Top-Down Proteomics of Medicinal Cannabis



Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Standards
2.1.1. Standard List
2.1.2. Standard Preparation
2.2. Plant Materials
2.2.1. Cannabis Sampling and Grinding
2.2.2. Cannabis Protein Extraction
2.2.3. Protein Assay and Cannabis Protein Alkylation
2.2.4. Cannabis Protein Desalting and Evaporation
2.3. Mass Spectrometry Analyses
2.3.1. Infusion of Protein Standards and Analyses by Mass Spectrometry
2.3.2. Separation of Cannabis Intact Proteins by Ultra-Performance Liquid Chromatography (UPLC)
2.3.3. Analyses of Cannabis Intact Protein Extracts using Mass Spectrometry online with UPLC
Full Scan FTMS1
FTMS2
2.4. Data Files Analysis
2.4.1. Analysis of Infusion MS/MS Spectra
Manual Annotations of Standards
Automatic Annotations of Standards
2.4.2. Analysis of LC-MS and LC-MS/MS Data from Cannabis Samples
Statistical Analyses of Cannabis Samples
Identifications of Cannabis Protein by Mascot
3. Results and Discussion
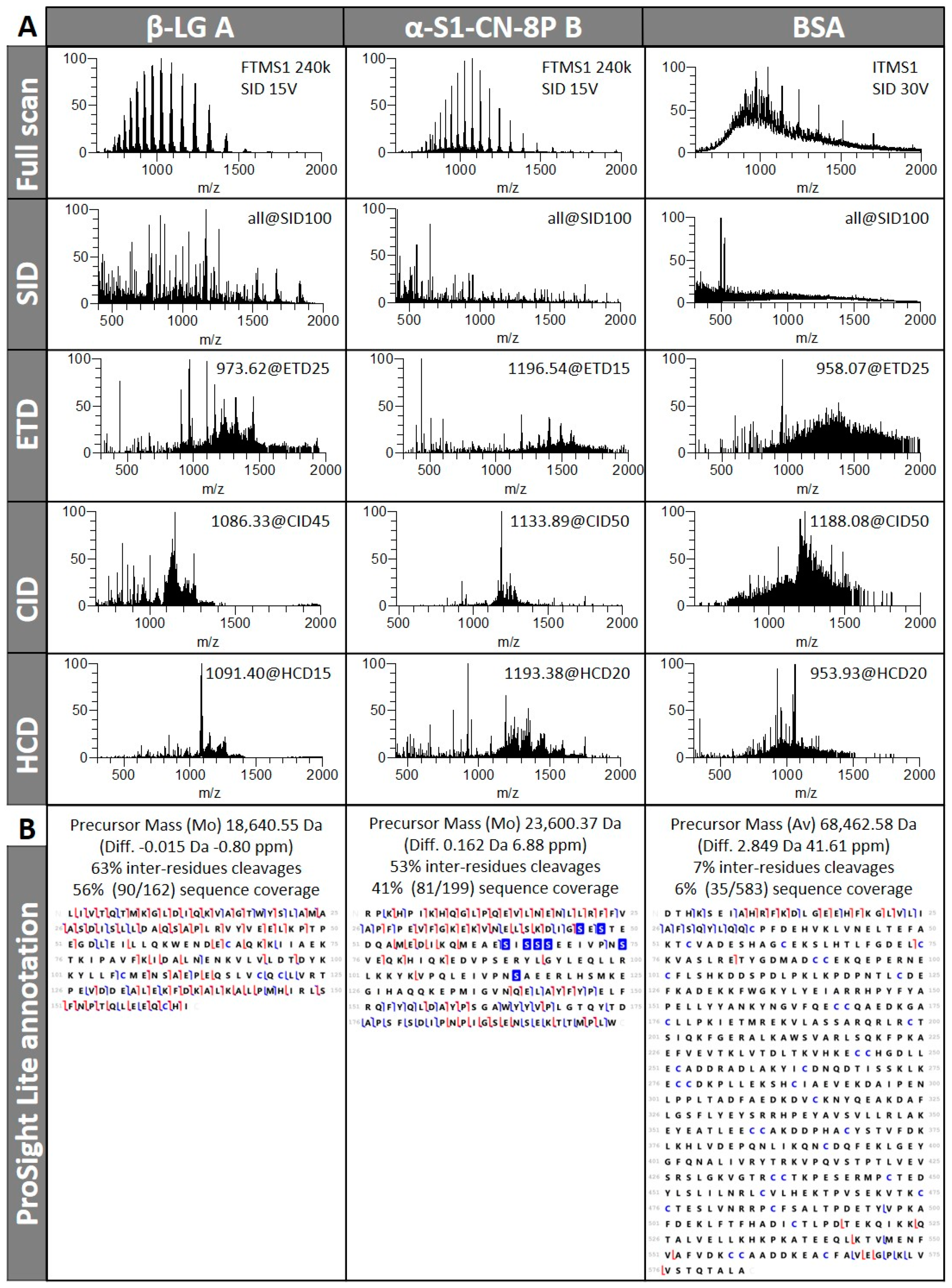
3.1. TDS of Infused Protein Standards
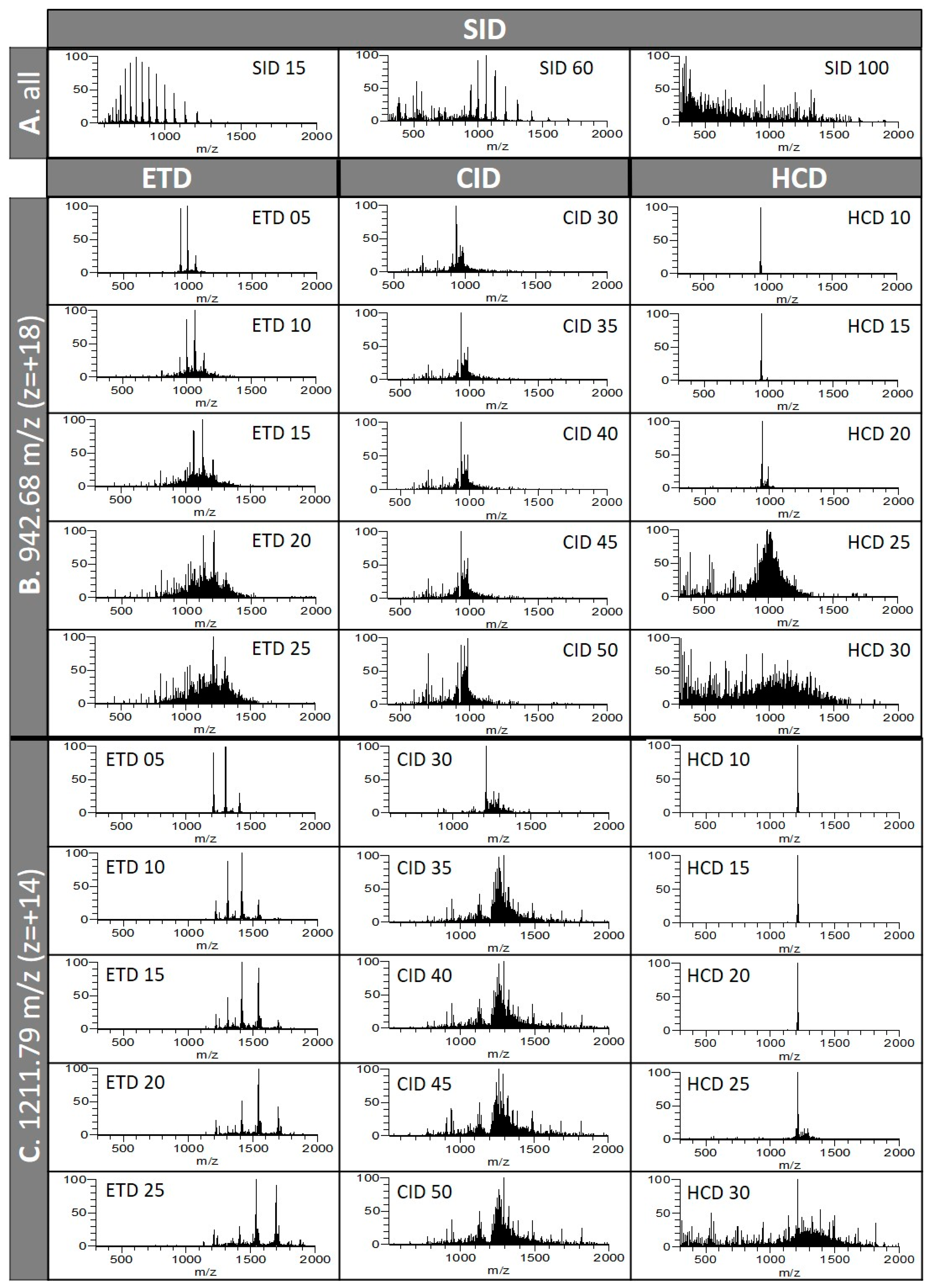
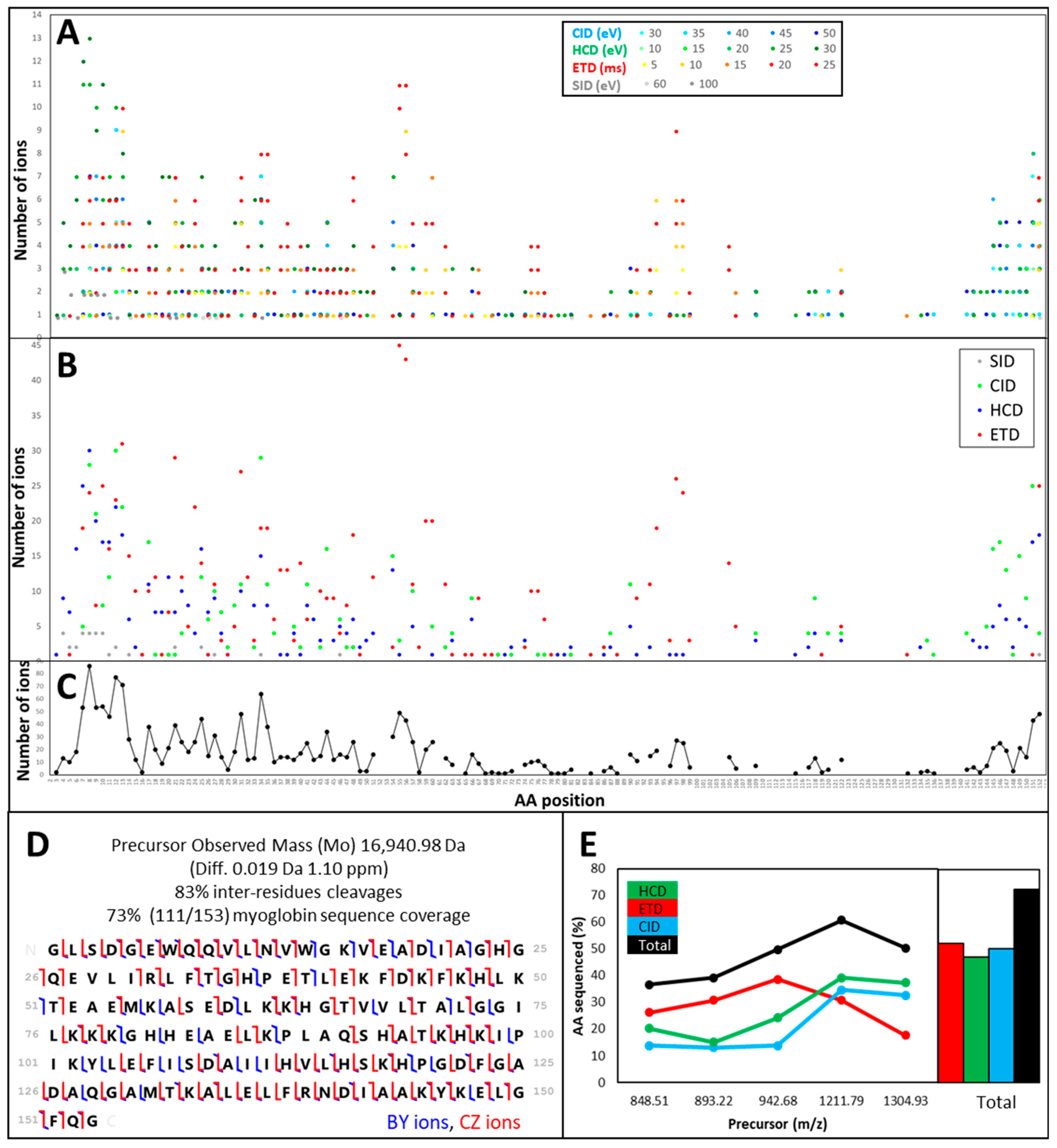
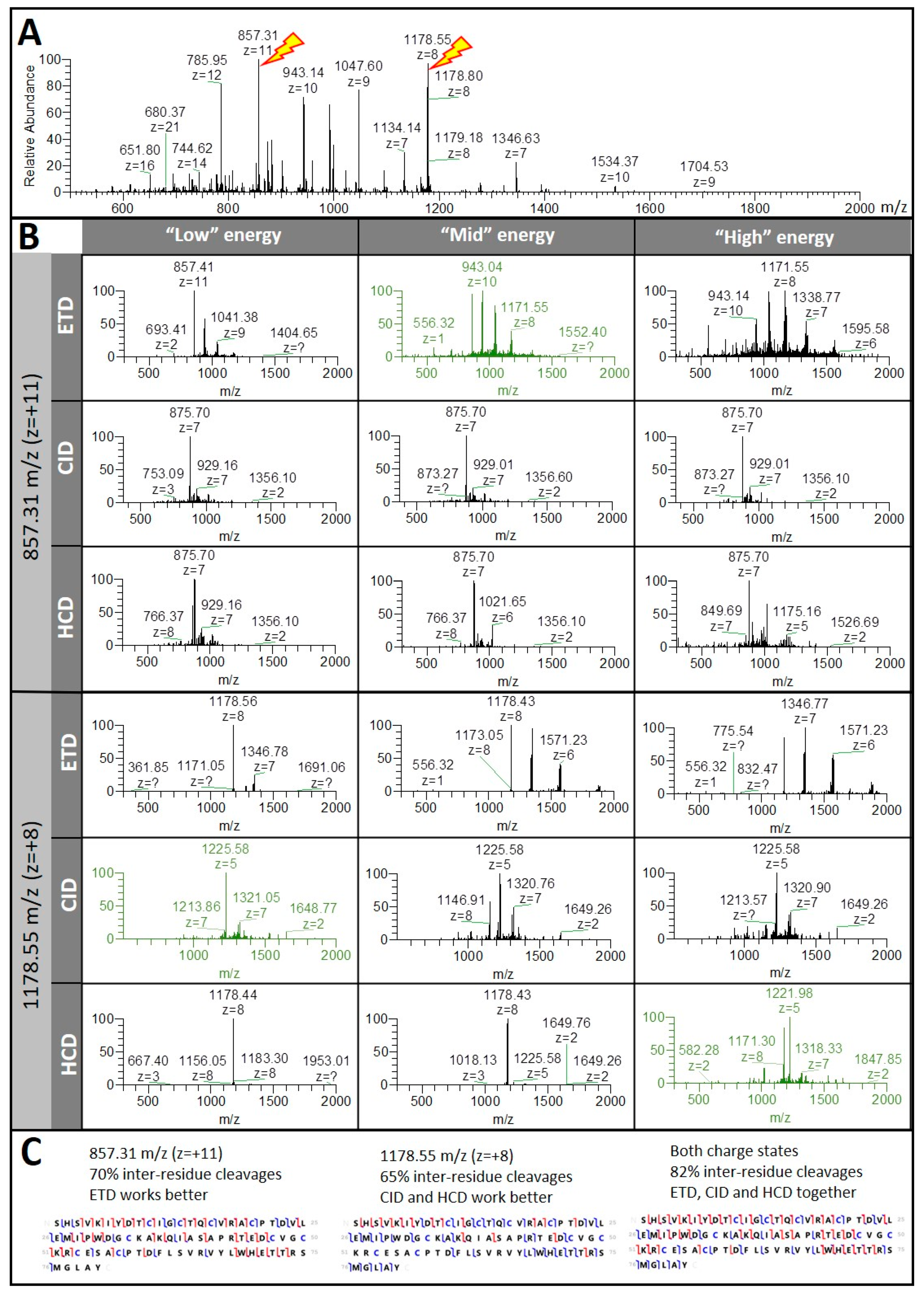
3.1.1. Different Fragmentation Modes Produce Different Spectral Patterns in a Precursor-Dependent Manner
3.1.2. The Central Part of a Protein is Difficult to Fragment, Therefore Recalcitrant to Top-Down Sequencing
3.1.3. Fragmentation Efficiency Varies from Protein to Protein in a Size-Dependent Fashion
3.1.4. Automatic Workflow Success Depends on Database Searched and Tolerance Parameters
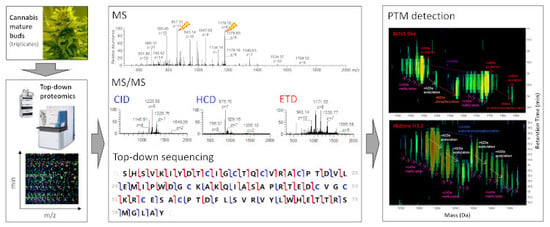
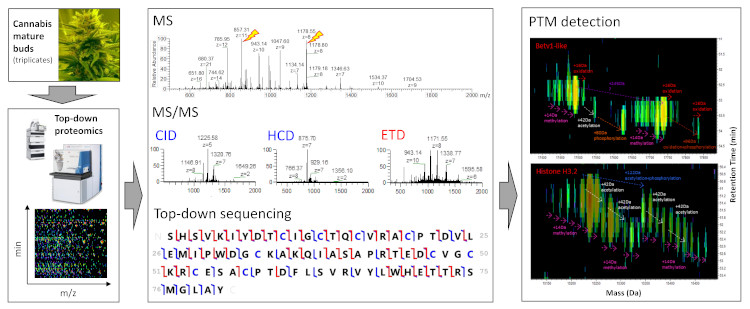
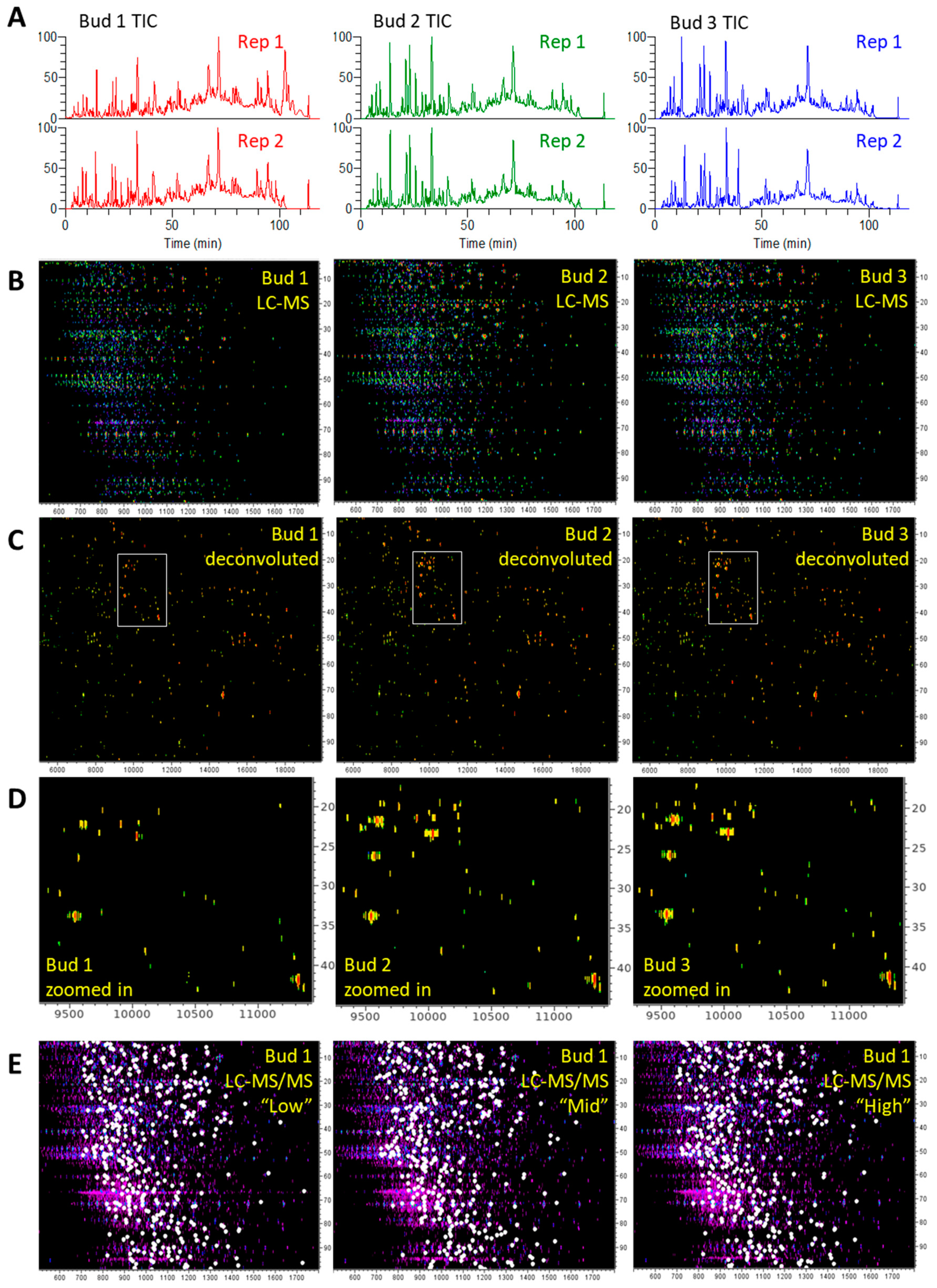
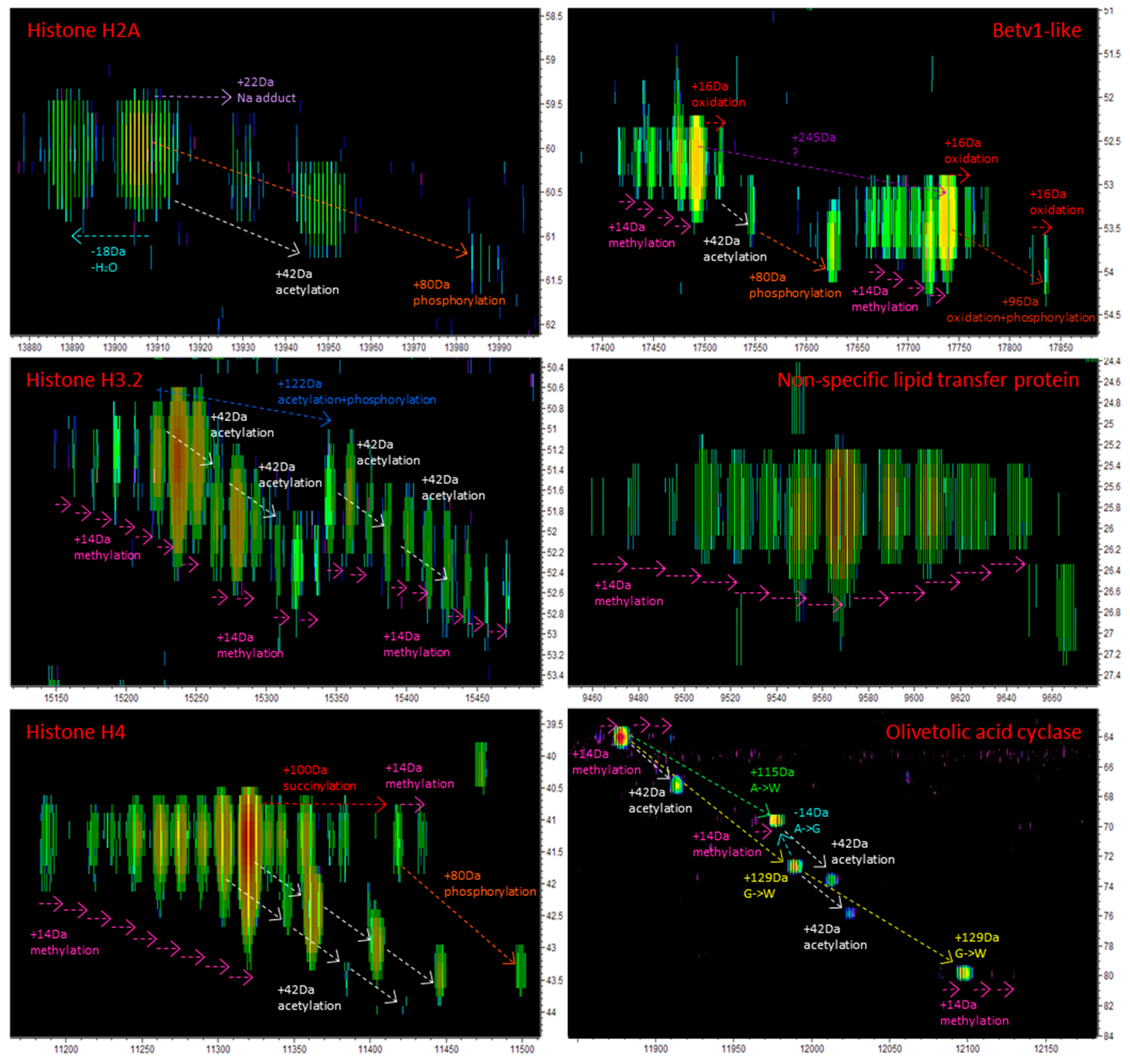
3.2. TDS of Cannabis Proteins
3.2.1. LC-MS and LC-MS/MS Patterns of Cannabis Protein Extracts are Very Reproducible
3.2.2. Proteins from Cannabis Buds are Small
3.2.3. The Vast Majority of MS/MS Data from Cannabis Samples Remains Unannotated
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vincent:, D.; Rochfort, S.; Spangenberg, G. Optimisation of Protein Extraction from Medicinal Cannabis Mature Buds for Bottom-Up Proteomics. Molecules 2019, 24, 659. [Google Scholar] [CrossRef] [PubMed]
