode interface within several seconds. However, the total measurement time is much longer because involves several steps of reagents dilution, washing and incubation necessary to reach Ag–Ab (aptamer–mycotoxin) equilibrium on the electrode interface. From this point of view, the information on the residuals of mycotoxins is mostly available in 60–90 min. The following progress in the area of biosensors for mycotoxin determination is directed to both improvement of operational characteristics and biosensor design. Most important goals of such improvements are briefly considered below.
Bioreceptor. Although most of the mycotoxins have obtained appropriate antibodies and aptamers providing their sensitive determination, the variety of bioreceptors is expected to grow to cover the problems of multi-analyte analysis and simplification of the surface layer assembly. The use of reduced antibodies (scFv fragments), rather popular in immunoassay schemes, will be extended to biosensor format. This might be useful for measurements in extreme media, for example, in organic solvents or in the presence of strong acids/bases used for the extraction of analytes. The progress in the selection of bioreceptors can accelerate the design of single use biosensors based on paper, as well as microfluidic devices.
Transducer mode. Both immunosensors and aptasensors mostly follow trends previously established for their common alternatives, e.g., ELISA. Such an approach does not take into account in a full-scale advantages of heterogeneous interface transducer–biorecognition layer–sample tested. Thus, application of mesoporous supports with channels providing fast transfer of charge carriers not only reduces measurement time but also can affect equilibrium stage and hence shift the theoretical limit of detectable amounts of mycotoxins against macrosystems. Similar consequences can be obtained by combination of ultra-small electrodes and microelectrode arrays with flow-through systems of the controllable transfer of reactants. Such electrochemical devices would be useful not only for measurements in aqueous medial but also as detectors of electrophoretic and chromatographic sensors like electroluminescence technique or electrochemical quartz crystal microbalances (surface plasmon resonance) analyzers.
It should be also mentioned that alternatives to electrochemical transducers can be successfully used for mycotoxin determination. They offer a variety of opportunities both in the measurement mode and reliability of response [177,178] especially in analysis of food and biological tissues. Although they are considered less appropriate for the design of portable (bio)sensors and yet less sensitive toward mycotoxins than electrochemical devices, it can be expected that the gap between the electrochemical and optical technique will disappear in the near future.
Analytes. The number of potential analytes for biosensors used in food safety and human health control is increasing and requirements for their threshold levels being clearer. From this point of view, universal systems able to work with different bioreceptors and multiplex systems should get priority in future efforts. Similarly to optic sensors, electrochemical biosensors should offer universal technical decisions easily adapted to novel analytes by the replacement of immobilized biocomponents. In this case, the number of new biosensors will be dictated by market necessities.
Author Contributions
Conceptualization, writing—review and editing, T.H.; methodology, project administration, writing—original draft preparation, G.E.
Funding
T.H. is grateful for financial support to the European Commission within the project Innovative technology for the detection of enzyme activity in milk (FORMILK) under grant agreement number 690898 /H2020-MSCA-RISE-2015. G.E. acknowledges financial support for investigations performed with electropolymerized and supramolecular supports from the Russian Science Foundation (grant No 17-13-01208).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Alshannaq, A.; Yu, J.-H. Occurrence, toxicity, and analysis of major mycotoxins in food. Int. J. Environ. Res. Public Health 2017, 14, 632. [Google Scholar] [CrossRef] [PubMed]
- Stoev, S.D. Food safety and increasing hazard of mycotoxin occurrence in foods and feeds. Crit. Rev. Food Sci. Nutr. 2013, 53, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.I.; Miller, J.D. A concise history of mycotoxin research. J. Agric. Food Chem. 2017, 65, 7021–7033. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Benford, D.; Boobis, A.; Eskola, M.; Fink-Gremmels, J.; Fürst, P.; Heppner, C.; Schlatter, J.; van Leeuwen, R. Risk assessment of contaminants in food and feed. EFSA J. 2002, 10, s1004. [Google Scholar] [CrossRef]
- Ragona, M. Mycotoxins, the unknowns: Trends in food availability and consumer perceptions. World Mycotoxin J. 2016, 9, 813–830. [Google Scholar] [CrossRef]
- Medina, A.; Akbar, A.; Baazem, A.; Rodriguez, A.; Maga, N. Climate change, food security and mycotoxins: Do we know enough? Fungal Biol. Rev. 2017, 31, 143–154. [Google Scholar] [CrossRef]
- Pitt, J.I. Toxigenic fungi: Which are important? Med. Mycol. 2000, 38, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.L. Some major mycotoxins and their mycotoxicoses—An overview. Int. J. Food Microbiol. 2007, 119, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, R.; Sing, J.; Sachdev, T.; Basu, T.; Malhotra, B.D. Recent advances in mycotoxins detection. Biosens. Bioelectron. 2016, 81, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Skottrup, P.D.; Nicolaisen, M.; Justesen, A.F. Towards on-site pathogen detection using antibody-based sensors. Biosens. Bioelectron. 2008, 24, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.C.; Bonel, L.; Ezquerra, A.; Hernández, S.; Bertolín, J.R.; Cubel, C.; Castillo, J.R. Electrochemical affinity biosensors for detection of mycotoxins: A review. Biosens. Bioelectron. 2013, 49, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Thevenot, D.R.; Toth, K.; Durst, R.A.; Wilson, G.S. Electrochemical biosensors: Recommended definitions and classification. Pure Appl. Chem. 1999, 71, 2333–2348. [Google Scholar] [CrossRef]
