Unbiased Thiol-Labeling and Top-Down Proteomic Analyses Implicate Multiple Proteins in the Late Steps of Regulated Secretion



Abstract
1. Introduction
2. Materials and Methods
2.1. CV Isolation and Fusion Assays
2.2. Proteome Resolution by 2DE
2.3. Mass Spectrometry
3. Results
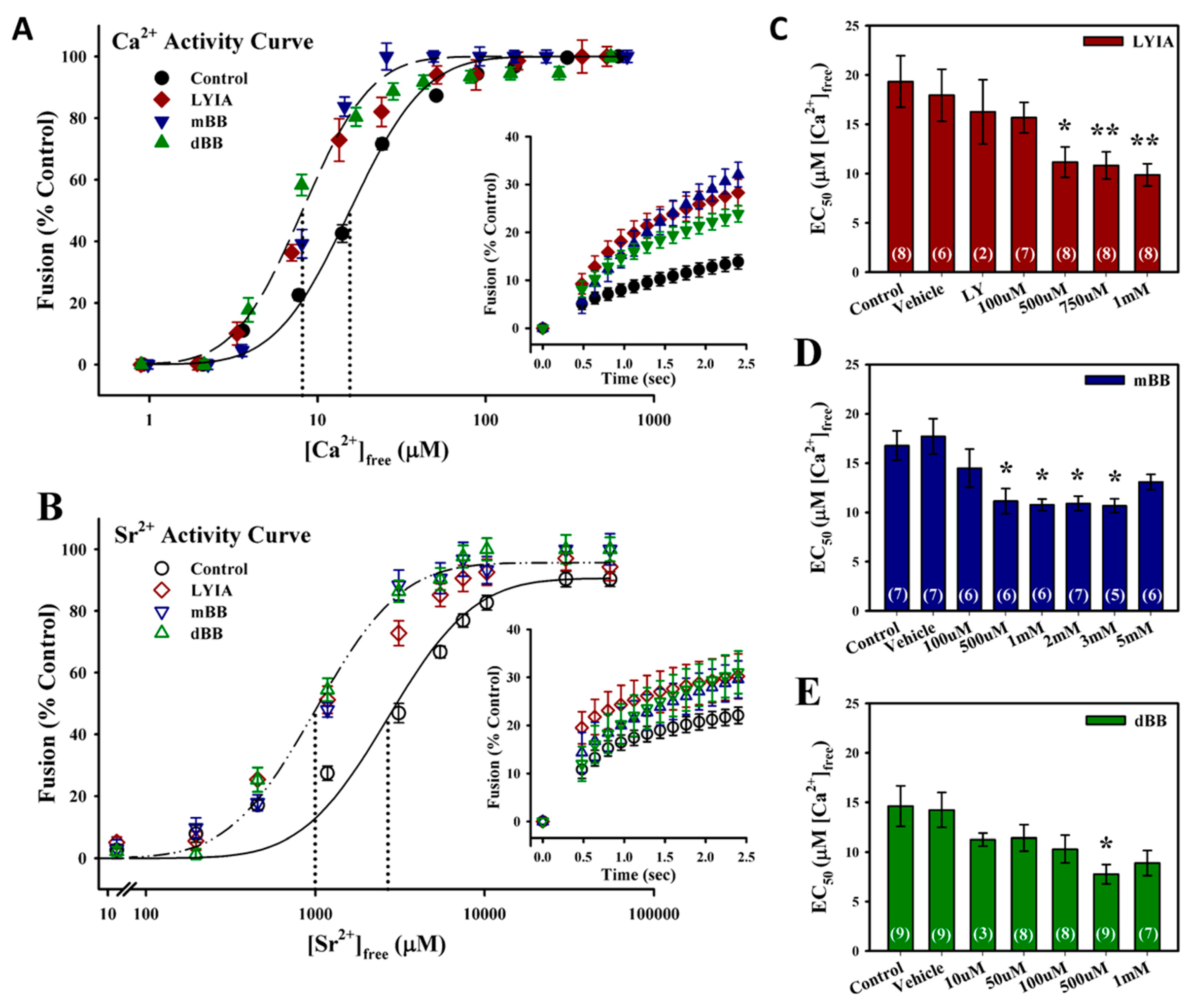
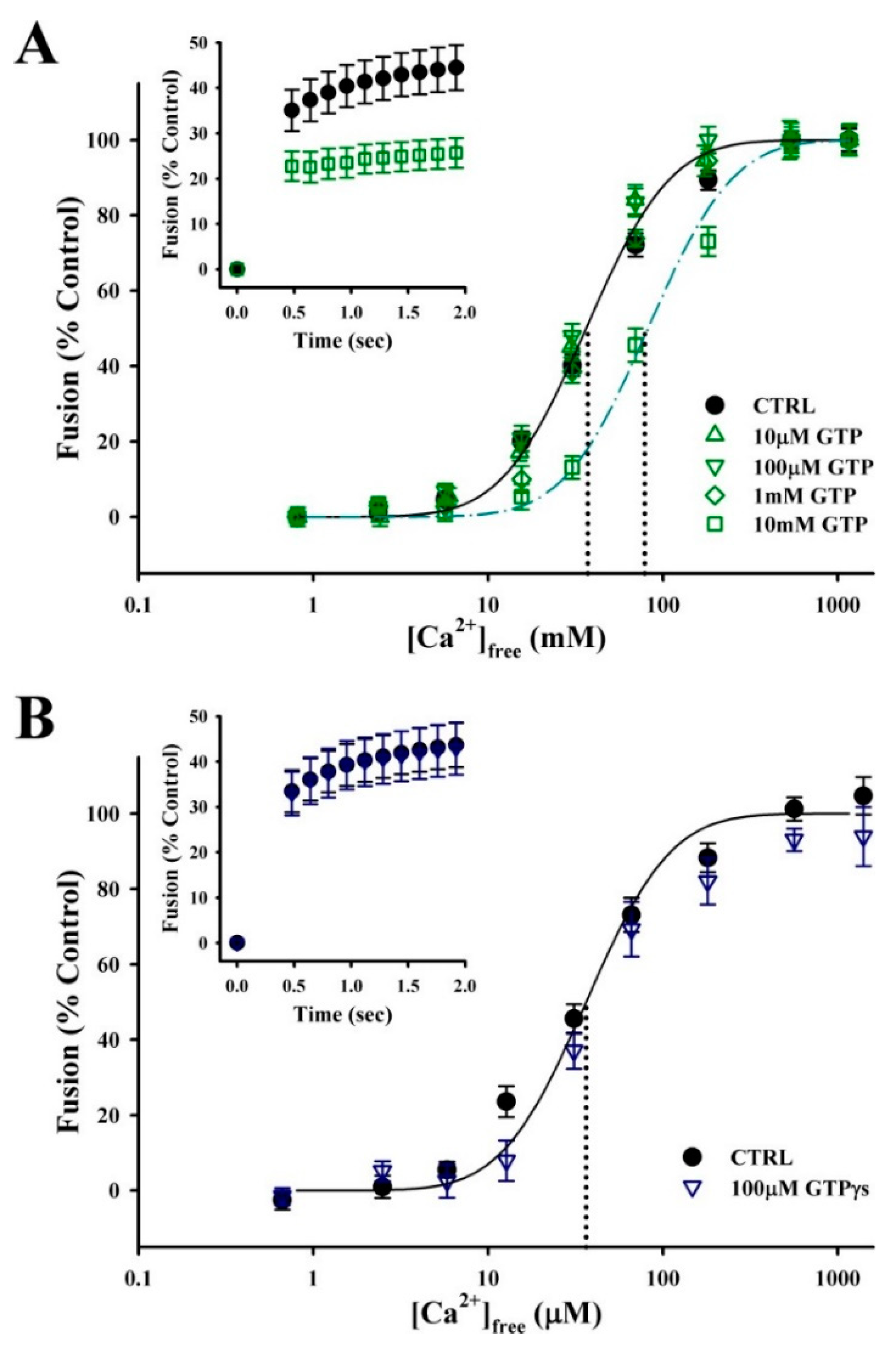
3.1. Fluorescent Thiol-Reactive Reagents Modulate the Ca2+ Sensitivity and Rate of Membrane fusion
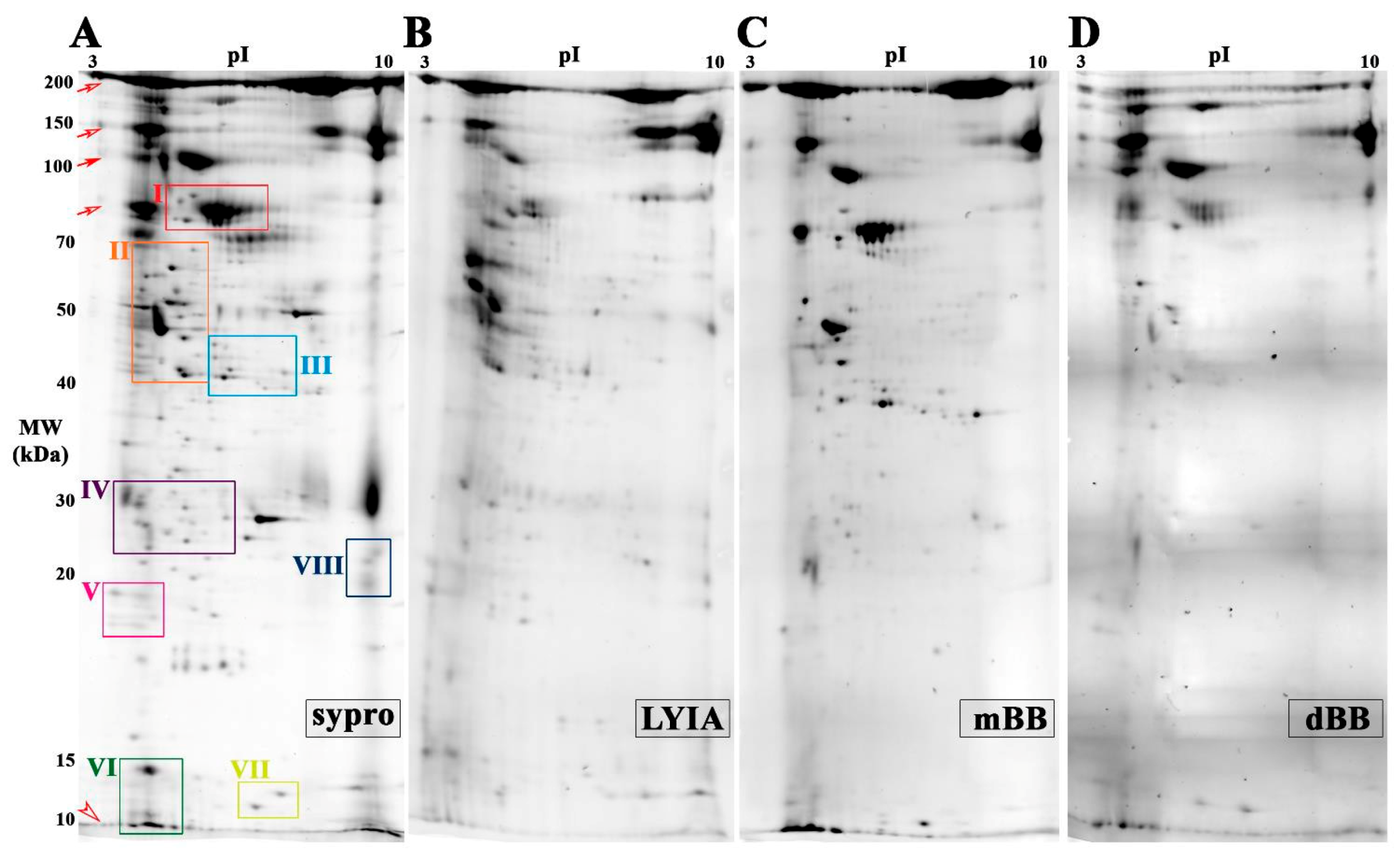
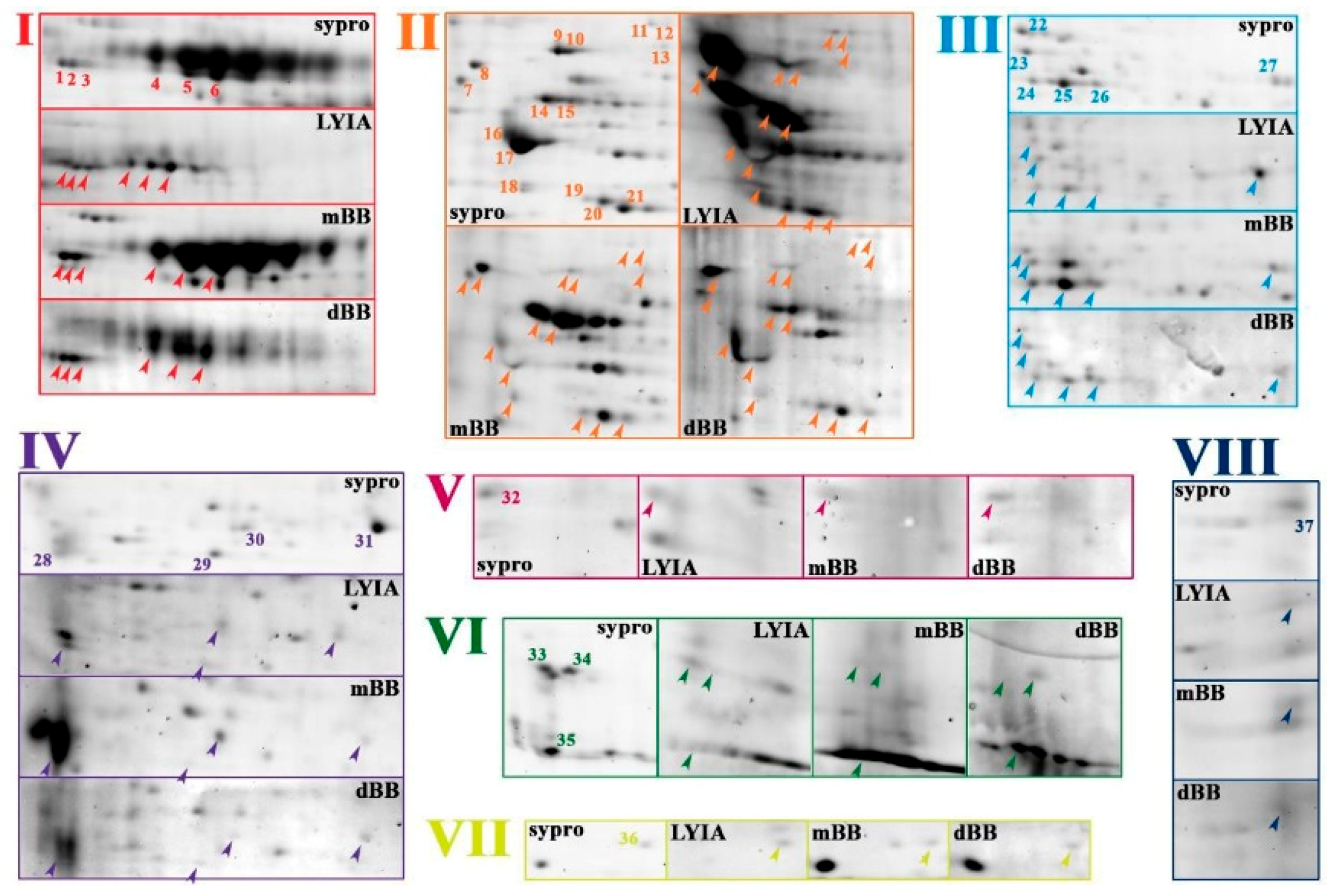
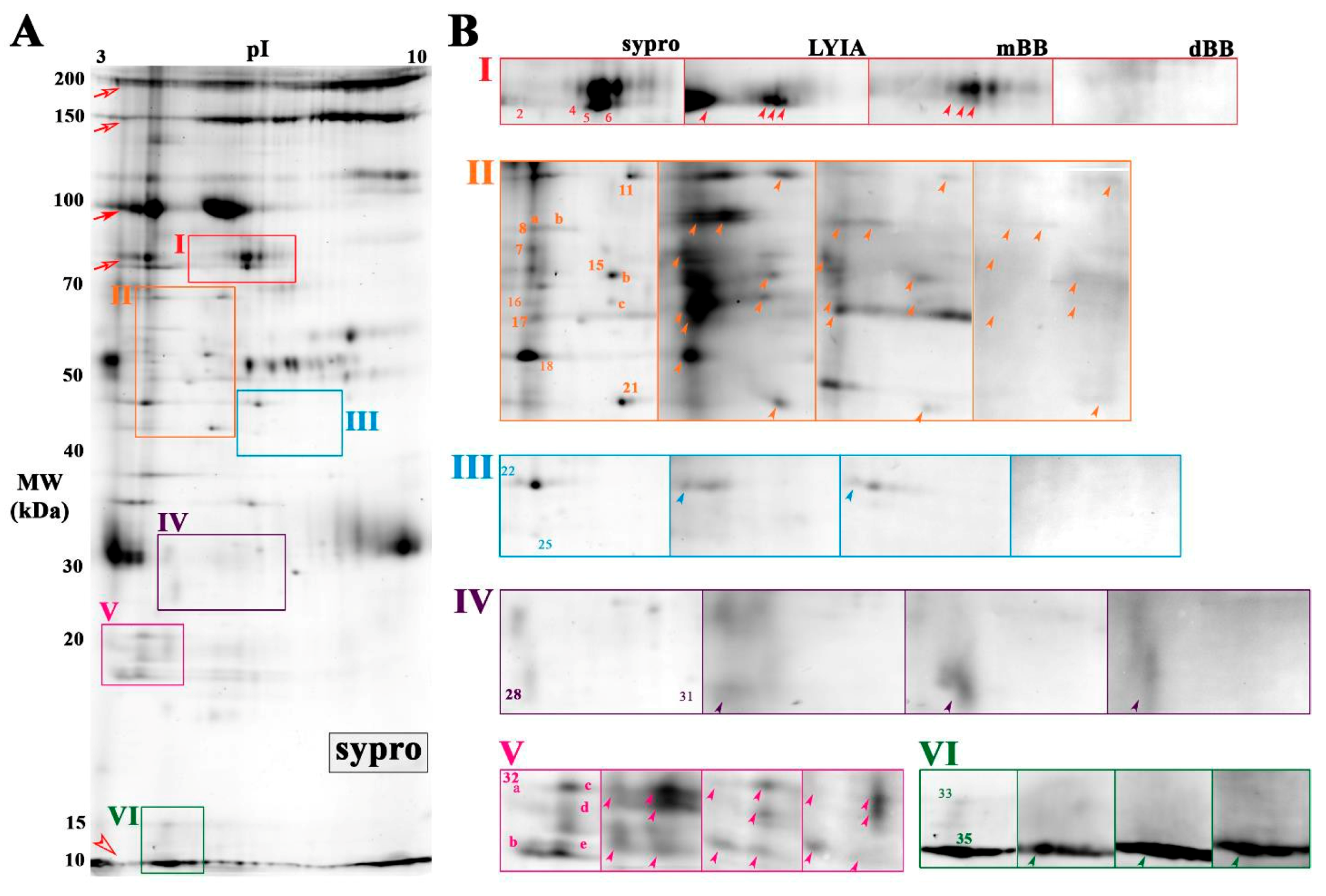
3.2. Identification of Fluorescently Labeled Candidate Proteoforms
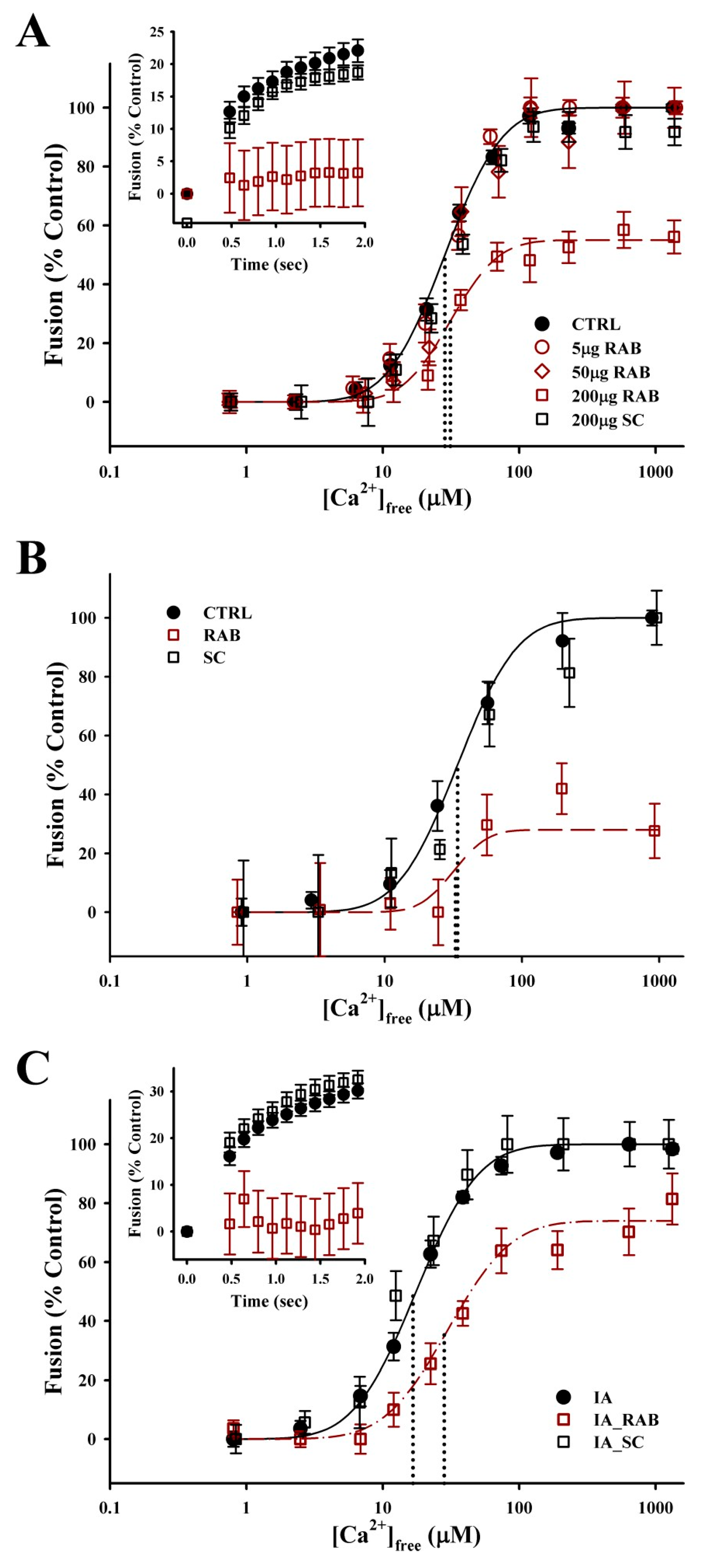
3.3. Rab-GTPases Modulate the Efficiency of Exocytosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dabral, D.; Coorssen, J.R. Arachidonic acid and lysophosphatidylcholine inhibit multiple late steps of regulated exocytosis. Biochem. Biophys. Res. Commun. 2019, 515, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Dabral, D.; Coorssen, J.R. Combined targeted Omic and Functional Assays Identify Phospholipases A(2) that Regulate Docking/Priming in Calcium-Triggered Exocytosis. Cells 2019, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- Rogasevskaia, T.P.; Coorssen, J.R. The Role of Phospholipase D in Regulated Exocytosis. J. Biol. Chem. 2015, 290, 28683–28696. [Google Scholar] [CrossRef] [PubMed]
- James, D.J.; Khodthong, C.; Kowalchyk, J.A.; Martin, T.F. Phosphatidylinositol 4,5-bisphosphate regulates SNARE-dependent membrane fusion. J. Cell Biol. 2008, 182, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Lang, T.; Bruns, D.; Wenzel, D.; Riedel, D.; Holroyd, P.; Thiele, C.; Jahn, R. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 2001, 20, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Rogasevskaia, T.P.; Churchward, M.A.; Coorssen, J.R. Anionic lipids in Ca(2+)-triggered fusion. Cell Calcium 2012, 52, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Rogasevskaia, T.P.; Coorssen, J.R. A new approach to the molecular analysis of docking, priming, and regulated membrane fusion. J. Chem. Biol. 2011, 4, 117–136. [Google Scholar] [CrossRef][Green Version]
- Abbineni, P.S.; Coorssen, J.R. Sphingolipids modulate docking, Ca(2+) sensitivity and membrane fusion of native cortical vesicles. Int. J. Biochem. Cell Biol. 2018, 104, 43–54. [Google Scholar] [CrossRef]
- Churchward, M.A.; Rogasevskaia, T.; Hofgen, J.; Bau, J.; Coorssen, J.R. Cholesterol facilitates the native mechanism of Ca2+-triggered membrane fusion. J. Cell. Sci. 2005, 118, 4833–4848. [Google Scholar] [CrossRef]
- Mahadeo, M.; Furber, K.L.; Lam, S.; Coorssen, J.R.; Prenner, E.J. Secretory vesicle cholesterol: Correlating lipid domain organization and Ca2+ triggered fusion. Biochim. Biophys. Acta 2015, 1848, 1165–1174. [Google Scholar] [CrossRef]
- Rogasevskaia, T.; Coorssen, J.R. Sphingomyelin-enriched microdomains define the efficiency of native Ca(2+)-triggered membrane fusion. J. Cell Sci. 2006, 119, 2688–2694. [Google Scholar] [CrossRef]
- Churchward, M.A.; Rogasevskaia, T.; Brandman, D.M.; Khosravani, H.; Nava, P.; Atkinson, J.K.; Coorssen, J.R. Specific lipids supply critical negative spontaneous curvature—An essential component of native Ca2+-triggered membrane fusion. Biophys. J. 2008, 94, 3976–3986. [Google Scholar] [CrossRef]
- Chernomordik, L.V.; Vogel, S.S.; Sokoloff, A.; Onaran, H.O.; Leikina, E.A.; Zimmerberg, J. Lysolipids reversibly inhibit Ca(2+)-, GTP- and pH-dependent fusion of biological membranes. FEBS Lett. 1993, 318, 71–76. [Google Scholar] [CrossRef]
- Kreutzberger, A.J.B.; Kiessling, V.; Liang, B.; Yang, S.T.; Castle, J.D.; Tamm, L.K. Asymmetric Phosphatidylethanolamine Distribution Controls Fusion Pore Lifetime and Probability. Biophys. J. 2017, 113, 1912–1915. [Google Scholar] [CrossRef]