- Elkins, A.C.; Deseo, M.A.; Rochfort, S.; Ezernieks, V.; Spangenberg, G. Development of a validated method for the qualitative and quantitative analysis of cannabinoids in plant biomass and medicinal cannabis resin extracts obtained by super-critical fluid extraction. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2019, 1109, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Asati, A.K.; Sahoo, H.B.; Sahu, S.; Dwivedi, A. Phytochemical and pharmacological profile of Cannabis sativa L. Int. J. Ind. Herbs Drugs 2017, 2, 37–45. [Google Scholar]
- ElSohly, M.A.; Radwan, M.M.; Gul, W.; Chandra, S.; Galal, A. Phytochemistry of Cannabis sativa L. Prog. Chem. Org. Nat. Prod. 2017, 103, 1–36. [Google Scholar] [CrossRef] [PubMed]
- McPartland, J.M. Cannabis Systematics at the Levels of Family, Genus, and Species. Cannabis Cannabinoid Res. 2018, 3.1, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.; Hunt, M.; Clark, J.H. Structure of cannabidiol, a product isolated from the marihuana extract of Minnesota wild hemp. J. Am. Chem. Soc. 1940, 62, 196–200. [Google Scholar] [CrossRef]
- ElSohly, M.A.; Slade, D. Chemical constituents of marijuana: The complex mixture of natural cannabinoids. Life Sci. 2005, 78, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Gaoni, Y. Recent advances in the chemistry of hashish. In Progress in the Chemistry of Organic Natural Products; Springer: Vienna, Austria, 1967; Volume 25, pp. 175–213. [Google Scholar]
- Andre, C.M.; Hausman, J.-F.; Guerriero, G. Cannabis sativa: The Plant of the Thousand and One Molecules. Front. Plant. Sci. 2016, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Sirikantaramas, S.; Taura, F.; Morimoto, S.; Shoyama, Y. Recent advances in Cannabis sativa research: Biosynthetic studies and its potential in biotechnology. Curr. Pharm. Biotechnol. 