- Arduini, F.; Amine, A.; Mosconee, D.; Palleschi, G. Biosensors based on cholinesterase inhibition for insecticides, nerve agents and aflatoxin B1 detection (review). Microchim. Acta 2010, 170, 193–214. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, G.; Mao, X.; Yang, S.; De Ruyck, K.; Wu, Y. High sensitivity immunoassays for small molecule compounds detection – Novel noncompetitive immunoassay designs. TrAC – Trends Anal. Chem. 2018, 103, 198–208. [Google Scholar] [CrossRef]
- Peltomaa, R.; Benito-Peña, E.; Moreno-Bondi, M.C. Bioinspired recognition elements for mycotoxin sensors. Anal. Bioanal. Chem. 2018, 410, 747–771. [Google Scholar] [CrossRef] [PubMed]
- Rhouati, A.; Bulbul, B.; Latif, U.; Hayat, A.; Li, Z.-H.; Marty, J.L. Nano-aptasensing in mycotoxin analysis: Recent updates and progress. Toxins 2017, 9, 349. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Goud, K.Y.; Hayat, A.; Bhand, S.; Marty, J.L. Recent advances in electrochemical-based sensing platforms for aflatoxins detection. Chemosensors 2017, 5, 1. [Google Scholar] [CrossRef]
- Alizadeh, N.; Salimi, A. Ultrasensitive bioaffinity electrochemical sensors: Advances and new perspectives. Electroanalysis 2018, 30, 2803–2840. [Google Scholar] [CrossRef]
- Peraica, M.; Radić, B.; Lucić, A.; Pavlović, M. Toxic effects of mycotoxins in humans. WHO Bull. 1999, 77, 754–766. [Google Scholar]
- Shephard, G.S.; Berthiller, F.; Burdaspal, P.A.; Crews, C.; Jonker, M.A.; Krska, P.; Mc Donald, S.; Malone, R.J.; Maragos, C.; Sabino, M.; et al. Developments in mycotoxin analysis: An update for 2010–2011. World Mycotoxin J. 2012, 5, 3–30. [Google Scholar] [CrossRef]
- Do, J.H.; Choi, D.-K. Aflatoxins: Detection, toxicity and biosynthesis. Biotechnol. Bioprocess Eng. 2007, 12, 585–593. [Google Scholar] [CrossRef]
- Zheng, M.Z.; Richard, J.L.; Binder, J. A review of rapid methods for the analysis of mycotoxins. Mycopathologia 2006, 161, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.W.; Klich, M. Mycotoxins. Clin. Microbiol. Rev. 2003, 16, 497–516. [Google Scholar] [CrossRef] [PubMed]
- Rocha, O.; Ansaril, K.; Doohan, F.M. Effects of trichothecene mycotoxins on eukaryotic cells: A review. Food Addiv. Contaminant. 2005, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.F.; Li, R.; Yang, H.; Qi, P.P.; Xiao, Y.P.; Qian, M.R. Occurrence of patulin in various fruit products and dietary exposure assessment for consumers in China. Food Control 2017, 78, 100–107. [Google Scholar] [CrossRef]
- Puel, O.; Galtier, P.; Oswald, I.P. Biosynthesis and toxicological effects of patulin. Toxins 2010, 2, 613–631. [Google Scholar] [CrossRef] [PubMed]
- Zinedine, A.; Soriano, J.M.; Moltó, J.C.; Mañes, J. Review on the toxicity, occurrence, metabolism, detoxification, regulations and intake of zearalenone: An oestrogenic mycotoxin. Food Chem. Toxicol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Mitterbauer, R.; Weindrofer, H.; Safaie, N.; Krska, R.; Lemmens, M.; Ruckenbauer, P.; Kuchler, K.; Adam, G. A sensitive and inexpensive yeast bioassay for the mycotoxin zearalenone and other compounds with estrogenic activity. Appl. Environ. Microbiol. 2003, 69, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, A.; Jinap, S.; Soleimany, F. Qualitative and quantitative analysis of mycotoxins. Compr. Rev. Food Sci. Food Saf. 2009, 8, 202–251. [Google Scholar] [CrossRef]
- Vidal, A.; Marín, S.; Ramos, A.J.; Cano-Sancho, G.; Sanchis, V. Determination of aflatoxins, deoxynivalenol, ochratoxin A and zearalenone in wheat and oat based bran supplements sold in the Spanish market. Food Chem. Toxicol. 2013, 53, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.W.; Bramhmbhatt, H.; Szabo-Vezsem, M.; Pomam, A.; Coker, R.; Piletsky, S.A. Analytical methods for determination of mycotoxins: An update (2009–2014). Anal. Chim. Acta 2015, 901, 12–33. [Google Scholar] [CrossRef] [PubMed]
- Cigić, I.K.; Prosen, H. An overview of conventional and emerging analytical methods for the determination of mycotoxins. Int. J. Mol. Sci. 2009, 10, 62–115. [Google Scholar] [CrossRef] [PubMed]
- Nardiello, D.; Magro, S.L.; Iammarino, M.; Palermo, C.; Muscarella, M.; Centonze, D. Recent advances in the post-column derivatization for the determination of mycotoxins in food products and feed materials by liquid chromatography and fluorescence detection. Curr. Anal. Chem. 2014, 10, 355–365. [Google Scholar] [CrossRef]
- Hickert, S.; Gerding, J.; Ncube, E.; Hübner, F.; Flett, B.; Cramer, B.; Humpf, H.-U. A new approach using micro HPLC-MS/MS for multi-mycotoxin analysis in maize samples. Mycotoxin Res. 2015, 31, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Man, Y.; Liang, G.; Li, A.; Pan, L. Recent advances in mycotoxin determination for food monitoring via microchip. Toxins 2017, 9, 324. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, R.J. A theory of antibody-antigen reactions. I. Theory for reactions of multivalent antigen with bivalent and univalent antibody. J. Am. Chem. Soc. 1952, 74, 5715–5725. [Google Scholar] [CrossRef]
- He, Q.; Xu, Y. Antibody developments and immunoassays for mycotoxins. Curr. Org. Chem. 2017, 21, 2622–2631. [Google Scholar] [CrossRef]