- Burgoyne, R.D.; Morgan, A. Secretory granule exocytosis. Physiol. Rev. 2003, 83, 581–632. [Google Scholar] [CrossRef]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Gronborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brugger, B.; Ringler, P.; et al. Molecular anatomy of a trafficking organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef]
- Churchward, M.A.; Brandman, D.M.; Rogasevskaia, T.; Coorssen, J.R. Copper (II) sulfate charring for high sensitivity on-plate fluorescent detection of lipids and sterols: Quantitative analyses of the composition of functional secretory vesicles. J. Chem. Biol. 2008, 1, 79–87. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Blank, P.S.; Albertorio, F.; Bezrukov, L.; Kolosova, I.; Backlund, P.S., Jr.; Zimmerberg, J. Quantitative femto- to attomole immunodetection of regulated secretory vesicle proteins critical to exocytosis. Anal. Biochem. 2002, 307, 54–62. [Google Scholar] [CrossRef]
- Burré, J.; Beckhaus, T.; Schägger, H.; Corvey, C.; Hofmann, S.; Karas, M.; Zimmermann, H.; Volknandt, W. Analysis of the synaptic vesicle proteome using three gel-based protein separation techniques. Proteomics 2006, 6, 6250–6262. [Google Scholar] [CrossRef]
- Bennett, M.K.; Scheller, R.H. The molecular machinery for secretion is conserved from yeast to neurons. Proc. Natl. Acad. Sci. USA 1993, 90, 2559–2563. [Google Scholar] [CrossRef]
- Fasshauer, D.; Sutton, R.B.; Brunger, A.T.; Jahn, R. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc. Natl. Acad. Sci. USA 1998, 95, 15781–15786. [Google Scholar] [CrossRef]
- Abbineni, P.S.; Hibbert, J.E.; Coorssen, J.R. Critical role of cortical vesicles in dissecting regulated exocytosis: Overview of insights into fundamental molecular mechanisms. Biol. Bull. 2013, 224, 200–217. [Google Scholar] [CrossRef]
- Block, M.R.; Glick, B.S.; Wilcox, C.A.; Wieland, F.T.; Rothman, J.E. Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc. Natl. Acad. Sci. USA 1988, 85, 7852–7856. [Google Scholar] [CrossRef]
- Weidman, P.J.; Melancon, P.; Block, M.R.; Rothman, J.E. Binding of an N-ethylmaleimide-sensitive fusion protein to Golgi membranes requires both a soluble protein(s) and an integral membrane receptor. J. Cell Biol. 1989, 108, 1589–1596. [Google Scholar] [CrossRef]
- Söllner, T.; Whiteheart, S.W.; Brunner, M.; Erdjument-Bromage, H.; Geromanos, S.; Tempst, P.; Rothman, J.E. SNAP receptors implicated in vesicle targeting and fusion. Nature 1993, 362, 318–324. [Google Scholar] [CrossRef]
- Sudhof, T.C. Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron 2013, 80, 675–690. [Google Scholar] [CrossRef]
- He, E.; Wierda, K.; van Westen, R.; Broeke, J.H.; Toonen, R.F.; Cornelisse, L.N.; Verhage, M. Munc13-1 and Munc18-1 together prevent NSF-dependent de-priming of synaptic vesicles. Nat. Commun. 2017, 8, 15915. [Google Scholar] [CrossRef]
- Szule, J.A.; Coorssen, J.R. Revisiting the role of SNAREs in exocytosis and membrane fusion. Biochim. Biophys. Acta 2003, 1641, 121–135. [Google Scholar] [CrossRef][Green Version]
- Rizo, J.; Sudhof, T.C. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices—Guilty as charged? Annu. Rev. Cell Dev. Biol. 2012, 28, 279–308. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Blank, P.S.; Tahara, M.; Zimmerberg, J. Biochemical and functional studies of cortical vesicle fusion: The SNARE complex and Ca2+ sensitivity. J. Cell Biol. 1998, 143, 1845–1857. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Blank, P.S.; Albertorio, F.; Bezrukov, L.; Kolosova, I.; Chen, X.; Backlund, P.S., Jr.; Zimmerberg, J. Regulated secretion: SNARE density, vesicle fusion and calcium dependence. J. Cell Sci. 2003, 116, 2087–2097. [Google Scholar] [CrossRef]