2007, 8, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Bona, E.; Marsano, F.; Cavaletto, M.; Berta, G. Proteomic characterization of copper stress response in Cannabis sativa roots. Proteomics 2007, 7, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Behr, M.; Sergeant, K.; Leclercq, C.C.; Planchon, S.; Guignard, C.; Lenouvel, A.; Renaut, J.; Hausman, J.F.; Lutts, S.; Guerriero, G. Insights into the molecular regulation of monolignol-derived product biosynthesis in the growing hemp hypocotyl. BMC Plant. Biol. 2018, 18, 1. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Hong, L.; Yang, Y.; Yanping, X.; Xing, H.; Gang, D. Protein Changes in Response to Lead Stress of Lead-Tolerant and Lead-Sensitive Industrial Hemp Using SWATH Technology. Genes 2019, 10, 396. [Google Scholar] [CrossRef] [PubMed]
- Aiello, G.; Fasoli, E.; Boschin, G.; Lammi, C.; Zanoni, C.; Citterio, A.; Arnoldi, A. Proteomic characterization of hempseed (Cannabis sativa L.). J. Proteom. 2016, 147, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Seo, J.B.; Lee, M.Y. Proteomic profiling of hempseed proteins from Cheungsam. Biochim. Biophys. Acta 2012, 1824, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Raharjo, T.J.; Widjaja, I.; Roytrakul, S.; Verpoorte, R. Comparative proteomics of Cannabis sativa plant tissues. J. Biomol. Tech. 2004, 15, 97–106. [Google Scholar] [PubMed]
- Happyana, N. Metabolomics, Proteomics, and Transcriptomics of Cannabis sativa L. Trichomes. PhD Thesis, University in Dortmund, Dortmund, Germany, 2014. [Google Scholar]
- Jenkins, C.; Orsburn, B. Constructing a Draft Map of the Cannabis Proteome. Biorxiv 2019. [Google Scholar] [CrossRef]
- Toby, T.K.; Fornelli, L.; Kelleher, N.L. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. [Google Scholar] [CrossRef]
- Schaffer, L.V.; Millikin, R.J.; Miller, R.M.; Anderson, L.C.; Fellers, R.T.; Ge, Y.; Kelleher, N.L.; LeDuc, R.D.; Liu, X.; Payne, S.H.; et al. Identification and Quantification of Proteoforms by Mass Spectrometry. Proteomics 2019, 19, e1800361. [Google Scholar] [CrossRef]
- Kelleher, N.L. Top-down proteomics. Anal. Chem. 2004, 76, 197A–203A. [Google Scholar] [CrossRef]