- Soares, R.R.G.; Ricelli, A.; Fanelli, C.; Caputo, D.; Cesare, G.; Chu, V.; Aires-Barros, M.R.; Conde, J.P. Advances, challenges and opportunities for point-of-need screening of mycotoxins in foods and feeds. Analyst 2018, 143, 1015–1035. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.R.A. Mycotoxin immunoassays (with special reference to elisas). Tetrahedron 1989, 45, 2237–2249. [Google Scholar] [CrossRef]
- Vdovenko, M.M.; Lu, C.-C.; Yu, F.-Y.; Sakharov, I.Y. Development of ultrasensitive direct chemiluminescent enzyme immunoassay for determination of aflatoxin M1 in milk. Food Chem. 2014, 158, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Usleber, E.; Dade, M.; Schneider, E.; Dietrich, R.; Bauer, J.; Märtlbauer, E. Enzyme immunoassay for mycophenolic acid in milk and cheese. J. Agric. Food Chem. 2008, 56, 6857–6862. [Google Scholar] [CrossRef] [PubMed]
- Arévalo, F.J.; Granero, A.M.; Fernández, H.; Raba, J.; Zón, M.A. Citrinin (CIT) determination in rice samples using a micro fluidic electrochemical immunosensor. Talanta 2011, 83, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Shiu, C.-M.; Wang, J.-J.; Yua, F.-Y. Sensitive enzyme-linked immunosorbent assay and rapid one-step immunochromatographic strip for fumonisin B1 in grain-based food and feed samples. Sci. Food Agric. 2010, 90, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Basova, E.Y.; Goryacheva, I.Y.; Rusanova, T.Y.; Burmistrova, N.A.; Dietrich, R.; Märtlbauer, E.; Detavernier, C.; Van Peteghem, C.; De Saeger, S. An immunochemical test for rapid screening of zearalenone and T-2 toxin. Anal. Bioanal. Chem. 2010, 397, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Hervás, M.; López, M.A.; Escarpa, A. Integrated electrokinetic magnetic bead-based electrochemical immunoassay on microfluidic chips for reliable control of permitted levels of zearalenone in infant foods. Analyst 2011, 136, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Eguílaz, M.; Moreno-Guzmán, M.; Campuzano, S.; González-Cortés, A.; Yáñez-Sedeño, P.; Pingarrón, J.M. An electrochemical immunosensor for testosterone using functionalized magnetic beads and screen-printed carbon electrodes. Biosens. Bioelectron. 2010, 26, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Y.; Feng, H.; Wu, Y.; Joshi, L.; Zeng, X.; Li, J. Layer-by-layer assembly of chemical reduced graphene and carbon nanotubes for sensitive electrochemical immunoassay. Biosens. Bioelectron. 2012, 35, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Ghindilis, A.L.; Atanasov, P.; Wilkins, E. Fast amperometric immunoassay utilizing highly dispersed electrode material. Anal. Lett. 1995, 28, 2459–2474. [Google Scholar] [CrossRef]
- Li, Y.-F.; Sun, Y.-M.; Beier, R.C.; Lei, H.-T.; Gee, S.; Hammock, B.D.; Wang, H.; Wang, Z.; Sun, X.; Shen, Y.-D.; et al. Immunochemical techniques for multianalyte analysis of chemical residues in food and the environment: A review. Trends Anal. Chem. 2017, 88, 25–40. [Google Scholar] [CrossRef]
- Anfossi, L.; Giovannoli, C.; Baggiani, C. Mycotoxin detection. Curr. Opin. Biotechnol. 2015, 37, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Shephard, G.S. Determination of mycotoxins in human foods. Chem. Soc. Rev. 2008, 37, 2468–2477. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Byun, J.-Y.; Kim, B.B.; Shin, Y.-B.; Kim, M.-G. Label-free homogeneous FRET immunoassay for the detection of mycotoxins that utilizes quenching of the intrinsic fluorescence of antibodies. Biosens. Bioelectron. 2013, 42, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J. Recent advances in analytical methods for mycotoxins. Food Addit. Contamin. 1993, 10, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Kokkinos, C.; Economou, A.; Prodromidis, M.I. Electrochemical immunosensors: Critical survey of different architectures and transduction strategies. Trends Anal. Chem. 2016, 79, 88–105. [Google Scholar] [CrossRef]
- Pagkali, V.; Petrou, P.S.; Salapatas, A.; Makaronab, E.; Peters, J.; Haasnoot, W.; Jobst, G.; Economou, A.; Misiakos, K.; Raptis, I.; et al. Detection of ochratoxin A in beer samples with a label-free monolithically integrated optoelectronic biosensor. J. Hazard. Mater. 2017, 323, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Nabok, A.; Al-Jawdah, A.M.; Tsargorodska, A. Development of planar waveguide-based immunosensor for detection of low molecular weight molecules such as mycotoxins. Sens. Actuators B Chem. 2017, 247, 975–980. [Google Scholar] [CrossRef]
- Al-Rubaye, A.; Nabok, A.; Catanante, G.; Marty, J.-L.; Takacs, E.; Szekacs, A. Detection of ochratoxin A in aptamer assay using total internal reflection ellipsometry. Sens. Actuators B Chem. 2018, 263, 248–251. [Google Scholar] [CrossRef]
- Al-Rubaye, A.G.; Nabok, A.; Catanante, G.; Marty, J.-L.; Takács, E.; Székács, A. Label-free optical detection of mycotoxins using specific aptamers immobilized on gold nanostructures. Toxins 2018, 10, 291. [Google Scholar] [CrossRef] [PubMed]
- Tsounidi, D.; Koukouvinos, G.; Petrou, P.; Misiakos, K.; Zisis, G.; Goustouridis, D.; Raptis, I.; Kakabakos, S.E. Rapid and sensitive label-free determination of aflatoxin M1 levels in milk through a White Light Reflectance Spectroscopy immunosensor. Sens. Actuators B Chem. 2019, 282, 104–111. [Google Scholar] [CrossRef]
- Karczmarczyk, A.; Haupt, K.; Feller, K.-H. Development of a QCM-D biosensor for Ochratoxin A detection in red wine. Talanta 2017, 166, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Bučko, M.; Mislovičová, M.; Nahálka, J.; Vikartovská, A.; Šefčovičová, J.; Katrlik, J.; Tkač, J.; Gemeiner, P.; Lacík, I.; Štefuca, V.; et al. Immobilization in biotechnology and biorecognition: From macro- to nanoscale systems. Chem. Pap. 2012, 66, 983–998. [Google Scholar] [CrossRef]
- Ricci, F.; Adornetto, G.; Palleschi, G. A review of experimental aspects of electrochemical immunosensors. Electrochim. Acta 2012, 84, 74–83. [Google Scholar] [CrossRef]
