Using Fluorescence Intensity of Enhanced Green Fluorescent Protein to Quantify Pseudomonas aeruginosa

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. FAB Minimal Media Preparation
2.2. M9 Minimal Media Preparation
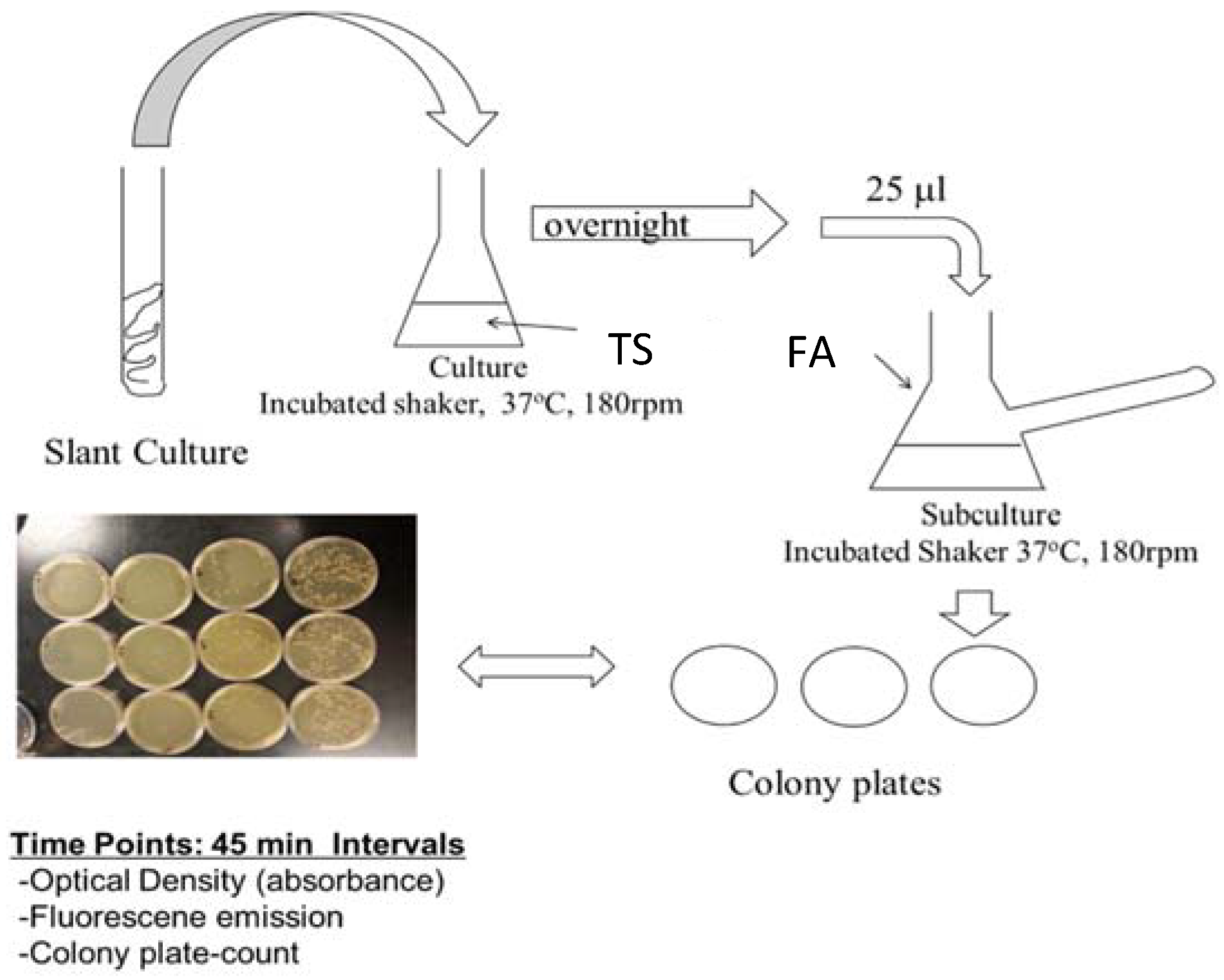
2.3. PA14/EGFP Planktonic and Biofilm Culture—Procedure 1
2.4. PA14/EGFP Planktonic and Biofilm Culture Confirmation Study—Procedure 2
2.5. Assays Procedure
2.6. Data Analysis
3. Results
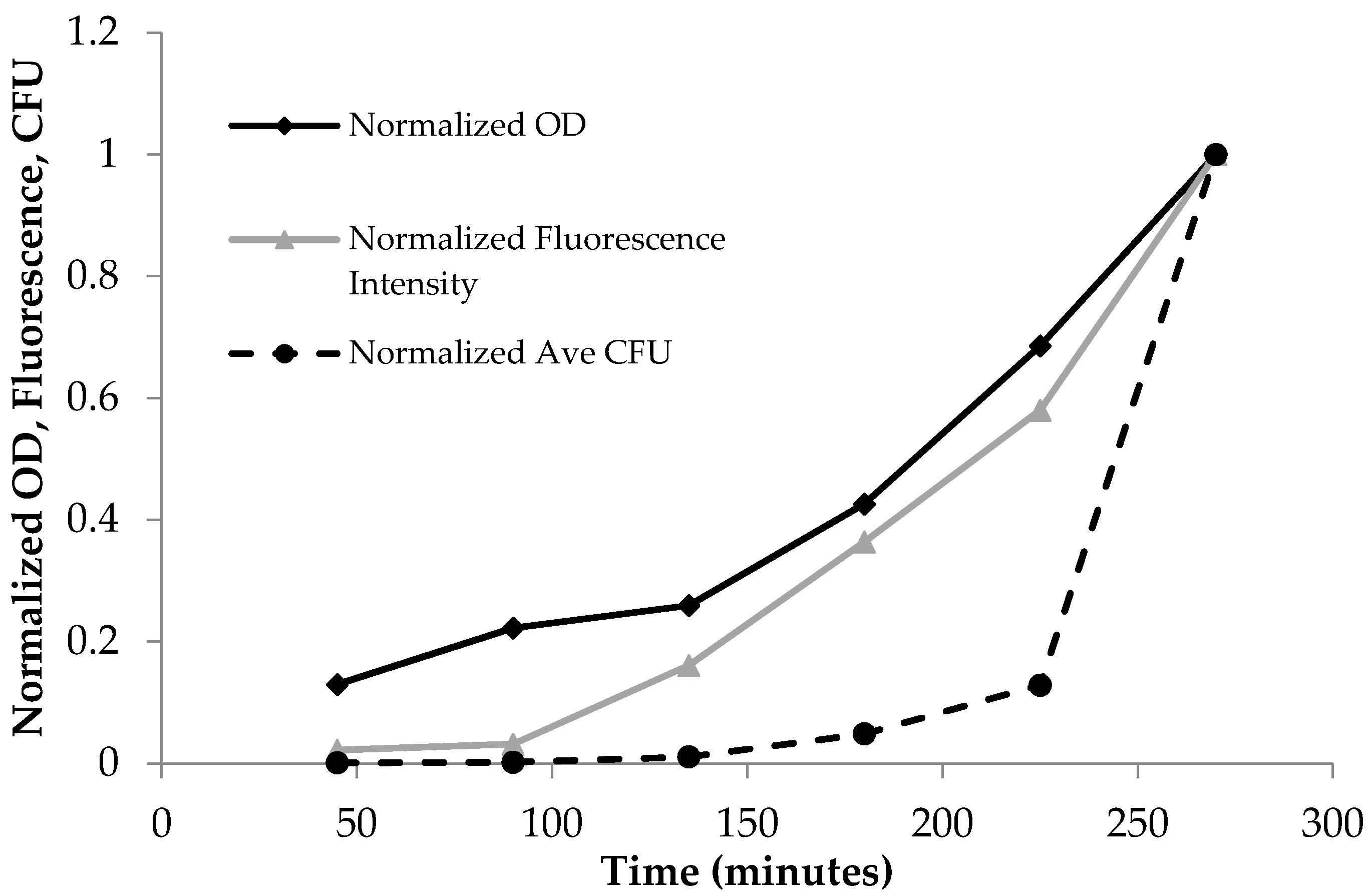
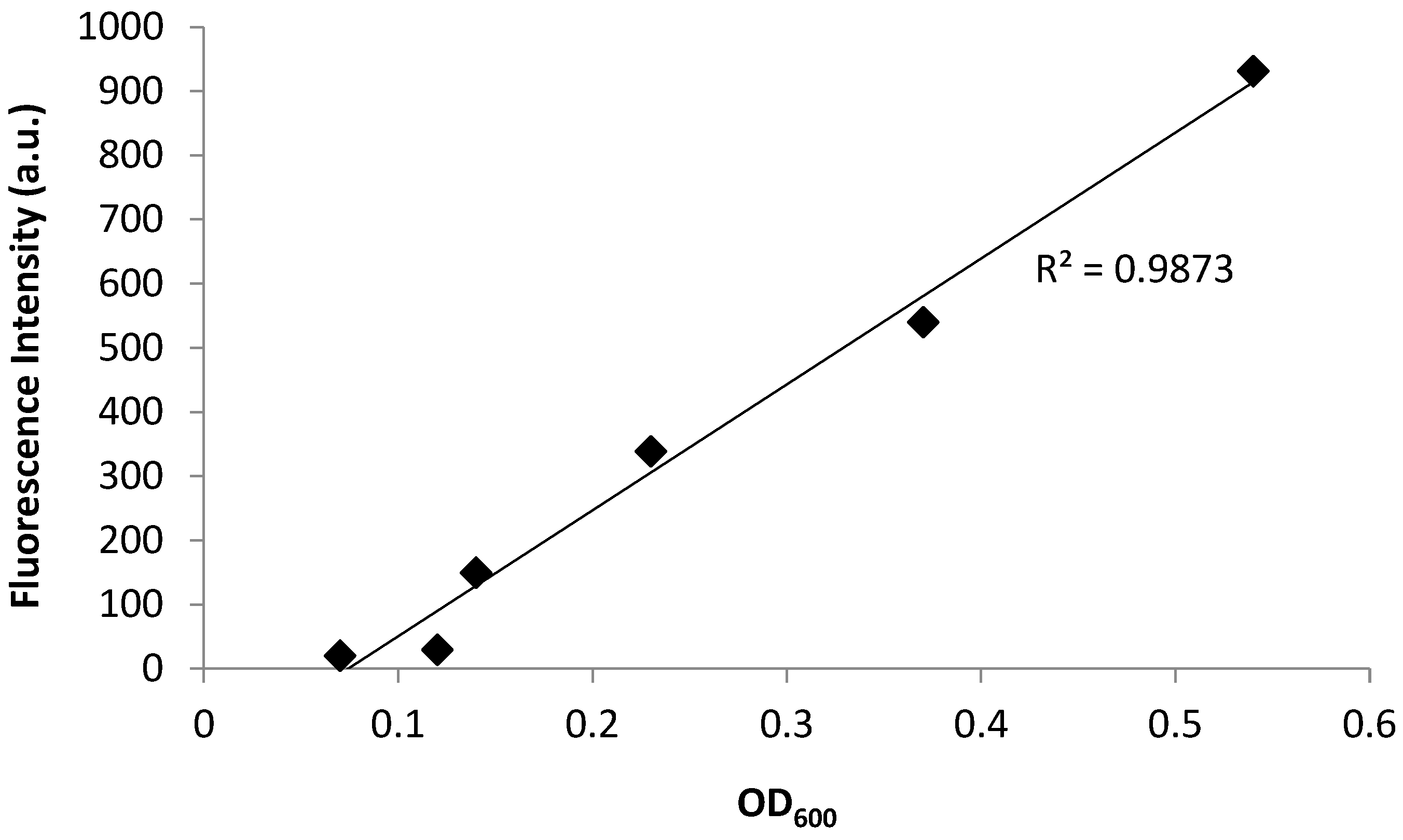
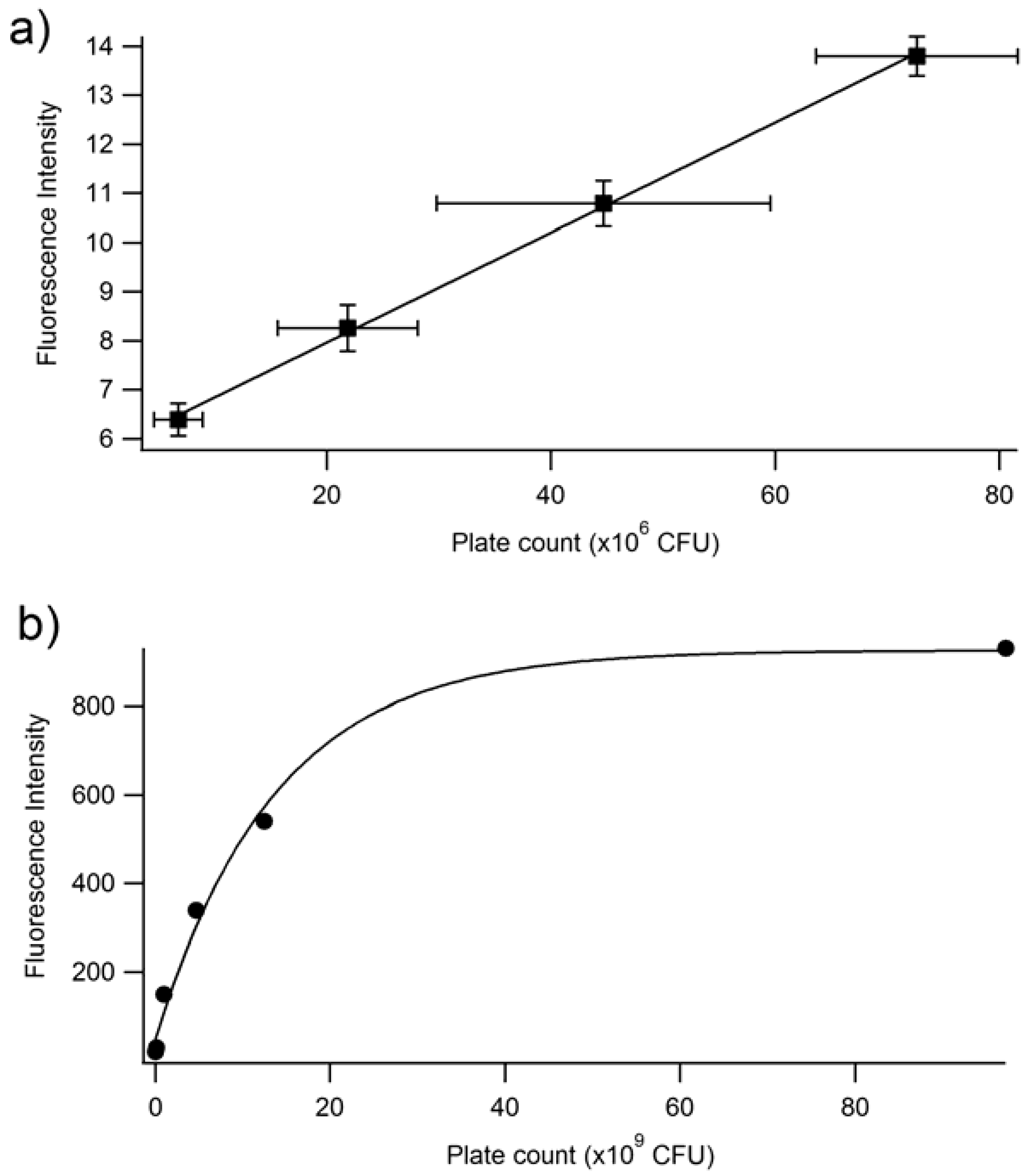
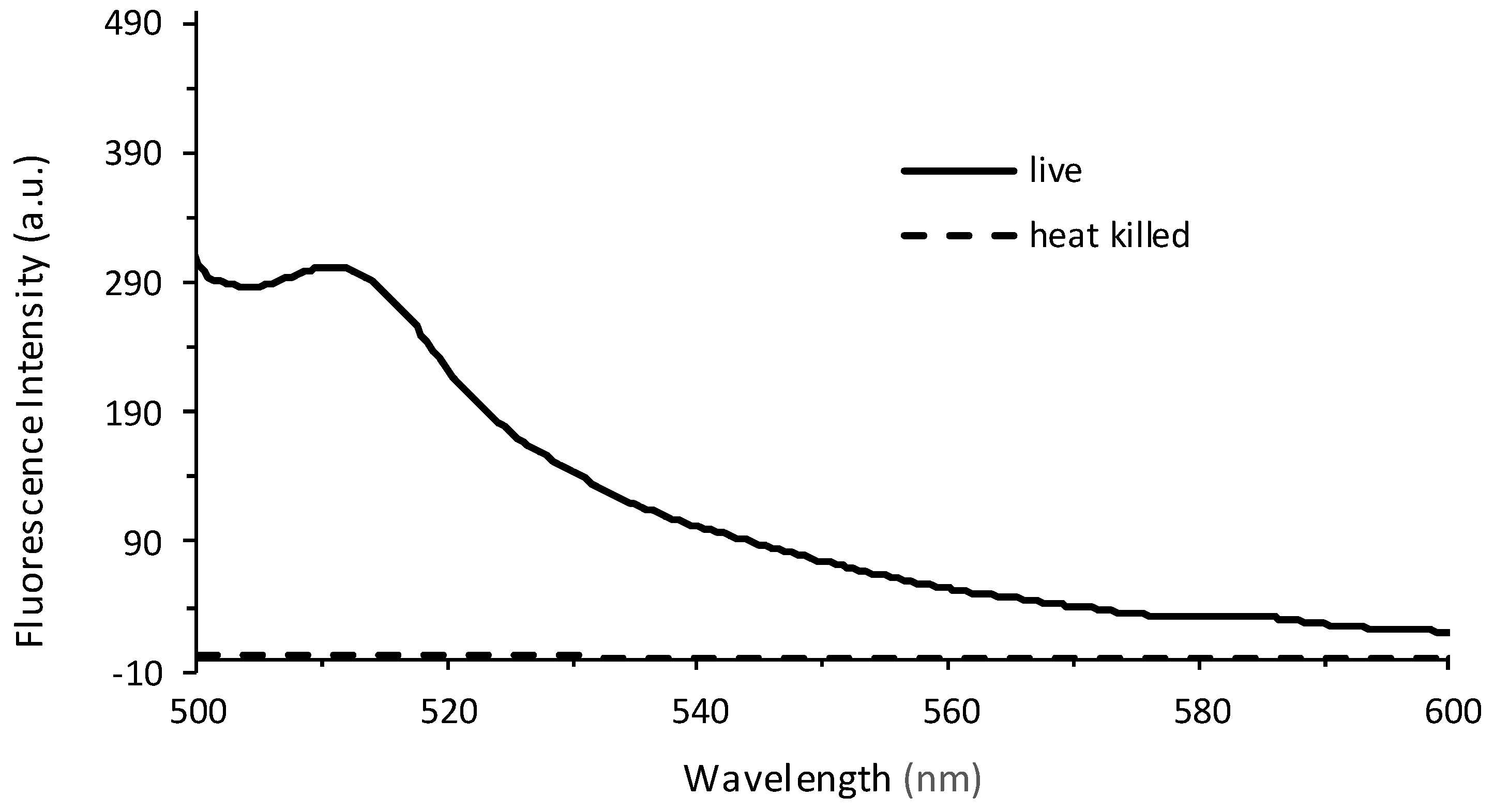
3.1. Fluorescence Spectroscopy as a Tool to Quantify the Planktonic Bacteria of PA14/EFGP
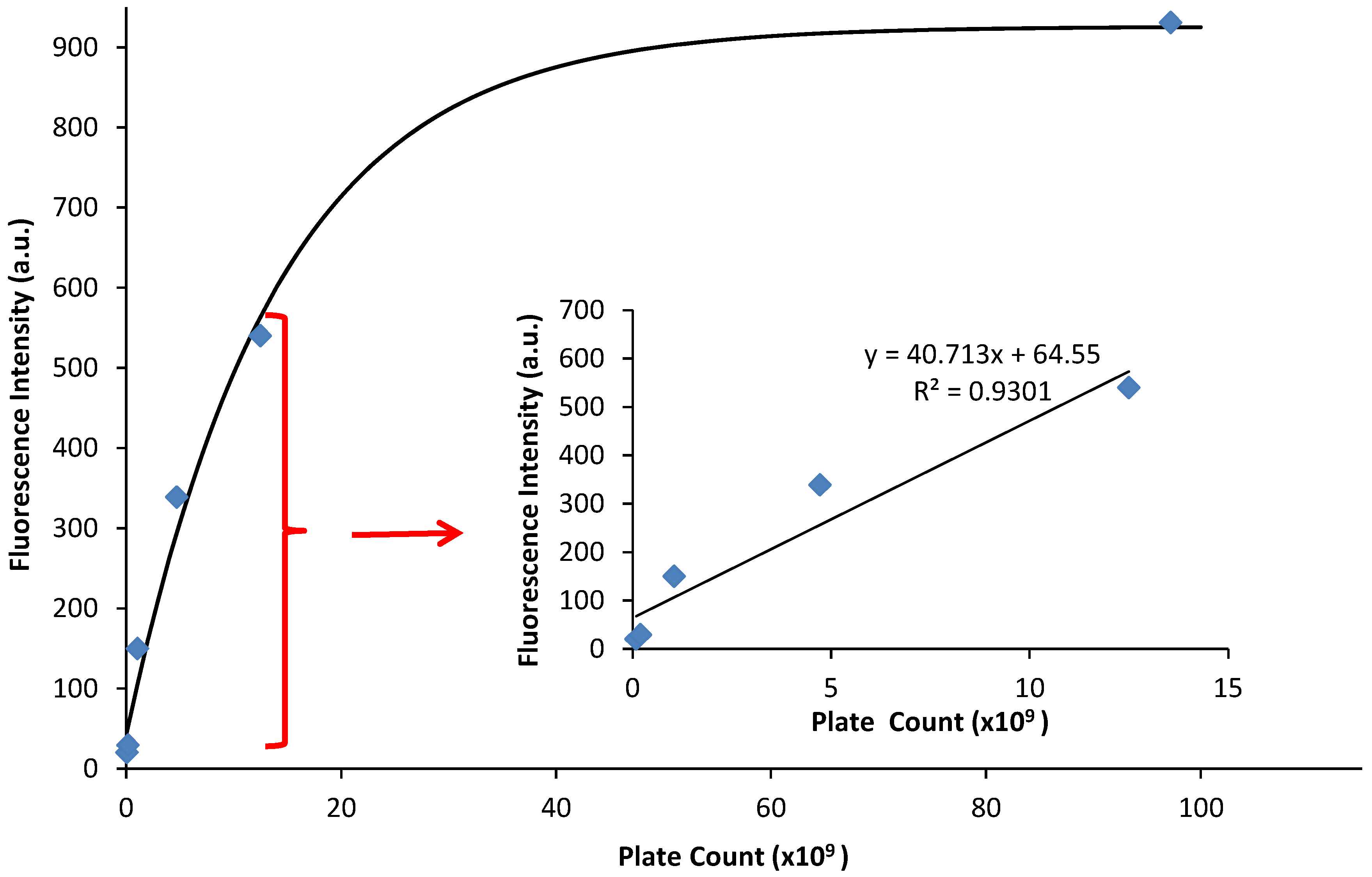
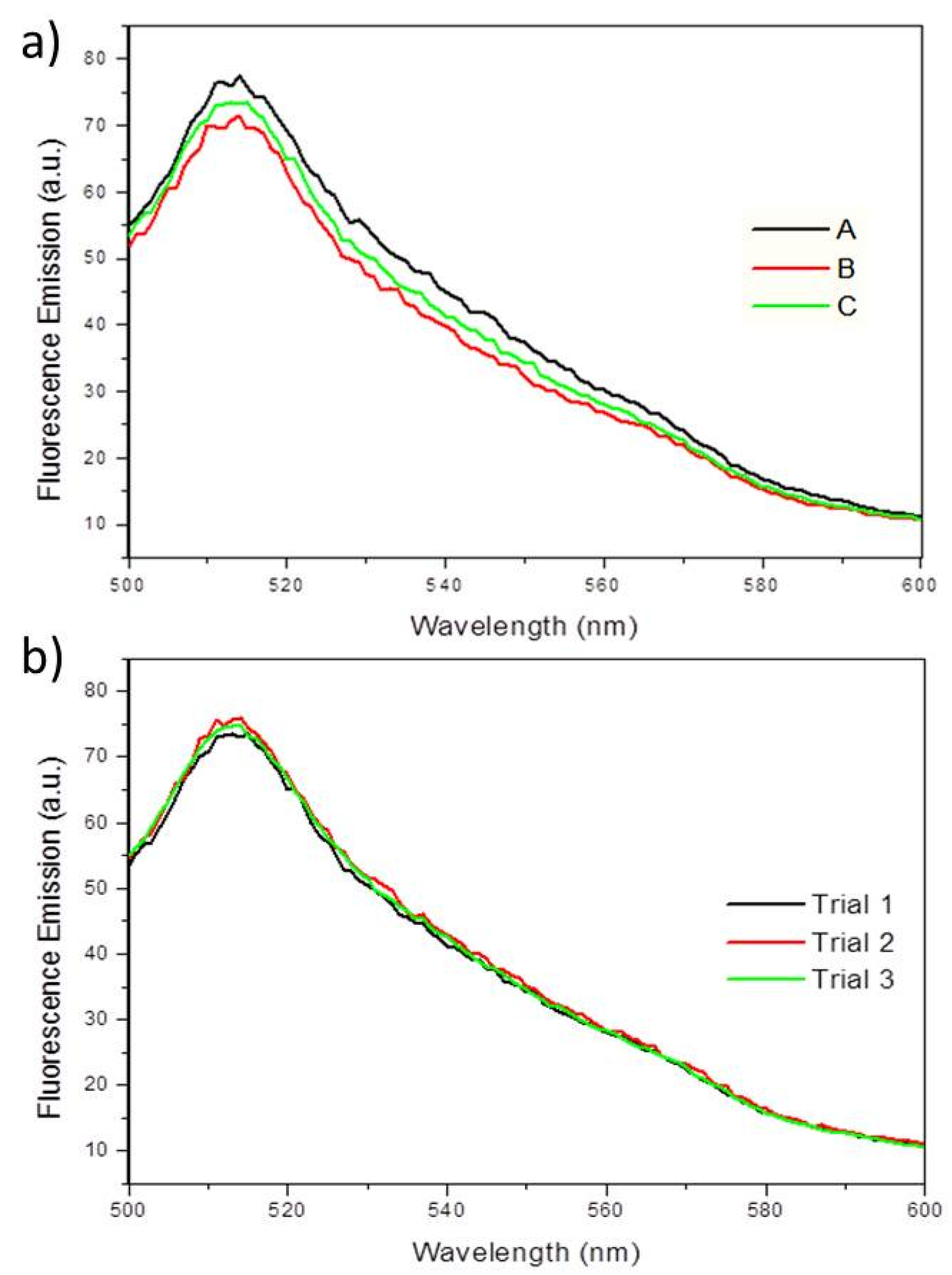
3.2. Confirmation of Fluorescence Data
3.3. Fluorescence Spectroscopy for Quantification of PA14/EFGP Bacteria in Biofilms
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Driscoll, J.A.; Brody, S.L.; Kollef, M.H. The epidemiology, pathogenesis and treatment of pseudomonas aeruginosa infection. Drugs 2007, 67, 351–368. [Google Scholar] [CrossRef] [PubMed]
- Skindersoe, M.E.; Alhede, M.; Phipps, R.; Yang, L.; Jensen, P.O.; Rasmussen, T.B.; Bjarnsholt, T.; Tolker-Nielsen, T.; Høiby, N.; Givskov, M. Effects of antibiotics on quorum sensing in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2008, 52, 3648–3663. [Google Scholar] [CrossRef] [PubMed]
- Smyth, A.R.; Cifelli, P.M.; Ortori, C.A.; Righetti, K.; Lewis, S.; Erskine, P.; Holland, E.D.; Givskov, M.; Williams, P.; Cámara, M.; et al. Garlic as an inhibitor of pseudomonas aeruginosa quorum sensing in cystic fibrosis—A pilot randomized controlled trial. Pediatr. Pulmonol. 2010, 45, 356–362. [Google Scholar] [PubMed]