- Dang, X.; Scotcher, J.; Wu, S.; Chu, R.K.; Tolic, N.; Ntai, I.; Thomas, P.M.; Fellers, R.T.; Early, B.P.; Zheng, Y.; et al. The first pilot project of the consortium for top-down proteomics: A status report. Proteomics 2014, 14, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- LeDuc, R.D.; Schwammle, V.; Shortreed, M.R.; Cesnik, A.J.; Solntsev, S.K.; Shaw, J.B.; Martin, M.J.; Vizcaino, J.A.; Alpi, E.; Danis, P.; et al. ProForma: A Standard Proteoform Notation. J. Proteome Res. 2018, 17, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, D.P.; Rawlins, C.M.; DeHart, C.J.; Fornelli, L.; Schachner, L.F.; Lin, Z.; Lippens, J.L.; Aluri, K.C.; Sarin, R.; Chen, B.; et al. Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat. Methods 2019, 16, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Shliaha, P.V.; Gibb, S.; Gorshkov, V.; Jespersen, M.S.; Andersen, G.R.; Bailey, D.; Schwartz, J.; Eliuk, S.; Schwammle, V.; Jensen, O.N. Maximizing Sequence Coverage in Top-Down Proteomics By Automated Multimodal Gas-Phase Protein Fragmentation. Anal. Chem. 2018, 90, 12519–12526. [Google Scholar] [CrossRef] [PubMed]
- Fornelli, L.; Toby, T.K.; Schachner, L.F.; Doubleday, P.F.; Srzentic, K.; DeHart, C.J.; Kelleher, N.L. Top-down proteomics: Where we are, where we are going? J. Proteom. 2018, 175, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Raynes, J.K.; Vincent, D.; Zawadzki, J.L.; Savin, K.; Mertens, D.; Logan, A.; Williams, R.P.W. Investigation of Age Gelation in UHT Milk. Beverages 2018, 4, 95. [Google Scholar] [CrossRef]
- Vincent, D.; Elkins, A.; Condina, M.R.; Ezernieks, V.; Rochfort, S. Quantitation and Identification of Intact Major Milk Proteins for High-Throughput LC-ESI-Q-TOF MS Analyses. PLoS ONE 2016, 11, e0163471. [Google Scholar] [CrossRef] [PubMed]
- Vincent, D.; Mertens, D.; Rochfort, S. Optimisation of Milk Protein Top-Down Sequencing Using In-Source Collision-Induced Dissociation in the Maxis Quadrupole Time-of-Flight Mass Spectrometer. Molecules 2018, 23, 2777. [Google Scholar] [CrossRef]
- Vincent, D.; Ezernieks, V.; Elkins, A.; Nguyen, N.; Moate, P.J.; Cocks, B.G.; Rochfort, S. Milk Bottom-Up Proteomics: Method Optimization. Front. Genet. 2015, 6, 360. [Google Scholar] [CrossRef]