- Haruyama, T. Design and fabrication of a molecular interface on an electrode with functional protein molecules for bioelectronic properties. Electrochemistry 2010, 78, 888–895. [Google Scholar] [CrossRef]
- Dolatabadi, J.E.N.; de la Guardia, M. Nanomaterial-based electrochemical immunosensors as advanced diagnostic tools. Anal. Methods 2014, 6, 3891–3900. [Google Scholar] [CrossRef]
- Veetil, J.V.; Yea, K. Development of immunosensors using carbon nanotubes. Biotechnol. Prog. 2007, 23, 517–731. [Google Scholar] [CrossRef] [PubMed]
- Chauhana, R.; Singh, I.; Solanki, P.R.; Manaka, T.; Iwamoto, M.; Basu, T.; Malhotra, B.D. Label-free piezoelectric immunosensor decorated with gold nanoparticles: Kinetic analysis and biosensing application. Sens. Actuators B Chem. 2016, 222, 804–814. [Google Scholar] [CrossRef]
- Vashist, S.K.; Lam, E.; Hrapovic, S.; Male, K.B.; Luong, J.H.T. Immobilization of antibodies and enzymes on 3-aminopropyltriethoxysilane-functionalized bioanalytical platforms for biosensors and diagnostics. Chem. Rev. 2014, 114, 11083–11130. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Simón, B.; Campàs, M.; Marty, J.-L.; Noguer, T. Novel highly-performing immunosensor-based strategy for ochratoxin A detection in wine samples. Biosens. Bioelectron. 2008, 23, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Y.; Yuan, R.; Chai, Y.; Zhang, Y.; Li, X.; Wang, N.; Zhu, Q. Amperometric enzyme immunosensors based on layer-by-layer assembly of gold nanoparticles and thionine on Nafion modified electrode surface for α-1-fetoprotein determinations. Sens. Actuators B Chem. 2006, 114, 631–639. [Google Scholar] [CrossRef]
- Cosnier, S.; Holzinger, M. Electrosynthesized polymers for biosensing. Chem. Soc. Rev. 2011, 40, 2146–2156. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ahmed, N.B.; Alvarez, G.S.; Tuttolomondo, M.; Helary, C.; Desimone, M.F.; Coradin, T. Sol-gel encapsulation of biomolecules and cells for medicinal applications. Curr. Top. Med. Chem. 2015, 15, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Iijima, I.; Kuroda, S. Scaffolds for oriented and close-packed immobilization of immunoglobulins. Biosens. Bioelectron. 2017, 89, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Turková, J. Oriented immobilization of biologically active proteins as a tool for revealing protein interactions and function. J. Chromatogr. B 1999, 722, 11–31. [Google Scholar] [CrossRef]
- Bertok, T.; Klukova, L.; Sediva, A.; Kasák, P.; Semak, V.; Micusik, M.; Omastova, M.; Chovanová, L.; Vlček, M.; Imrich, R.; et al. Ultrasensitive impedimetric lectin biosensors with efficient antifouling properties applied in glycoprofiling of human serum samples. Anal. Chem. 2013, 85, 7324–7332. [Google Scholar] [CrossRef] [PubMed]
- Kabir, S. Immunoglobulin purification by affinity chromatography using protein a mimetic ligands prepared by combinatorial chemical synthesis. J. Mol. Cell. Immun. 2002, 31, 263–278. [Google Scholar] [CrossRef]
- Zeng, X.; Shen, Z.; Mernaugh, R. Recombinant antibodies and their use in biosensors. Anal. Bioanal. Chem. 2012, 402, 3027–3038. [Google Scholar] [CrossRef] [PubMed]
- Maragos, C.M.; Li, L.; Chen, D. Production and characterization of a single chain variable fragment (scFv) against the mycotoxin deoxynivalenol. Food Agric. Immunol. 2012, 23, 57–61. [Google Scholar] [CrossRef]
- Romanazzo, D.; Ricci, F.; Volpe, G.; Elliott, C.T.; Vesco, S.; Kroeger, K.; Moscone, D.; Stroka, J.; Van Egmond, H.; Vehniäinen, M.; et al. Development of a recombinant Fab-fragment based electrochemical immunosensor for deoxynivalenol detection in food samples. Biosens. Bioelectron. 2010, 25, 2615–2621. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Clarke, J.R.; Zhou, H.R.; Linz, J.E.; Pestka, J.J.; Hart, L.P. Molecular cloning, expression, and characterization of a functional single-chain Fv antibody to the mycotoxin zearalenone. Appl. Environ. Microbiol. 1997, 63, 263–269. [Google Scholar] [PubMed]
- Yan, Q.; Hu, W.; Pestka, J.J.; He, S.Y.; Hart, L.P. Expression of a functional antizearalenone single-chain Fv antibody in transgenic Arabidopsis plants. Appl. Environ. Microbiol. 2000, 66, 3499–3505. [Google Scholar] [CrossRef]
- Wang, S.-H.; Du, X.-Y.; Lin, L.; Huang, Y.-M.; Wang, Z.-H. Zearalenone (ZEN) detection by a single chain fragment variable (scFv) antibody. World J. Microb. Biotechnol. 2008, 24, 1681–1685. [Google Scholar] [CrossRef]
- Edupuganti, S.R.; Edupuganti, O.P.; O’Kennedy, R. Generation of anti-zearalenone scFv and its incorporation into surface plasmon resonance-based assay for the detection of zearalenone in sorghum. Food Control 2013, 34, 668–674. [Google Scholar] [CrossRef]
- Wang, S.-H.; Du, X.-Y.; Huang, Y.-M.; Lin, D.-S.; Hart, P.L.; Wang, Z.-H. Detection of deoxynivalenol based on a single-chain fragment variable of the antideoxynivalenol antibody. FEMS Microb. Lett. 2007, 272, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.-H.; Lee, D.-H.; Min, W.-K.; Cho, Y.-I.; Kweon, D.-H.; Son, D.-H.; Park, J.-H.K.; Seo, J.-H. Cloning, expression, and characterization of single-chain variable fragment antibody against mycotoxin deoxynivalenol in recombinant Escherichia coli. Protein Express. Purif. 2004, 35, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Li, Y.; He, Q.; Chen, B.; Wang, D. Development of a single-chain variable fragment antibody-based enzyme-linked immunosorbent assay for determination of fumonisin B1 in corn samples. J. Sci. Food Agric. 2013, 94, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Min, W.-K.; Cho, Y.-J.; Park, J.B.; Bae, Y.-H.; Kim, E.-J.; Park, K.; Park, Y.-C.; Seo, J.-H. Production and characterization of monoclonal antibody and its recombinant single chain variable fragment specific for a food-born mycotoxin, fumonisin B1. Bioprocess Biosyst. Eng. 2010, 33, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-Q.; Li, H.-P.; Wu, P.; Li, Y.-B.; Zhou, Z.-Q.; Zhang, J.-B.; Liu, J.-L.; Li, Y.-C. An affinity improved single-chain antibody from phage display of a library derived from monoclonal antibodies detects fumonisins by immunoassay. Anal. Chim. Acta 2015, 867, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Rangnoi, K.; Choowongkomon, K.; O’Kennedy, R.; Rüker, F.; Yamabhai, M. Enhancement and analysis of human antiaflatoxin b1 (AFB1) scFv antibody–ligand interaction using chain shuffling. J. Agric. Food Chem. 2018, 66, 5713–5722. [Google Scholar] [CrossRef] [PubMed]