- Szule, J.A.; Jarvis, S.E.; Hibbert, J.E.; Spafford, J.D.; Braun, J.E.; Zamponi, G.W.; Wessel, G.M.; Coorssen, J.R. Calcium-triggered membrane fusion proceeds independently of specific presynaptic proteins. J. Biol. Chem. 2003, 278, 24251–24254. [Google Scholar] [CrossRef]
- Zimmerberg, J.; Blank, P.S.; Kolosova, I.; Cho, M.S.; Tahara, M.; Coorssen, J.R. A stage-specific preparation to study the Ca(2+)-triggered fusion steps of exocytosis: Rationale and perspectives. Biochimie 2000, 82, 303–314. [Google Scholar] [CrossRef]
- Abbineni, P.S.; Wright, E.P.; Rogasevskaia, T.P.; Killingsworth, M.C.; Malladi, C.S.; Coorssen, J.R. The Sea Urchin Egg and Cortical Vesicles as Model Systems to Dissect the Fast, Ca2+-Triggered Steps of Regulated Exocytosis. Neuromethods 2014, 83. [Google Scholar] [CrossRef]
- Vogel, S.S.; Chernomordik, L.V.; Zimmerberg, J. Calcium-triggered fusion of exocytotic granules requires proteins in only one membrane. J. Biol. Chem. 1992, 267, 25640–25643. [Google Scholar]
- Whalley, T.; Timmers, K.; Coorssen, J.; Bezrukov, L.; Kingsley, D.H.; Zimmerberg, J. Membrane fusion of secretory vesicles of the sea urchin egg in the absence of NSF. J. Cell Sci. 2004, 117, 2345–2356. [Google Scholar] [CrossRef]
- Haggerty, J.G.; Jackson, R.C. Release of granule contents from sea urchin egg cortices. New assay procedures and inhibition by sulfhydryl-modifying reagents. J. Biol. Chem. 1983, 258, 1819–1825. [Google Scholar]
- Vogel, S.S.; Blank, P.S.; Zimmerberg, J. Poisson-distributed active fusion complexes underlie the control of the rate and extent of exocytosis by calcium. J. Cell Biol. 1996, 134, 329–338. [Google Scholar] [CrossRef]
- Furber, K.L.; Brandman, D.M.; Coorssen, J.R. Enhancement of the Ca(2+)-triggering steps of native membrane fusion via thiol-reactivity. J. Chem. Biol. 2009, 2, 27–37. [Google Scholar] [CrossRef][Green Version]
- Furber, K.L.; Dean, K.T.; Coorssen, J.R. Dissecting the mechanism of Ca2+-triggered membrane fusion: Probing protein function using thiol reactivity. Clin. Exp. Pharmacol. Physiol. 2010, 37, 208–217. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Yergey, A.L. Proteomics Is Analytical Chemistry: Fitness-for-Purpose in the Application of Top-Down and Bottom-Up Analyses. Proteomes 2015, 3, 440–453. [Google Scholar] [CrossRef]
- Padula, M.P.; Berry, I.J.; MB, O.R.; Raymond, B.B.; Santos, J.; Djordjevic, S.P. A Comprehensive Guide for Performing Sample Preparation and Top-Down Protein Analysis. Proteomes 2017, 5, 11. [Google Scholar] [CrossRef]
- Zhan, X.; Yang, H.; Peng, F.; Li, J.; Mu, Y.; Long, Y.; Cheng, T.; Huang, Y.; Li, Z.; Lu, M.; et al. How many proteins can be identified in a 2DE gel spot within an analysis of a complex human cancer tissue proteome? Electrophoresis 2018, 39, 965–980. [Google Scholar] [CrossRef]
- Thiede, B.; Koehler, C.J.; Strozynski, M.; Treumann, A.; Stein, R.; Zimny-Arndt, U.; Schmid, M.; Jungblut, P.R. High resolution quantitative proteomics of HeLa cells protein species using stable isotope labeling with amino acids in cell culture (SILAC), two-dimensional gel electrophoresis (2DE) and nano-liquid chromatograpohy coupled to an LTQ-OrbitrapMass spectrometer. Mol. Cell Proteomics 2013, 12, 529–538. [Google Scholar] [CrossRef]
- Furber, K.L.; Churchward, M.A.; Rogasevskaia, T.P.; Coorssen, J.R. Identifying critical components of native Ca2+-triggered membrane fusion. Integrating studies of proteins and lipids. Ann. N. Y. Acad. Sci. 2009, 1152, 121–134. [Google Scholar] [CrossRef]
- Churchward, M.A.; Coorssen, J.R. Cholesterol, regulated exocytosis and the physiological fusion machine. Biochem. J. 2009, 423, 1–14. [Google Scholar] [CrossRef]
- Conner, S.; Wessel, G.M. rab3 mediates cortical granule exocytosis in the sea urchin egg. Dev. Biol. 1998, 203, 334–344. [Google Scholar] [CrossRef][Green Version]