- DeHart, C.J.; Fellers, R.T.; Fornelli, L.; Kelleher, N.L.; Thomas, P.M. Bioinformatics Analysis of Top-Down Mass Spectrometry Data with ProSight Lite. Methods Mol. Biol. 2017, 1558, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Drabik, A.; Bodzon-Kulakowska, A.; Suder, P. Application of the ETD/PTR reactions in top-down proteomics as a faster alternative to bottom-up nanoLC-MS/MS protein identification. J. Mass Spectrom. 2012, 47, 1347–1352. [Google Scholar] [CrossRef] [PubMed]
- Riley, N.M.; Mullen, C.; Weisbrod, C.R.; Sharma, S.; Senko, M.W.; Zabrouskov, V.; Westphall, M.S.; Syka, J.E.; Coon, J.J. Enhanced Dissociation of Intact Proteins with High Capacity Electron Transfer Dissociation. J. Am. Soc. Mass Spectrom. 2016, 27, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Riley, N.M.; Westphall, M.S.; Coon, J.J. Activated Ion-Electron Transfer Dissociation Enables Comprehensive Top-Down Protein Fragmentation. J. Proteome Res. 2017, 16, 2653–2659. [Google Scholar] [CrossRef] [PubMed]
- Weisbrod, C.R.; Kaiser, N.K.; Syka, J.E.P.; Early, L.; Mullen, C.; Dunyach, J.J.; English, A.M.; Anderson, L.C.; Blakney, G.T.; Shabanowitz, J.; et al. Front-End Electron Transfer Dissociation Coupled to a 21 Tesla FT-ICR Mass Spectrometer for Intact Protein Sequence Analysis. J. Am. Soc. Mass Spectrom. 2017, 28, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Fort, K.L.; Cramer, C.N.; Voinov, V.G.; Vasil’ev, Y.V.; Lopez, N.I.; Beckman, J.S.; Heck, A.J.R. Exploring ECD on a Benchtop Q Exactive Orbitrap Mass Spectrometer. J. Proteome Res. 2018, 17, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Karabacak, N.M.; Li, L.; Tiwari, A.; Hayward, L.J.; Hong, P.; Easterling, M.L.; Agar, J.N. Sensitive and specific identification of wild type and variant proteins from 8 to 669 kDa using top-down mass spectrometry. Mol. Cell Proteom. 2009, 8, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Zubarev, R. Protein primary structure using orthogonal fragmentation techniques in Fourier transform mass spectrometry. Expert Rev. Proteom. 2006, 3, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.S.; Easterling, M.L.; Agar, J.N. Structural characterization of intact proteins is enhanced by prevalent fragmentation pathways rarely observed for peptides. J. Am. Soc. Mass Spectrom. 2010, 21, 949–959. [Google Scholar] [CrossRef]