- Min, W.-K.; Na, K.-I.; Yoon, J.-H.; Heo, Y.-J.; Lee, D.; Kim, S.-G.; Heo, J.-H. Affinity improvement by fine tuning of single-chain variable fragment against aflatoxin B1. Food Chem. 2016, 209, 312–317. [Google Scholar] [CrossRef]
- Min, W.-K.; Kim, S.-G.; Seo, J.-H. Affinity maturation of single-chain variable fragment specific for aflatoxin B1 using yeast surface display. Food Chem. 2015, 188, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Ye, Y.; Chen, W.; Wang, X.; Chen, F. Expression of VH-linker-VL orientation-dependent single-chain Fv antibody fragment derived from hybridoma 2E6 against aflatoxin B1 in Escherichia coli. J. Ind. Microb. Biotechnol. 2015, 42, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Min, W.-K.; Kweon, D.-H.; Park, K.; Park, Y.-C.; Seo, J.-H. Characterisation of monoclonal antibody against aflatoxin B1 produced in hybridoma 2C12 and its single-chain variable fragment expressed in recombinant Escherichia Coli. Food Chem. 2011, 126, 1316–1323. [Google Scholar] [CrossRef]
- Li, X.; Li, P.W.; Lei, J.W.; Zhang, Q.; Zhang, W.; Li, C.M. A simple strategy to obtain ultra-sensitive single-chain fragment variable antibodies for aflatoxin detection. RSC Adv. 2013, 3, 22367–22372. [Google Scholar] [CrossRef]
- Pansri, P.; Jaruseranee, N.; Rangnoi, K.; Kristensen, K.; Yamabhai, M. A compact phage display human scFv library for selection of antibodies to a wide variety of antigens. BMC Biotechnol. 2009, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhang, Y.; Hu, C.; Wu, H.; Yang, Y.; Huang, C.; Jia, N. Highly sensitive electrochemical impedance spectroscopy immunosensor for the detection of AFB1 in olive oil. Food Chem. 2015, 176, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Basu, J.; Datta, S.; Chaudhuri, S.R. A graphene field effect capacitive immunosensor for sub-femtomolar food toxin detection. Biosens. Bioelectron. 2015, 68, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Abraham, S.; Singh, C.; Ali, M.A.; Srivastava, A.; Sumana, A.; Malhotra, B.D. Protein conjugated carboxylated gold@reduced graphene oxide for aflatoxin B1 detection. RSC Adv. 2015, 5, 5406–5414. [Google Scholar] [CrossRef]
- Wang, D.; Hu, W.; Xiong, Y.; Xu, Y.; Li, C.M. Multifunctionalized reduced graphene oxide-doped polypyrrole/pyrrolepropylic acid nanocomposite impedimetric immunosensor to ultra-sensitively detect small molecular aflatoxin B1. Biosens. Bioelectron. 2015, 63, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Yagati, A.K.; Chavan, S.G.; Baek, C.; Lee, M.-H.; Min, J. Label-free impedance sensing of aflatoxin B1 with polyaniline nanofibers/Au nanoparticle electrode array. Sensors 2018, 18, 1320. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.P.; Frías, I.A.M.; Andrade, C.A.S.; Oliveira, M.D.L. Impedimetric immunoassay for aflatoxin B1 using a cysteine modified gold electrode with covalently immobilized carbon nanotubes. Microchim. Acta 2017, 184, 3205–3213. [Google Scholar] [CrossRef]
- Ma, H.; Sun, J.; Zhang, Y.; Bian, C.; Xia, S.; Zhen, T. Label-free immunosensor based on one-step electrodeposition of chitosan-gold nanoparticles biocompatible film on Au microelectrode for determination of aflatoxin B1 in maize. Biosens. Bioelectron. 2016, 80, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Sun, J.; Zhang, Y.; Xia, S. Disposable amperometric immunosensor for simple and sensitive determination of aflatoxin B1 in wheat. Biochem. Eng. J. 2016, 115, 38–46. [Google Scholar] [CrossRef]
- Solanki, P.R.; Singh, J.; Rupavali, B.; Tiwari, S.; Malhotra, B.D. Bismuth oxide nanorods based immunosensor for mycotoxin detection. Mater. Sci. Eng. C 2017, 70, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Kumar, A.; Khan, R. A highly sensitive amperometric immunosensor probe based on gold nanoparticle functionalized poly (3, 4-ethylenedioxythiophene) doped with graphene oxide for efficient detection of aflatoxin B1. Synth. Met. 2018, 235, 136–144. [Google Scholar] [CrossRef]
- Azri, F.A.; Selamat, J.; Sukor, R. Electrochemical immunosensor for the detection of aflatoxin B1 in palm kernel cake and feed samples. Sensors 2017, 17, 2776. [Google Scholar] [CrossRef] [PubMed]
- Malvano, F.; Albanese, D.; Crescitelli, A.; Pilloton, R.; Esposito, E. Impedimetric label-free immunosensor on disposable modified screen-printed electrodes for ochratoxin A. Biosensors 2016, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Chrouda, A.; Sbartai, A.; Bessueille, F.; Renaud, L.; Maarefa, A.; Jaffrezic-Renault, N. Electrically addressable deposition of diazonium-functionalized antibodies on boron-doped diamond microcells for the detection of ochratoxin A. Anal. Methods 2015, 7, 2444–2451. [Google Scholar] [CrossRef]
- Jodra, A.; Hervás, M.; Lopez, M.Á.; Escarpa, A. Disposable electrochemical magneto immunosensor for simultaneous simplified calibration and determination of Ochratoxin A in coffee samples. Sens. Actuators B Chem. 2015, 221, 777–783. [Google Scholar] [CrossRef]