- Blank, P.S.; Cho, M.S.; Vogel, S.S.; Kaplan, D.; Kang, A.; Malley, J.; Zimmerberg, J. Submaximal responses in calcium-triggered exocytosis are explained by differences in the calcium sensitivity of individual secretory vesicles. J. Gen. Physiol. 1998, 112, 559–567. [Google Scholar] [CrossRef]
- Hibbert, J.E.; Butt, R.H.; Coorssen, J.R. Actin is not an essential component in the mechanism of calcium-triggered vesicle fusion. Int. J. Biochem. Cell Biol. 2006, 38, 461–471. [Google Scholar] [CrossRef]
- Butt, R.H.; Coorssen, J.R. Postfractionation for enhanced proteomic analyses: Routine electrophoretic methods increase the resolution of standard 2D-PAGE. J. Proteome Res. 2005, 4, 982–991. [Google Scholar] [CrossRef]
- Jiang, X.S.; Backlund, P.S.; Wassif, C.A.; Yergey, A.L.; Porter, F.D. Quantitative proteomics analysis of inborn errors of cholesterol synthesis: Identification of altered metabolic pathways in DHCR7 and SC5D deficiency. Mol. Cell Proteomics 2010, 9, 1461–1475. [Google Scholar] [CrossRef] [PubMed]
- Hellman, U.; Wernstedt, C.; Gonez, J.; Heldin, C.H. Improvement of an “In-Gel” digestion procedure for the micropreparation of internal protein fragments for amino acid sequencing. Anal. Biochem. 1995, 224, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Vacquier, V.D. The isolation of intact cortical granules from sea urchin eggs: Calcium ions trigger granule discharge. Dev. Biol. 1975, 43, 62–74. [Google Scholar] [CrossRef]
- Decker, S.J.; Kinsey, W.H. Characterization of cortical secretory vesicles from the sea urchin egg. Dev. Biol. 1983, 96, 37–45. [Google Scholar] [CrossRef]
- Roux, M.M.; Radeke, M.J.; Goel, M.; Mushegian, A.; Foltz, K.R. 2DE identification of proteins exhibiting turnover and phosphorylation dynamics during sea urchin egg activation. Dev. Biol. 2008, 313, 630–647. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sodergren, E.; Weinstock, G.M.; Davidson, E.H.; Cameron, R.A.; Gibbs, R.A.; Angerer, R.C.; Coffman, J.A. The genome of the sea urchin Strongylocentrotus purpuratus. Science 2006, 314, 941–952. [Google Scholar] [CrossRef]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Oliveira, B.M.; Coorssen, J.R.; Martins-de-Souza, D. 2DE: The phoenix of proteomics. J. Proteomics 2014, 104, 140–150. [Google Scholar] [CrossRef]
- Naryzhny, S. Inventory of proteoforms as a current challenge of proteomics: Some technical aspects. J. Proteomics 2019, 191, 22–28. [Google Scholar] [CrossRef]
- Pfeffer, S.R. Rab GTPases: Master regulators that establish the secretory and endocytic pathways. Mol. Biol. Cell 2017, 28, 712–715. [Google Scholar] [CrossRef]
- Trifaró, J.M.; Gasman, S.; Gutiérrez, L.M. Cytoskeletal control of vesicle transport and exocytosis in chromaffin cells. Acta Physiol. 2008, 192, 165–172. [Google Scholar] [CrossRef]
- Li, P.; Bademosi, A.T.; Luo, J.; Meunier, F.A. Actin Remodeling in Regulated Exocytosis: Toward a Mesoscopic View. Trends Cell Biol. 2018, 28, 685–697. [Google Scholar] [CrossRef]
- Papadopulos, A. Membrane shaping by actin and myosin during regulated exocytosis. Mol. Cell Neurosci. 2017, 84, 93–99. [Google Scholar] [CrossRef][Green Version]
- Noordstra, I.; Akhmanova, A. Linking cortical microtubule attachment and exocytosis. F1000 Res. 2017, 6, 469. [Google Scholar] [CrossRef]
- Ñeco, P.; Giner, D.; del Mar Francés, M.; Viniegra, S.; Gutiérrez, L.M. Differential participation of actin- and tubulin-based vesicle transport systems during secretion in bovine chromaffin cells. Eur. J. Neurosci. 2003, 18, 733–742. [Google Scholar] [CrossRef]
- Trifaró, J.M.; Rodríguez del Castillo, A.; Vitale, M.L. Dynamic changes in chromaffin cell cytoskeleton as prelude to exocytosis. Mol. Neurobiol. 1992, 6, 339–358. [Google Scholar] [CrossRef]