- Byrd, M.S.; Pang, B.; Mishra, M.; Swords, W.E.; Wozniak, D.J. The Pseudomonas aeruginosa Exopolysaccharide Psl Facilitates Surface Adherence and NF- B Activation in A549 Cells. mBio 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Arciola, C.R.; Campoccia, D.; Speziale, P.; Montanaro, L.; Costerton, J.W. Biofilm formation in Staphylococcus implant infections. A review of molecular mechanisms and implications for biofilm-resistant materials. Biomaterials 2012, 33, 5967–5982. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Rajurkar, M.N.; Attal, R.O.; Mallick, S.K. Biofilms: A challenge to medical fraternity in infection control. In Infection Control; Basak, S., Ed.; InTech: London, UK, 2013. [Google Scholar]
- Costerton, J.W. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Christensen, L.D.; Moser, C.; Jensen, P.Ø.; Rasmussen, T.B.; Christophersen, L.; Kjelleberg, S.; Kumar, N.; Høiby, N.; Givskov, M.; Bjarnsholt, T. Impact of Pseudomonas aeruginosa quorum sensing on biofilm persistence in an in vivo intraperitoneal foreign-body infection model. Microbiology 2007, 153, 2312–2320. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.J.; Lin, Y.C.; Wu, W.B.; Chiu, P.H.; Lin, B.J.; Hao, S.P. Biofilm formations in nasopharyngeal tissues of patients with nasopharyngeal osteoradionecrosis. Otolaryngol. Head Neck Surg. 2013, 148, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Donlan, R.M.; Piede, J.A.; Heyes, C.D.; Sanii, L.; Murga, R.; Edmonds, P.; El-Sayed, I.; El-Sayed, M.A. Model system for growing and quantifying streptococcus pneumoniae biofilms in situ and in real time. Appl. Environ. Microbiol. 2004, 70, 4980–4988. [Google Scholar] [CrossRef] [PubMed]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Establishment of Pseudomonas aeruginosa infection: Lessons from a versatile opportunist. Microbes Infect. 2000, 2, 1051–1060. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Prashar, D.; Luk, Y.-Y. Anti-fouling chemistry of chiral monolayers: Enhancing biofilm resistance on racemic surface. Langmuir 2011, 27, 6124–6131. [Google Scholar] [CrossRef] [PubMed]