- Badea, M.; Floroian, L.; Restani, P.; Codruta, S.; Cobzac, A.; Moga, M. Ochratoxin A detection on antibody immobilized on BSA-functionalized gold electrodes. PLoS ONE 2016, 11, e0160021. [Google Scholar] [CrossRef] [PubMed]
- Bougrini, V.; Baraket, A.; Jamshaid, T.; El Aissari, A.; Bausells, J.; Zabala, M.; El Bari, N.; Bouchikhi, B.; Jaffrezic-Renault, N.; Abdelhamid, E.; et al. Development of a novel capacitance electrochemical biosensor based on silicon nitride for ochratoxin A detection. Sens. Actuators B Chem. 2016, 234, 446–452. [Google Scholar] [CrossRef]
- Gupta, P.K.; Pachauri, N.; Khan, Z.H.; Solanki, P.R. One pot synthesized zirconia nanoparticles embedded in amino functionalized amorphous carbon for electrochemical immunosensor. J. Electroanal. Chem. 2017, 807, 59–69. [Google Scholar] [CrossRef]
- Liu, L.; Chao, Y.; Cao, W.; Wang, Y.; Luo, C.; Pang, X.; Fan, D.; Wei, Q. A label-free amperometric immunosensor for detection of zearalenone based on trimetallic Au-core/AgPt-shell nanorattles and mesoporous carbon. Anal. Chim. Acta 2014, 847, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Nie, D.; Tan, Y.; Zhao, Z.; Liao, Y.; Wang, H.; Sun, C.; Wu, A. An ultrasensitive amperometric immunosensor for zearalenones based on oriented antibody immobilization on a glassy carbon electrode modified with MWCNTs and AuPt nanoparticles. Microchim. Acta 2017, 184, 147–153. [Google Scholar] [CrossRef]
- Xu, W.; Qing, Y.; Chen, S.; Chen, J.; Qin, Z.; Qiu, J.F.; Li, C.R. Electrochemical indirect competitive immunoassay for ultrasensitive detection of zearalenone based on a glassy carbon electrode modified with carboxylated multi-walled carbon nanotubes and chitosan. Microchim. Acta 2017, 184, 3339–3347. [Google Scholar] [CrossRef]
- Regiart, M.; Fernández, O.; Vicario, A.; Villarroel-Rocha, J.; Sapag, K.; Messina, G.A.; Raba, J.; Bertolino, F.A. Mesoporous immunosensor applied to zearalenone determination in Amaranthus cruentus seeds. Microchem. J. 2018, 141, 388–394. [Google Scholar] [CrossRef]
- Sunday, C.E.; Masikini, M.; Wilson, L.; Rassie, C.; Waryo, T.; Baker, P.C.L.; Iwuoha, E.I. Application on gold nanoparticles-dotted 4-nitrophenylazo graphene in a label-free impedimetric deoxynivalenol immunosensor. Sensors 2015, 15, 3854–3871. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Seenivasan, R.; Wang, Y.-C.; Yu, J.-H.; Gunasekaran, S. An electrochemical immunosensor for rapid and sensitive detection of mycotoxins fumonisin B1 and deoxynivalenol. Electrochim. Acta 2016, 213, 89–97. [Google Scholar] [CrossRef]
- Qing, Y.; Li, C.R.; Yang, X.X.; Zhou, X.P.; Xue, J.; Luo, M.; Xu, X.; Chen, S.; Qiu, J.F. Electrochemical immunosensor using single-walled carbon nanotubes/chitosan for ultrasensitive detection of deoxynivalenol in food samples. J. Appl. Electrochem. 2016, 46, 1049–1057. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Peng, D.; Xie, S.; Chen, D.; Pan, Y.; Tao, Y.; Yuan, Z. Construction of electrochemical immunosensor based on gold-nanoparticles/carbon nanotubes/chitosan for sensitive determination of T-2 toxin in feed and swine meat. Int. J. Mol. Sci. 2018, 19, 3895. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.S.; Szostak, J.W. In vitro selection of functional nucleic acids. Annu. Rev. Biochem. 1999, 68, 611–647. [Google Scholar] [CrossRef] [PubMed]
- You, K.M.; Lee, S.H.; Im, A.; Lee, S.B. Aptamers as functional nucleic acids: In vitro selection and biotechnological applications. Biotechnol. Bioprocess Eng. 2003, 8, 64–75. [Google Scholar] [CrossRef]
- Gopinath, S.C.B. Methods developed for SELEX. Anal. Bioanal. Chem. 2007, 387, 171–182. [Google Scholar] [CrossRef] [PubMed]
- González-Fernández, E.; de-los-Santos-Álvarez, N.; Miranda-Ordieres, A.J.; Lobo-Castañón, M.J. SPR evaluation of binding kinetics and affinity study of modified RNA aptamers towards small molecules. Talanta 2012, 99, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Seok, Y.; Byun, J.-Y.; Shim, W.-B.; Kim, M.G. A structure-switchable aptasensor for aflatoxin B1 detection based on assembly of an aptamer/split DNAzyme. Anal. Chim. Acta 2015, 886, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Setlem, K.; Mondal, B.; Ramlal, S.; Kingston, J. Immuno affinity SELEX for simple, rapid, and cost-effective aptamer enrichment and identification against aflatoxin B1. Front. Microbiol. 2016, 7, 1909. [Google Scholar] [CrossRef] [PubMed]
- Goud, K.Y.; Hayatl, A.; Catanatel, G.; Satyanarayanal, M.; Gobil, K.V.; Martyl, J.L. An electrochemical aptasensor based on functionalized graphene oxide assisted electrocatalytic signal amplification of methylene blue for aflatoxin B1 detection. Electrochim. Acta 2017, 244, 96–103. [Google Scholar] [CrossRef]
- Goud, K.Y.; Sharma, A.; Hayat, A.; Catanante, G.; Gobi, K.W.; Gurban, A.M.; Marty, J.L. Tetramethyl-6-carboxyrhodamine quenching-based aptasensing platform for aflatoxin B1: Analytical performance comparison of two aptamers. Anal. Biochem. 2016, 508, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, W.; Chen, X.; Xia, Y.; Duan, N.; Wu, S.; Whang, Z. Selection, characterization and application of aptamers targeted to Aflatoxin B2. Food Control 2015, 47, 545–551. [Google Scholar] [CrossRef]
- Joo, M.; Baek, S.H.; Cheon, S.A.; Chun, H.S.; Choi, S.-W.; Park, T.J. Development of aflatoxin B1 aptasensor based on wide-range fluorescence detection using graphene oxide quencher. Colloids Surf. B 2017, 154, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Catanante, G.; Hayat, A.; Istamboulie, G.; Rejeb, I.B.; Bhand, S.; Marty, J.L. Development of structure switching aptamer assay for detection of aflatoxin M1 in milk sample. Talanta 2016, 158, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Hamula, C.L.A.; Guthrie, J.W.; Zhang, H.; Li, X.F.; Le, X.C. Selection and analytical applications of aptamers. Trends Anal. Chem. 2006, 25, 681–691. [Google Scholar] [CrossRef]