- Tran, D.T.; Ten Hagen, K.G. Real-time insights into regulated exocytosis. J. Cell Sci. 2017, 130, 1355–1363. [Google Scholar] [CrossRef]
- Wollman, R.; Meyer, T. Coordinated oscillations in cortical actin and Ca2+ correlate with cycles of vesicle secretion. Nat. Cell Biol. 2012, 14, 1261–1269. [Google Scholar] [CrossRef]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Fukuda, M. Regulation of secretory vesicle traffic by Rab small GTPases. Cell Mol. Life Sci. 2008, 65, 2801–2813. [Google Scholar] [CrossRef]
- Grosshans, B.L.; Ortiz, D.; Novick, P. Rabs and their effectors: Achieving specificity in membrane traffic. Proc. Natl. Acad. Sci. USA 2006, 103, 11821–11827. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.Y.; Lamer, S.; Schümann, M.; Schmidt, M.R.; Krause, E.; Haucke, V. Quantitative proteomics analysis of detergent-resistant membranes from chemical synapses: Evidence for cholesterol as spatial organizer of synaptic vesicle cycling. Mol. Cell. Proteomics 2006, 5, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Brunner, Y.; Couté, Y.; Iezzi, M.; Foti, M.; Fukuda, M.; Hochstrasser, D.F.; Wollheim, C.B.; Sanchez, J.C. Proteomics analysis of insulin secretory granules. Mol. Cell. Proteomics 2007, 6, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Pavlos, N.J.; Grønborg, M.; Riedel, D.; Chua, J.J.; Boyken, J.; Kloepper, T.H.; Urlaub, H.; Rizzoli, S.O.; Jahn, R. Quantitative analysis of synaptic vesicle Rabs uncovers distinct yet overlapping roles for Rab3a and Rab27b in Ca2+-triggered exocytosis. J. Neurosci. 2010, 30, 13441–13453. [Google Scholar] [CrossRef]
- Geppert, M.; Goda, Y.; Stevens, C.F.; Südhof, T.C. The small GTP-binding protein Rab3A regulates a late step in synaptic vesicle fusion. Nature 1997, 387, 810–814. [Google Scholar] [CrossRef]
- Holz, R.W.; Brondyk, W.H.; Senter, R.A.; Kuizon, L.; Macara, I.G. Evidence for the involvement of Rab3A in Ca(2+)-dependent exocytosis from adrenal chromaffin cells. J. Biol. Chem. 1994, 269, 10229–10234. [Google Scholar] [PubMed]
- Johannes, L.; Lledo, P.M.; Roa, M.; Vincent, J.D.; Henry, J.P.; Darchen, F. The GTPase Rab3a negatively controls calcium-dependent exocytosis in neuroendocrine cells. EMBO J. 1994, 13, 2029–2037. [Google Scholar] [CrossRef]
- Khvotchev, M.V.; Ren, M.; Takamori, S.; Jahn, R.; Südhof, T.C. Divergent functions of neuronal Rab11b in Ca2+-regulated versus constitutive exocytosis. J. Neurosci. 2003, 23, 10531–10539. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, K.; Shibasaki, T.; Mizoguchi, A.; Saito, T.; Seino, S. Rab11 and its effector Rip11 participate in regulation of insulin granule exocytosis. Genes Cells 2009, 14, 445–456. [Google Scholar] [CrossRef]
- Shirakawa, R.; Yoshioka, A.; Horiuchi, H.; Nishioka, H.; Tabuchi, A.; Kita, T. Small GTPase Rab4 regulates Ca2+-induced alpha-granule secretion in platelets. J. Biol. Chem. 2000, 275, 33844–33849. [Google Scholar] [CrossRef]
- Graham, M.E.; Handley, M.T.; Barclay, J.W.; Ciufo, L.F.; Barrow, S.L.; Morgan, A.; Burgoyne, R.D. A gain-of-function mutant of Munc18-1 stimulates secretory granule recruitment and exocytosis and reveals a direct interaction of Munc18-1 with Rab3. Biochem. J. 2008, 409, 407–416. [Google Scholar] [CrossRef]
- Johnson, J.L.; He, J.; Ramadass, M.; Pestonjamasp, K.; Kiosses, W.B.; Zhang, J.; Catz, S.D. Munc13-4 Is a Rab11-binding Protein That Regulates Rab11-positive Vesicle Trafficking and Docking at the Plasma Membrane. J. Biol. Chem. 2016, 291, 3423–3438. [Google Scholar] [CrossRef]
- Neeft, M.; Wieffer, M.; de Jong, A.S.; Negroiu, G.; Metz, C.H.; van Loon, A.; Griffith, J.; Krijgsveld, J.; Wulffraat, N.; Koch, H.; et al. Munc13-4 is an effector of rab27a and controls secretion of lysosomes in hematopoietic cells. Mol. Biol. Cell 2005, 16, 731–741. [Google Scholar] [CrossRef]