- Kreft, J.; Wimpenny, J. Effect of EPS on biofilm structure and function as revealed by anindividual-based model of biofilm growth. Water Sci. Technol. 2001, 43, 135–141. [Google Scholar] [PubMed]
- Wu, T.; Hu, W.; Guo, L.; Finnegan, M.; Bradshaw, D.J.; Webster, P.; Loewy, Z.G.; Zhou, X.; Shi, W.; Lux, R. Development of a new model system to study microbial colonization on dentures. J. Prosthodont. Off. J. Am. Coll. Prosthodont. 2013, 22, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Tawakoli, P.N.; Al-Ahmad, A.; Hoth-Hannig, W.; Hannig, M.; Hannig, C. Comparison of different live/dead stainings for detection and quantification of adherent microorganisms in the initial oral biofilm. Clin. Oral Investig. 2012, 17, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Rice, K.C.; Mann, E.E.; Endres, J.L.; Weiss, E.C.; Cassat, J.E.; Smeltzer, M.S.; Bayles, K.W. The cidA murein hydrolase regulator contributes to DNA release and biofilm development in Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2007, 104, 8113–8118. [Google Scholar] [CrossRef] [PubMed]
- Nett, J.E.; Cain, M.T.; Crawford, K.; Andes, D.R. Optimizing a Candida biofilm microtiter plate model for measurement of antifungal susceptibility by tetrazolium salt assay. J. Clin. Microbiol. 2011, 49, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; López, D.A.; Tillich, U.M.; Frohme, M. A simple viability analysis for unicellular cyanobacteria using a new autofluorescence assay, automated microscopy, and ImageJ. BMC Biotechnol. 2011, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ambriz-Aviña, V.; Contreras-Garduño, J.A.; Pedraza-Reyes, M. Applications of flow cytometry to characterize bacterial physiological responses. BioMed Res. Int. 2014, 2014, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chalfie, M.; Tu, Y.; Euskirche, G.; Ward, W.W.; Prashert, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Heim, R.; Prasher, D.; Tsien, R. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. USA 1994, 91, 12501–12504. [Google Scholar] [CrossRef] [PubMed]