- Cruz-Aguado, J.A.; Penner, G. Determination of ochratoxin A with a DNA aptamer. J. Agric. Food Chem. 2008, 56, 10456–10461. [Google Scholar] [CrossRef] [PubMed]
- Chrouda, A.; Sbartai, A.; Baraket, A.; Renaud, L.; Maaref, A.; Jaffrezic-Renault, N. An aptasensor for ochratoxin A based on grafting of polyethylene glycol on a boron-doped diamond microcell. Anal. Biochem. 2015, 488, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Ren, J.; Wang, J.; Wang, E. Single-walled carbon nanotubes based quenching of free FAM-aptamer for selective determination of ochratoxin A. Talanta 2011, 85, 2517–2521. [Google Scholar] [CrossRef] [PubMed]
- McKeague, M.; Bradley, C.R.; De Girolamo, A.; Visconti, A.; Miller, D.; De Rosa, M.C. Screening and initial binding assessment of fumonisin B1 aptamers. Int. J. Mol. Sci. 2010, 11, 4864–4881. [Google Scholar] [CrossRef] [PubMed]
- Goud, K.Y.; Hayat, A.; Satyanarayana, M.; Kumar, V.S.; Gobi, K.V.; Marty, J.-L. Aptamer-based zearalenone assay based on the use of a fluorescein label and a functional graphene oxide as a quencher. Microchim. Acta 2017, 184, 4401–4408. [Google Scholar] [CrossRef]
- Chen, X.; Bai, X.; Li, H.; Zhang, B. Aptamer-based microcantilever array biosensor for detection of fumonisin B1. RSC Adv. 2015, 5, 35448–35452. [Google Scholar] [CrossRef]
- Khan, I.M.; Zhao, S.; Niazi, S.; Mohsin, A.; Shoaib, M.; Duan, N.; Wu, S.; Wang, Z. Silver nanoclusters based FRET aptasensor for sensitive and selective fluorescent detection of T-2 toxin. Sens. Actuators B Chem. 2018, 277, 328–335. [Google Scholar] [CrossRef]
- Werner, A.; Konarev, P.V.; Svergun, D.I.; Hahn, U. Characterization of a fluorophore binding RNA aptamer by fluorescence correlation spectroscopy and small angle X-ray scattering. Anal. Biochem. 2009, 389, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, Y.; Huang, X.; Chen, Y.; Gao, W. Combining a loop-stem aptamer sequence with methylene blue: A simple assay for thrombin detection by resonance light scattering technique. RSC Adv. 2015, 38, 30268–30274. [Google Scholar] [CrossRef]
- Evtugyn, G.; Porfireva, A.; Stoikov, I. Electrochemical DNA sensors based on spatially distributed redox mediators: Challenges and promises. Pure Appl. Chem. 2017, 89, 1471–1490. [Google Scholar] [CrossRef]
- Deng, C.; Pi, X.; Qian, P.; Chen, X.; Wu, W.; Xiang, J. High-performance ratiometric electrochemical method based on the combination of signal probe and inner reference probe in one hairpin-structured DNA. Anal. Chem. 2017, 89, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Zhang, Y.; Liu, G.; Du, L.; Wang, S. Aptamer-based fluorescent detection of ochratoxin A by quenching of gold nanoparticles. RSC Adv. 2017, 7, 16290–16294. [Google Scholar] [CrossRef]
- Sharma, A.; Hayat, A.; Mishra, R.K.; Catanante, G.; Bhand, S.; Marty, J.L. Titanium dioxide nanoparticles (TiO2) quenching based aptasensing platform: Application to ochratoxin A detection. Toxins 2015, 7, 3771–3784. [Google Scholar] [CrossRef] [PubMed]
- Rapini, R.; Marrazza, G. Electrochemical aptasensors for contaminants detection in food and environment: Recent advances. Bioelectrochemistry 2017, 118, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhang, Z.W.; Zhang, Q.; Li, P.W. Mycotoxin determination in foods using advanced sensors based on antibodies or aptamers. Toxins 2016, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Evtugyn, G.; Porfireva, A.; Stepanova, V.; Sitdikov, R.; Stoikov, I.; Nikolelis, D.; Hianik, T. Electrochemical aptasensor based on polycarboxylic macrocycle modified with Neutral red for aflatoxin B1 detection. Electroanalysis 2014, 26, 2100–2109. [Google Scholar] [CrossRef]
- Wu, L.; Ding, F.; Yin, W.; Ma, J.; Wang, B.; Nie, A.; Han, H. From electrochemistry to electroluminescence: Development and application in a ratiometric aptasensor for aflatoxin B1. Anal. Chem. 2017, 89, 7578–7585. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Teng, J.; Cheng, L.; Ye, Y.; Pan, D.; Wu, J.; Xue, F.; Liu, G.; Chen, W. Hetero-enzyme-based two-round signal amplification strategy for trace detection of aflatoxin B1 using an electrochemical aptasensor. Biosens. Bioelectron. 2016, 80, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Goud, K.Y.; Catanate, G.; Hayat, A.; Satyanarayana, M.; Gobi, K.V.; Marty, J.-L. Disposable and portable electrochemical aptasensor for label free detection of aflatoxin B1 in alcoholic beverages. Sens Actuators B Chem. 2016, 235, 466–473. [Google Scholar] [CrossRef]
- Geleta, G.S.; Zhao, Z.; Wang, Z. A novel reduced graphene oxide/molybdenum disulfide/polyaniline nanocomposite-based electrochemical aptasensor for detection of aflatoxin B1. Analyst 2018, 143, 1644–1649. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Li, X.; Cui, F.; Qiu, Q.; Chen, X.; Huang, H. Aflatoxin B1 electrochemical aptasensor based on tetrahedral DNA nanostructures functionalized three dimensionally ordered macroporous MoS2-AuNPs film. ACS Appl. Mater. Interfaces 2018, 10, 17551–17559. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Qian, J.; An, K.; Ren, C.; Lu, X.; Hao, N.; Liu, Q.; Li, H.; Huang, X.; Wang, K. Fabrication of magnetically assembled aptasensing device for label-free determination of aflatoxin B1 based on EIS. Biosens. Bioelectron. 2018, 108, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Istamboulié, G.; Paniel, N.; Zara, L.; Granados, L.R.; Barthelmebs, L.; Noguer, T. Development of an impedimetric aptasensor for the determination of aflatoxin M1 in milk. Talanta 2016, 146, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Smolko, V.; Shurpik, D.; Porfireva, A.; Evtugyn, G.; Stoikov, I.; Hianik, T. Electrochemical aptasensor based on poly(Neutral red) and carboxylated pillar[5]arene for sensitive determination of aflatoxin M1. Electroanalysis 2018, 30, 486–496. [Google Scholar] [CrossRef]