- Tsuboi, T.; Fukuda, M. The C2B domain of rabphilin directly interacts with SNAP-25 and regulates the docking step of dense core vesicle exocytosis in PC12 cells. J. Biol. Chem. 2005, 280, 39253–39259. [Google Scholar] [CrossRef]
- Ferrer-Orta, C.; Perez-Sanchez, M.D.; Coronado-Parra, T.; Silva, C.; Lopez-Martinez, D.; Baltanas-Copado, J.; Gomez-Fernandez, J.C.; Corbalan-Garcia, S.; Verdaguer, N. Structural characterization of the Rabphilin-3A-SNAP25 interaction. Proc. Natl. Acad. Sci. USA 2017, 114, E5343–E5351. [Google Scholar] [CrossRef]
- Gomi, H.; Mizutani, S.; Kasai, K.; Itohara, S.; Izumi, T. Granuphilin molecularly docks insulin granules to the fusion machinery. J. Cell Biol. 2005, 171, 99–109. [Google Scholar] [CrossRef]
- Torii, S.; Takeuchi, T.; Nagamatsu, S.; Izumi, T. Rab27 effector granuphilin promotes the plasma membrane targeting of insulin granules via interaction with syntaxin 1a. J. Biol. Chem. 2004, 279, 22532–22538. [Google Scholar] [CrossRef]
- Deak, F.; Shin, O.H.; Tang, J.; Hanson, P.; Ubach, J.; Jahn, R.; Rizo, J.; Kavalali, E.T.; Sudhof, T.C. Rabphilin regulates SNARE-dependent re-priming of synaptic vesicles for fusion. EMBO J. 2006, 25, 2856–2866. [Google Scholar] [CrossRef]
- Shin, H.W.; Hayashi, M.; Christoforidis, S.; Lacas-Gervais, S.; Hoepfner, S.; Wenk, M.R.; Modregger, J.; Uttenweiler-Joseph, S.; Wilm, M.; Nystuen, A.; et al. An enzymatic cascade of Rab5 effectors regulates phosphoinositide turnover in the endocytic pathway. J. Cell Biol. 2005, 170, 607–618. [Google Scholar] [CrossRef]
- Christoforidis, S.; Miaczynska, M.; Ashman, K.; Wilm, M.; Zhao, L.; Yip, S.; Waterfield, M.D.; Backer, J.M.; Zerial, M. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat. Cell Biol. 1999, 1, 249–252. [Google Scholar] [CrossRef]
- Martin, T.F. PI(4,5)P2-binding effector proteins for vesicle exocytosis. Biochim. Biophys. Acta 2015, 1851, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Deeney, J.T.; Gromada, J.; Høy, M.; Olsen, H.L.; Rhodes, C.J.; Prentki, M.; Berggren, P.O.; Corkey, B.E. Acute stimulation with long chain acyl-CoA enhances exocytosis in insulin-secreting cells (HIT T-15 and NMRI beta-cells). J. Biol. Chem. 2000, 275, 9363–9368. [Google Scholar] [CrossRef] [PubMed]
- Zinsmaier, K.E.; Bronk, P. Molecular chaperones and the regulation of neurotransmitter exocytosis. Biochem. Pharmacol. 2001, 62, 1–11. [Google Scholar] [CrossRef]
- Gorenberg, E.L.; Chandra, S.S. The Role of Co-chaperones in Synaptic Proteostasis and Neurodegenerative Disease. Front. Neurosci. 2017, 11, 248. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spot # | Accession # | Protein Description | Organism | Total CVmem | Chol- CVmem | Theoretical | Experimental c | Biological Function d,e | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mascot b | Peptides | Mascot b | Peptides | pI | MW | pI | MW | |||||
| 1 | unidentified | n/s | - | 5.3 | 85.4 | |||||||
| 2 | XP_011662952.1 | succinate dehydrogenase, flavoprotein subunit a | S. purpuratus | 299 | 5 | 5.15 | 61.5 | 5.3 | 84.9 | metabolic process | ||
| 3 | XP_011662952.1 | succinate dehydrogenase, flavoprotein subunit a | S. purpuratus | 137 | 3 | 5.15 | 61.5 | 5.4 | 84.9 | metabolic process | ||
| 4 | unidentified | n/s | - | 5.7 | 84.2 | |||||||
| 5 | unidentified | n/s | - | 5.8 | 83.0 | |||||||
| 6 | XP_779941.1 | long-chain-fatty-acid--CoA ligase 1 a | S. purpuratus | 201 | 3 | 195 | 6 | 5.58 | 73.5 | 5.9 | 82.4 | metabolic process |
| 7 | NP_999643.1 | calreticulin precursor | S. purpuratus | 303 | 7 | 116 | 2 | 4.57 | 95.5 | 3.9 | 65.0 | protein folding |
| XP_782447.2 | dihydrolipoyl dehydrogenase a | S. purpuratus | 111 | 2 | 7.10 | 101.3 | metabolic process | |||||
| 8 | NP_999697.1 | S. purpuratus | 257 | 3 | 4.38 | 54.9 | 4.1 | 68.1 | redox/protein folding | |||