- Neu, T.R.; Lawrence, J.R. Development and structure of microbial biofilms in river water studied by confocal laser scanning microscopy. FEMS Microbiol. Ecol. 1997, 24, 11–25. [Google Scholar] [CrossRef]
- Berney, M.; Hammes, F.; Bosshard, F.; Weilenmann, H.; Egli, T. Assessment and interpretation of bacterial viability by using the LIVE/DEAD BacLight kit in combination with flow cytometry. Appl. Environ. Microbiol. 2007, 73, 3283–3290. [Google Scholar] [CrossRef] [PubMed]
- Adetunji, V.; Odetokun, I.A. Assessment of biofilm in E. coli O157:H7 and Salmonella strains: Influence of cultural conditions. Am. J. Food Technol. 2012, 7, 582–595. [Google Scholar] [CrossRef]
- Cloete, T.E.; Brozel, V.S.; Von Holy, A. Practical aspects of biofouling control in industrial water systems. Int. Biodeterior. Biodegrad. 1992, 29, 299–341. [Google Scholar] [CrossRef]
- Bakke, R.; Olsson, P.Q. Biofilm thickness measurements by light microscopy. J. Microbiol. Methods 1986, 5, 93–98. [Google Scholar] [CrossRef]
- Bakke, R.; Kommedal, R.; Kalvenes, S. Quantification of biofilm accumulation by an optical approach. J. Microbiol. Methods 2001, 44, 13–26. [Google Scholar] [CrossRef]
- Romanova, N.A.; Gawande, P.V.; Brovko, L.Y.; Griffiths, M.W. Rapid methods to assess sanitizing efficacy of benzalkonium chloride to Listeria monocytogenes biofilms. J. Microbiol. Methods 2007, 71, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Trulear, M.G.; Characklis, W.G. Dynamics of biofilm processes. J. Water Pollut. Control Fed. 1982, 54, 1288–1301. [Google Scholar]
- Kolecka, A.; Chorvat, D., Jr.; Bujdakova, H. The impact of growth conditions on biofilm formation and the cell surface hydrophobicity in fluconazole susceptible and tolerant Candida albicans. Folia Microbiol. 2015, 60, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Paul, E.; Ochoa, J.C.; Pechaud, Y.; Liu, Y.; Line, A. Effect of shear stress and growth conditions on detachment and physical properties of biofilms. Water Res. 2012, 46, 5499–5508. [Google Scholar] [CrossRef] [PubMed]