- Macek, B.; Waanders, L.F.; Olsen, J.V.; Mann, M. Top-down protein sequencing and MS3 on a hybrid linear quadrupole ion trap-orbitrap mass spectrometer. Mol. Cell Proteom. 2006, 5, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Schey, K.L. Fragmentation of multiply-charged intact protein ions using MALDI TOF-TOF mass spectrometry. J. Am. Soc. Mass Spectrom. 2008, 19, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Suckau, D.; Resemann, A. T3-sequencing: Targeted characterization of the N- and C-termini of undigested proteins by mass spectrometry. Anal. Chem. 2003, 75, 5817–5824. [Google Scholar] [CrossRef] [PubMed]
- Sellami, L.; Belgacem, O.; Villard, C.; Openshaw, M.E.; Barbier, P.; Lafitte, D. In-source decay and pseudo tandem mass spectrometry fragmentation processes of entire high mass proteins on a hybrid vacuum matrix-assisted laser desorption ionization-quadrupole ion-trap time-of-flight mass spectrometer. Anal. Chem. 2012, 84, 5180–5185. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Tolic, N.; Purvine, S.O.; Smith, R.D. Improving CID, HCD, and ETD FT MS/MS degradome-peptidome identifications using high accuracy mass information. J. Proteome Res. 2012, 11, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Geer, L.Y.; Markey, S.P.; Kowalak, J.A.; Wagner, L.; Xu, M.; Maynard, D.M.; Yang, X.; Shi, W.; Bryant, S.H. Open mass spectrometry search algorithm. J. Proteome Res. 2004, 3, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sirotkin, Y.; Shen, Y.; Anderson, G.; Tsai, Y.S.; Ting, Y.S.; Goodlett, D.R.; Smith, R.D.; Bafna, V.; Pevzner, P.A. Protein identification using top-down. Mol. Cell Proteom. 2012, 11, M111-008524. [Google Scholar] [CrossRef] [PubMed]
- Ursem, S.R.; Vervloet, M.G.; Hillebrand, J.J.G.; de Jongh, R.T.; Heijboer, A.C. Oxidation of PTH: In vivo feature or effect of preanalytical conditions? Clin. Chem. Lab. Med. 2018, 56, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ponniah, G.; Neill, A.; Patel, R.; Andrien, B. Accurate determination of protein methionine oxidation by stable isotope labeling and LC-MS analysis. Anal. Chem. 2013, 85, 11705–11709. [Google Scholar] [CrossRef]