| 8a | NP_999697.1 | ER calcistorin precursor | S. purpuratus | 126 | 3 | 4.38 | 54.9 | 4.7 | 71.1 | redox/protein folding | ||
| XP_795205.2 | 60 kDa heat shock protein a | S. purpuratus | 119 | 3 | 5.24 | 62.2 | protein folding | |||||
| XP_011677933.1 | disulfide-isomerase 2-like a | S. purpuratus | 114 | 3 | 5.14 | 45.8 | redox/protein folding | |||||
| 8b | NP_999697.1 | ER calcistorin precursor | S. purpuratus | 220 | 6 | 4.57 | 95.5 | 5.0 | 71.1 | redox/protein folding | ||
| XP_779941.1 | long-chain-fatty-acid--CoA ligase 1 a | S. purpuratus | 60 | 2 | 5.58 | 73. 5 | metabolic process | |||||
| 9 | XP_795205.2 | 60 kDa heat shock protein a | S. purpuratus | 685 | 9 | 5.24 | 62.2 | 4.9 | 70.4 | protein folding | ||
| 10 | XP_795205.2 | 60 kDa heat shock protein a | S. purpuratus | 612 | 7 | 5.24 | 62.2 | 5.0 | 70.7 | protein folding | ||
| 11 | XP_003726658.1 | V-type proton ATPase subunit A isoform X4 a | S. purpuratus | 131 | 3 | 561 | 16 | 5.32 | 67.9 | 5.4 | 75.5 | ion transport |
| 12 | XP_003726658.1 | V-type proton ATPase subunit A isoform X4 a | S. purpuratus | 183 | 3 | 5.32 | 67.9 | 5.5 | 75.3 | ion transport | ||
| 13 | unidentified | n/s | - | 5.5 | 71.5 | |||||||
| 14 | XP_791790.1 | tubulin beta chain a | S. purpuratus | 443 | 9 | 4.73 | 50.1 | 4.8 | 62.1 | cellular organization | ||
| 15 | XP_789821.1 | tubulin beta chain a | S. purpuratus | 172 | 4 | 4.61 | 50.1 | 5.0 | 61.8 | cellular organization | ||
| XP_003731782.2 | aldehyde dehydrogenase a | S. purpuratus | 597 | 13 | 5.32 | 41.2 | metabolic process | |||||
| 15b | XP_011662394.1 | V-type proton ATPase subunit B isoform X1 a | S. purpuratus | 341 | 8 | 5.21 | 55.1 | 5.5 | 64.6 | ion transport | ||
| AFG26286.1 | aldolase class-1 protein | A. japonicus | 82 | 2 | 8.30 | 39.4 | metabolic process | |||||
| 15c | XP_011661972.1 | protein disulfide-isomerase A3 a | S. purpuratus | 184 | 6 | 5.44 | 65.5 | 5.5 | 60.8 | redox/protein folding | ||
| 16 | n/s | - | 4.6 | 57.3 | ||||||||
| 17 | NP_001116974.1 | ATP synthase beta subunit | S. purpuratus | 1084 | 13 | 5.14 | 56.0 | 4.6 | 55.1 | metabolic process | ||
| Q25117.1 | ATP synthase subunit beta | H. pulcherrimus | 249 | 6 | 5.10 | 56.1 | metabolic process | |||||
| XP_786080.3 | arginine kinase a | S. purpuratus | 76 | 2 | 5.26 | 46.5 | signal transduction | |||||
| 18 | XP_786753.1 | succinyl-CoA ligase subunit beta a | S. purpuratus | 213 | 4 | 5.16 | 49.1 | 4.7 | 49.3 | metabolic process | ||
| 19 | unidentified | n/s | - | 5.1 | 48.3 | |||||||
| 20 | XP_782503.3 | long-chain specific acyl-CoA dehydrogenase, isoform X1 a | S. purpuratus | 130 | 5 | 5.67 | 48.9 | 5.2 | 47.7 | metabolic process | ||
| 21 | XP_003725373.1 | actin, cytoskeletal 3 B a | S. purpuratus | 112 | 3 | 5.22 | 41.8 | 5.3 | 46.9 | cellular organization | ||
| 22 | XP_786922.3 | NADH dehydrogenase, iron-sulfur protein 2 a | S. purpuratus | 123 | 3 | 6.0 | 52.7 | 5.8 | 51.9 | metabolic process | ||
| 23 | XP_788937.1 | elongation factor Tu a | S. purpuratus | 333 | 5 | 6.3 | 50.0 | 5.9 | 49.3 | protein biosynthesis | ||
| /protein folding | ||||||||||||
| 24 | XP_011665962.1 | isocitrate dehydrogenase [NADP], isoform X1 a | S. purpuratus | 112 | 4 | 6.2 | 50.2 | 5.9 | 45.7 | metabolic process | ||
| 25 | XP_011665962.1 | isocitrate dehydrogenase [NADP], isoform X1 a | S. purpuratus | 305 | 5 | 6.2 | 50.2 | 6.0 | 45.2 | metabolic process | ||
| 26 | XP_011665962.1 | isocitrate dehydrogenase [NADP], isoform X1 a | S. purpuratus | 120 | 4 | 6.2 | 50.2 | 6.1 | 45.2 | metabolic process | ||
| 27 | XP_789891.3 | cytochrome b-c1 complex subunit 2 a | S. purpuratus | 76 | 3 | 8.7 | 50.8 | 6.8 | 45.5 | metabolic process | ||
| 28 | AAG15425.1 | 34kDa cortical vesicle protein, partial | S. purpuratus | 158 | 2 | 5.2 | 30.7 | 4.2 | 26.0 | uncharacterized | ||