- Hartree, E. Determination of protein: A modification of the Lowry method that gives a linear photometric response. Anal. Biochem. 1972, 48, 422–427. [Google Scholar] [CrossRef]
- He, Z.; Liang, J.; Huang, Z.; Jiang, Y.; Jiang, W. Comparison of different methods on extracting and measuring total protein of biofilms formed by Enterococcus faecalis. Kouqiang Yixue Yanjiu 2013, 29, 597–600. [Google Scholar]
- Carey, J.R.; Suslick, K.S.; Hulkower, K.I.; Imlay, J.A.; Imlay, K.R.C.; Ingison, C.K.; Ponder, J.B.; Sen, A.; Wittrig, A.E. Rapid identification of bacteria with a disposable colorimetric sensing array. J. Am. Chem. Soc. 2011, 133, 7571–7576. [Google Scholar] [CrossRef] [PubMed]
- Pomory, C.M. Color development time of the Lowry protein assay. Anal. Biochem. 2008, 378, 216–217. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Smith, W.L.; Gadd, G.M. Reduction and precipitation of chromate by mixed culture sulphate-reducing bacterial biofilms. J. Appl. Microbiol. 2000, 88, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Ragusa, S.R.; McNevin, D.; Qasem, S.; Mitchell, C. Indicators of biofilm development and activity in constructed wetlands microcosms. Water Res. 2004, 38, 2865–2873. [Google Scholar] [CrossRef] [PubMed]
- Hazan, R.; Que, Y.-A.; Maura, D.; Rahme, L.G. A method for high throughput determination of viable bacteria cell counts in 96-well plates. BMC Microbiol. 2012, 12, 259. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.; Mukherjee, A.; Sevgen, S.E.; Sanpitakseree, C.; Lee, J.; Schroeder, C.M.; Kenis, P.J.A. A multiplexed microfluidic platform for rapid antibiotic susceptibility testing. Biosens. Bioelectron. 