- Rogowska-Wrzesinska, A.; Wojdyla, K.; Nedic, O.; Baron, C.P.; Griffiths, H.R. Analysis of protein carbonylation--pitfalls and promise in commonly used methods. Free Radic. Res. 2014, 48, 1145–1162. [Google Scholar] [CrossRef]
- Ree, R.; Varland, S.; Arnesen, T. Spotlight on protein N-terminal acetylation. Exp. Mol. Med. 2018, 50, 90. [Google Scholar] [CrossRef]
- Falnes, P.O.; Jakobsson, M.E.; Davydova, E.; Ho, A.; Malecki, J. Protein lysine methylation by seven-beta-strand methyltransferases. Biochem. J. 2016, 473, 1995–2009. [Google Scholar] [CrossRef]
- Gong, F.; Miller, K.M. Histone methylation and the DNA damage response. Mutat. Res. 2019, 780, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Vlastaridis, P.; Kyriakidou, P.; Chaliotis, A.; Van de Peer, Y.; Oliver, S.G.; Amoutzias, G.D. Estimating the total number of phosphoproteins and phosphorylation sites in eukaryotic proteomes. Gigascience 2017, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Ford, K.L.; Bacic, A.; Heazlewood, J.L. N-linked Glycan Micro-heterogeneity in Glycoproteins of Arabidopsis. Mol. Cell Proteom. 2018, 17, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Lopez-Rodas, G.; Franco, L. Histone Post-Translational Modifications and Nucleosome Organisation in Transcriptional Regulation: Some Open Questions. Adv. Exp. Med. Biol. 2017, 966, 65–92. [Google Scholar] [CrossRef] [PubMed]
- Manning, L.R.; Manning, J.M. Contributions to nucleosome dynamics in chromatin from interactive propagation of phosphorylation/acetylation and inducible histone lysine basicities. Protein Sci. 2018, 27, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Decuyper, I.I.; Rihs, H.P.; Van Gasse, A.L.; Elst, J.; De Puysseleyr, L.; Faber, M.A.; Mertens, C.; Hagendorens, M.M.; Sabato, V.; Bridts, C.; et al. Cannabis allergy: What the clinician needs to know in 2019. Expert Rev. Clin. Immunol. 2019, 15, 599–606. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MS/MS Mode | SID | SID | SID | CID | CID | CID | CID | CID | ETD | ETD | ETD | ETD | ETD | HCD | HCD | HCD | HCD | HCD | min | max | mean | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NCE | 15 | 60 | 100 | 30 | 35 | 40 | 45 | 50 | 5 | 10 | 15 | 20 | 25 | 10 | 15 | 20 | 25 | 30 | ||||||