| 29 | unidentified | n/s | - | 5.3 | 25.8 | |||||||
| 30 | XP_003725895.1 | electron transfer flavoprotein subunit beta isoform X1 a | S. purpuratus | 174 | 5 | 5.5 | 27.8 | 5.5 | 27.6 | metabolic process | ||
| 31 | XP_780266.1 | voltage-dependent anion-selective channel protein 2 a | S. purpuratus | 227 | 3 | 6.3 | 30.4 | 6.0 | 27.6 | ion transport | ||
| 32 | unidentified | n/s | - | 3.2 | 21.8 | |||||||
| 32a | NP_001116984.1 | rab11 GTPase homolog SUrab11 | S. purpuratus | 107 | 3 | 6.1 | 24.5 | 3.6 | 22.2 | vesicular trafficking | ||
| 32b | NP_001116983.1 | rab7 GTPase homolog SUrab7 | S. purpuratus | 166 | 4 | 5.5 | 23.1 | 3.5 | 20.3 | vesicular trafficking | ||
| XP_783878.1 | ras-related protein Rab-5B a | S. purpuratus | 127 | 4 | 8.3 | 23.6 | vesicular trafficking | |||||
| XP_001201172.2 | ADP-ribosylation factor-like protein 8B-A a | S. purpuratus | 120 | 3 | 6.8 | 21.6 | vesicular trafficking | |||||
| 32c | XP_782537.1 | ras-related protein Rab-2A isoform X1 a | S. purpuratus | 91 | 2 | 6.2 | 23.7 | 4.6 | 23.3 | vesicular trafficking | ||
| 32d | n/s | - | 4.5 | 22.1 | ||||||||
| 32e | NP_001116983.1 | rab7 GTPase homolog SUrab7 | S. purpuratus | 234 | 6 | 5.5 | 23.1 | 4.4 | 20.3 | vesicular trafficking | ||
| PSN32081.1 | ras-related protein Rab-2A | B.germanica | 116 | 3 | 6.0 | 23.6 | vesicular trafficking | |||||
| 33 | NP_999641.1 | 18kDa egg cortical vesicle protein precursor | S. purpuratus | 208 | 2 | 4.8 | 20.6 | 4.1 | 12.7 | uncharacterized | ||
| 34 | NP_999641.1 | 18kDa egg cortical vesicle protein precursor cytochrome c oxidase subunit 5A a | S. purpuratus | 248 | 3 | 4.8 | 20.6 | 4.5 | 12.6 | uncharacterized | ||
| XP_784558.1 | S. purpuratus | 167 | 2 | 5.3 | 16.9 | metabolic process | ||||||
| 35 | ABO26625.1 | Ras-related protein Rab-1A | H. discus discus | 73 | 3 | 5.6 | 22.8 | 4.2 | 10.4 | vesicular trafficking | ||
| XP_015905011.1 | ras-related protein Rab-33B | P.tepidariorum | 72 | 2 | 7.6 | 25.6 | vesicular trafficking | |||||
| XP_020901910.1 | ras-related protein Rab-13 | E. pallida | 82 | 2 | 7.6 | 24.3 | vesicular trafficking | |||||
| 36 | XP_786378.1 | cytochrome c oxidase subunit 5B a | S. purpuratus | 231 | 5 | 8.3 | 14.2 | 6.8 | 11.4 | metabolic process | ||
| 37 | XP_794003.1 | succinate dehydrogenase, iron-sulfur subunit a | S. purpuratus | 145 | 4 | 8.8 | 31.8 | 9.7 | 24.8 | metabolic process | ||
| G2-Box/RabF1 Motif | S. purparatusa | H. sapiensb | ||
|---|---|---|---|---|
| % Identity | % Similarity | % Identity | ||
| Rab3 | TVGIDF | 100 | 100 | 100 |
| Rab2 | TIGVEF | 50 | 100 | 100 |
| Rab5 | TIGAAF | 50 | 67 | 100 |
| Rab7 | TIGADF | 67 | 83 | 100 |
| Rab11 | TIGVEF | 50 | 100 | 100 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furber, K.L.; Backlund, P.S.; Yergey, A.L.; Coorssen, J.R. Unbiased Thiol-Labeling and Top-Down Proteomic Analyses Implicate Multiple Proteins in the Late Steps of Regulated Secretion. Proteomes 2019, 7, 34. https://doi.org/10.3390/proteomes7040034
Furber KL, Backlund PS, Yergey AL, Coorssen JR. Unbiased Thiol-Labeling and Top-Down Proteomic Analyses Implicate Multiple Proteins in the Late Steps of Regulated Secretion. Proteomes. 2019; 7(4):34. https://doi.org/10.3390/proteomes7040034
Chicago/Turabian StyleFurber, Kendra L., Peter S. Backlund, Alfred L. Yergey, and Jens R. Coorssen. 2019. "Unbiased Thiol-Labeling and Top-Down Proteomic Analyses Implicate Multiple Proteins in the Late Steps of Regulated Secretion" Proteomes 7, no. 4: 34. https://doi.org/10.3390/proteomes7040034
APA StyleFurber, K. L., Backlund, P. S., Yergey, A. L., & Coorssen, J. R. (2019). Unbiased Thiol-Labeling and Top-Down Proteomic Analyses Implicate Multiple Proteins in the Late Steps of Regulated Secretion. Proteomes, 7(4), 34. https://doi.org/10.3390/proteomes7040034