2013, 49, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Filkins, L.M.; Graber, J.A.; Olson, D.G.; Dolben, E.L.; Lynd, L.R.; Bhuju, S.; O’Toole, G.A. Coculture of Staphylococcus aureus with Pseudomonas aeruginosa Drives S. aureus towards Fermentative Metabolism and Reduced Viability in a Cystic Fibrosis Model. J. Bacteriol. 2015, 197, 2252–2264. [Google Scholar] [CrossRef] [PubMed]
- Moreau-Marquis, S.; Redelman, C.V.; Stanton, B.A.; Anderson, G.G. Co-culture models of pseudomonas aeruginosa biofilms grown on live human airway cells. J. Vis. Exp. 2010. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.; Khan, M.; Kleiman, M.; Hochbaum, A.I. Effects of growth surface topography on bacterial signaling in coculture biofilms. ACS Appl. Mater. Interfaces 2017, 9, 18531–18539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Gurtu, V.; Kain, S.R. An enhanced green fluorescent protein allows sensitive detection of gene transfer in mammalian cells. Biochem. Biophys. Res. Commun. 1996, 227, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Patterson, G.H.; Knobel, S.M.; Sharif, W.D.; Kain, S.R.; Piston, D.W. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys. J. 1997, 73, 2782–2790. [Google Scholar] [CrossRef]
- Heydorn, A.; Ersboll, B.; Kato, J.; Hentzer, M.; Parsek, M.R.; Tolker-Nielsen, T.; Givskov, M.; Molin, S. Statistical analysis of Pseudomonas aeruginosa biofilm development: Impact of mutations in genes involved in twitching motility, cell-to-cell signaling, and stationary-phase sigma factor expression. Appl. Environ. Microbiol. 2002, 68, 2008–2017. [Google Scholar] [CrossRef] [PubMed]
- Bloemberg, G.V.; O’Toole, G.A.; Lugtenberg, B.J.; Kolter, R. Green fluorescent protein as a marker for Pseudomonas spp. Appl. Environ. Microbiol. 1997, 63, 4543–4551. [Google Scholar] [PubMed]