- Jalalian, S.H.; Ramezani, M.; Danesh, N.M.; Alibolandi, M.; Abnous, K.; Taghdisi, S.M. A novel electrochemical aptasensor for detection of aflatoxin M1 based on target-induced immobilization of gold nanoparticles on the surface of electrode. Biosens. Bioelectron. 2018, 117, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.K.; Rajput, Y.S.; Sharma, R.; Singh, D. Immobilized aptamer on gold electrode senses trace amount of aflatoxin M1. Appl. Nanosci. 2017, 7, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Karapetis, S.; Nikolelis, D.; Hianik, T. Label-free and redox markers-based electrochemical aptasensors for aflatoxin M1 detection. Sensors 2018, 18, 4218. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Qian, J.; Yang, X.; Yan, Y.; Liu, Q.; Wang, K.; Wang, K. Amplified impedimetric aptasensor based on gold nanoparticles covalently bound graphene sheet for the picomolar detection of ochratoxin A. Anal. Chim. Acta 2014, 806, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Jiang, L.; Yang, X.; Yan, Y.; Mao, H.; Wang, K. Highly sensitive impedimetric aptasensor based on covalent binding of gold nanoparticles on reduced graphene oxide with good dispersity and high density. Analyst 2014, 139, 5587–5593. [Google Scholar] [CrossRef] [PubMed]
- Qing, Y.; Li, X.; Chen, S.; Zhou, X.P.; Luo, M.; Xu, X.; Li, C.R.; Qiu, J.F. Differential pulse voltammetric ochratoxin A assay based on the use of an aptamer and hybridization chain reaction. Microchim. Acta 2017, 184, 863–870. [Google Scholar] [CrossRef]
- Wang, C.; Qian, J.; An, K.; Huang, X.; Zhao, L.; Liu, Q.; Hao, N.; Wang, K. Magneto-controlled aptasensor for simultaneous electrochemical detection of dual mycotoxins in maize using metal sulfide quantum dots coated silica as labels. Biosens. Bioelectron. 2017, 89, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.-L.; Zhang, Y.-F.; Sun, G.-P.; Wang, X.-N.; Tang, D. Homogeneous electrochemical detection of ochratoxin A in foodstuff using aptamer–graphene oxide nanosheets and DNase I-based target recycling reaction. Biosens. Bioelectron. 2017, 89, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Abnous, K.; Danesh, N.M.; Alibolandi, M.; Ramezani, M.; Taghdisi, S.M. Amperometric aptasensor for ochratoxin A based on the use of a gold electrode modified with aptamer, complementary DNA, SWCNTs and the redox marker Methylene Blue. Microchim. Acta 2017, 184, 1151–1159. [Google Scholar] [CrossRef]
- Wei, M.; Feng, S. A signal-off aptasensor for the determination of ochratoxin A by differential pulse voltammetry at a modified Au electrode using methylene blue as an electrochemical probe. Anal. Methods 2017, 9, 5449–5454. [Google Scholar] [CrossRef]
- Rivas, L.; Mayorga-Martinez, C.C.; Quesada-González, D.; Zamora-Gálvez, A.; de la Escosura-Muñiz, A.; Merkoçi, A. Label-free impedimetric aptasensor for ochratoxin-A detection using iridium oxide nanoparticles. Anal. Chem. 2015, 87, 5167–5172. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.K.; Hayat, A.; Catanante, G.; Ocaña, C.; Marty, J.-L. A label free aptasensor for ochratoxin A detection in cocoa beans: An application to chocolate industries. Anal. Chim. Acta 2015, 889, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.K.; Hayat, A.; Catanante, G.; Istamboulie, G.; Marty, J.-L. Sensitive quantitation of ochratoxin A in cocoa beans using differential pulse voltammetry based aptasensor. Food Chem. 2016, 192, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Mejri-Omrani, N.; Miodek, A.; Zribi, B.; Marrak, M.; Hamdi, M.; Marty, J.-L.; Korri-Youssoufi, H. Direct detection of OTA by impedimetric aptasensor based on modified polypyrrole-dendrimers. Anal. Chim. Acta 2016, 920, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, X.; Ma, Y.; Deng, Y.; Hu, R.; Yang, Y. Preparation of an OTA aptasensor based on a metal-organic framework. Anal. Methods 2018, 10, 3273–3279. [Google Scholar] [CrossRef]
- Zejli, K.; Goud, K.Y.; Marty, J.L. Label free aptasensor for ochratoxin A detection using polythiophene-3-carboxylic acid. Talanta 2018, 185, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ning, G.; Bi, H.; Wu, Y.; Liu, G.; Zhao, Y. A novel ratiometric electrochemical assay for ochratoxin A coupling Au nanoparticles decorated MoS2 nanosheets with aptamer. Electrochim. Acta 2018, 285, 120–127. [Google Scholar] [CrossRef]
- Chen, W.; Yan, C.; Cheng, L.; Yao, L.; Xue, F.; Xu, J. An ultrasensitive signal-on electrochemical aptasensor for ochratoxin A determination based on DNA controlled layer-by-layer assembly of dual gold nanoparticle conjugates. Biosens. Bioelectron. 2018, 117, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.Y.; Zheng, Y.T.; Zhang, H.B.; He, C.H.; Wu, W.D.; Zhang, H.B. DNA Electrochemical aptasensor for detecting fumonisins B1 based on graphene and thionine nanocomposite. Electroanalysis 2015, 27, 1097–1103. [Google Scholar] [CrossRef]
- Chen, X.; Huang, Y.; Ma, X.; Jia, F.; Guo, X.; Wang, Z. Impedimetric aptamer-based determination of the mold toxin fumonisin B1. Microchim. Acta 2015, 182, 1709–1714. [Google Scholar] [CrossRef]
- He, B.; Dong, X. Aptamer based voltammetric patulin assay based on the use of ZnO nanorods. Microchim. Acta 2018, 185, 462. [Google Scholar] [CrossRef] [PubMed]
- Zanchetta, G.; Lanfranco, R.; Giavazzi, F.; Bellini, T.; Buscaglia, M. Emerging applications of label-free optical biosensors. Nanophotonics 2017, 6, 627–645. [Google Scholar] [CrossRef]
- Khansili, N.; Rattu, G.; Krishna, P.M. Label-free optical biosensors for food and biological sensor applications. Sens. Actuators B Chem. 2018, 265, 35–49. [Google Scholar] [CrossRef]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).