- Goeres, D.M.; Hamilton, M.A.; Beck, N.A.; Buckingham-Meyer, K.; Hilyard, J.D.; Loetterle, L.R.; Lorenz, L.A.; Walker, D.K.; Stewart, P.S. A method for growing a biofilm under low shear at the air–liquid interface using the drip flow biofilm reactor. Nat. Protoc. 2009, 4, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Verheust, C.; Pauwels, K.; Mahillon, J.; Helinski, D.R.; Herman, P. Contained use of bacteriophages: Risk assessment and biosafety recommendations. Appl. Biosaf. 2010, 15, 32–44. [Google Scholar] [CrossRef]
- Miles, A.A.; Misra, S.S.; Irwin, J.O. The estimation of the bactericidal power of the blood. Epidemiol. Infect. 1938, 38, 732–749. [Google Scholar] [CrossRef]
- Wilson, C.; Lukowicz, R.; Merchant, S.; Valquier-Flynn, H.; Caballero, J.; Sandoval, J.; Okuom, M.; Huber, C.; Brooks, T.D.; Wilson, E.; et al. Quantitative and qualitative assessment methods for biofilm growth: A mini-review. Res. Rev. J. Eng. Technol. 2017, 6, 1–25. [Google Scholar]
- Hecht, A.; Endy, D.; Salit, M.; Munson, M.S. When wavelengths collide: Bias in cell abundance measurements due to expressed fluorescent proteins. ACS Synth. Biol. 2016, 5, 1024–1027. [Google Scholar] [CrossRef] [PubMed]
- Sismaet, H.J.; Pinto, A.J.; Goluch, E.D. Electrochemical sensors for identifying pyocyanin production in clinical Pseudomonas aeruginosa isolates. Biosens. Bioelectron. 2017, 97, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Koenig, K.; Schneckenburger, H. Laser-induced autofluorescence for medical diagnosis. J. Fluoresc. 1994, 4, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Snyder, T.L. The relative errors of bacteriological plate counting methods. J. Bacteriol. 1947, 54, 641–654. [Google Scholar] [PubMed]
- Pettit, R.K.; Weber, C.A.; Kean, M.J.; Hoffman, H.; Pettit, G.R.; Tan, R.; Franks, K.S.; Horton, M.L. Microplate alamar blue assay for Staphylococcus epidermidis biofilm susceptibility testing. Antimicrob. Agents Chemother. 2005, 49, 2612–2617. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, F.; Harms, H.; Maskow, T. Biofilm research using calorimetry—A marriage made in heaven? Biotechnol. J. 2010, 5, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.A.; Rees, W.T. Fluorescence spectrometry. A review. Analyst 1962, 87, 83–111. [Google Scholar] [CrossRef]
- Dartnell, L.R.; Roberts, T.A.; Moore, G.; Ward, J.M.; Muller, J.-P. Fluorescence characterization of clinically-important bacteria. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | Absorbance | Fluorescence | Ave Plate Count (CFU) |
|---|---|---|---|
| 45 | 0.07 | 20.24 | 8.99 × 107 |
| 90 | 0.12 | 29.48 | 1.96 × 108 |
| 135 | 0.14 | 149.87 | 1.05 × 109 |
| 180 | 0.23 | 338.66 | 4.72 × 109 |
| 225 | 0.37 | 539.96 | 1.25 × 1010 |
| 270 | 0.54 | 931.05 | 9.72 × 1010 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, E.; Okuom, M.; Kyes, N.; Mayfield, D.; Wilson, C.; Sabatka, D.; Sandoval, J.; Foote, J.R.; Kangas, M.J.; Holmes, A.E.; et al. Using Fluorescence Intensity of Enhanced Green Fluorescent Protein to Quantify Pseudomonas aeruginosa. Chemosensors 2018, 6, 21. https://doi.org/10.3390/chemosensors6020021
Wilson E, Okuom M, Kyes N, Mayfield D, Wilson C, Sabatka D, Sandoval J, Foote JR, Kangas MJ, Holmes AE, et al. Using Fluorescence Intensity of Enhanced Green Fluorescent Protein to Quantify Pseudomonas aeruginosa. Chemosensors. 2018; 6(2):21. https://doi.org/10.3390/chemosensors6020021
Chicago/Turabian StyleWilson, Erin, Macduff Okuom, Nathan Kyes, Dylan Mayfield, Christina Wilson, Derek Sabatka, Jasmin Sandoval, Jared R. Foote, Michael J. Kangas, Andrea E. Holmes, and et al. 2018. "Using Fluorescence Intensity of Enhanced Green Fluorescent Protein to Quantify Pseudomonas aeruginosa" Chemosensors 6, no. 2: 21. https://doi.org/10.3390/chemosensors6020021
APA StyleWilson, E., Okuom, M., Kyes, N., Mayfield, D., Wilson, C., Sabatka, D., Sandoval, J., Foote, J. R., Kangas, M. J., Holmes, A. E., & Sutlief, A. L. (2018). Using Fluorescence Intensity of Enhanced Green Fluorescent Protein to Quantify Pseudomonas aeruginosa. Chemosensors, 6(2), 21. https://doi.org/10.3390/chemosensors6020021