| Protein | m/z | z | RI (%) 1 | |||||||||||||||||||||
| Myoglobin | all | n.a. | n.a. | 171 | 725 | 656 | 171 | 656 | 517 | |||||||||||||||
| Myoglobin | 848.51 | 20 | 100 | 210 | 255 | 223 | 226 | 233 | 220 | 66 | 120 | 135 | 89 | 102 | 146 | 250 | 253 | 303 | 66 | 303 | 189 | |||
| Myoglobin | 893.22 | 19 | 98 | 174 | 180 | 176 | 219 | 227 | 229 | 172 | 190 | 457 | 431 | 71 | 148 | 244 | 301 | 260 | 71 | 457 | 232 | |||
| Myoglobin | 942.68 | 18 | 96 | 194 | 233 | 243 | 227 | 209 | 356 | 470 | 504 | 516 | 468 | 116 | 175 | 280 | 511 | 376 | 116 | 516 | 325 | |||
| Myoglobin | 1211.79 | 14 | 38 | 241 | 369 | 389 | 385 | 402 | 143 | 392 | 455 | 411 | 365 | 60 | 105 | 252 | 529 | 462 | 60 | 529 | 331 | |||
| Myoglobin | 1304.93 | 13 | 24 | 180 | 389 | 411 | 383 | 368 | 79 | 282 | 273 | 309 | 263 | 42 | 118 | 262 | 499 | 572 | 42 | 572 | 295 | |||
| Myoglobin | mean | 171 | 725 | 656 | 200 | 285 | 288 | 288 | 288 | 205 | 276 | 308 | 366 | 323 | 78 | 138 | 258 | 419 | 395 | 274 | ||||
| b-LG A | all | n.a. | n.a. | 543 | 2160 | 3882 | 543 | 3882 | 2195 | |||||||||||||||
| b-LG A | 972.19 | 19 | 46 | 336 | 392 | 333 | 358 | 343 | 379 | 375 | 325 | 412 | 242 | 155 | 395 | 504 | 310 | 298 | 155 | 504 | 344 | |||
| b-LG A | 1026.15 | 18 | 74 | 344 | 412 | 397 | 439 | 387 | 220 | 271 | 137 | 170 | 102 | 230 | 469 | 588 | 449 | 350 | 102 | 588 | 331 | |||
| b-LG A | 1091.40 | 17 | 80 | 397 | 507 | 474 | 511 | 440 | 252 | 608 | 815 | 634 | 443 | 252 | 815 | 508 | ||||||||
| b-LG A | 1232.84 | 15 | 100 | 481 | 529 | 571 | 531 | 544 | 160 | 456 | 433 | 431 | 443 | 119 | 517 | 664 | 737 | 419 | 119 | 737 | 469 | |||
| b-LG A | mean | 543 | 2160 | 3882 | 390 | 460 | 444 | 460 | 429 | 253 | 367 | 298 | 338 | 262 | 189 | 497 | 643 | 533 | 378 | 413 | ||||
| a-S1-CN | all | n.a. | n.a. | 414 | 728 | 891 | 414 | 891 | 678 | |||||||||||||||
| a-S1-CN | 1139.60 | 21 | 94 | 159 | 455 | 401 | 455 | 435 | 112 | 660 | 660 | 431 | 289 | 112 | 660 | 406 | ||||||||
| a-S1-CN | 1193.38 | 20 | 100 | 166 | 460 | 466 | 389 | 375 | 111 | 424 | 352 | 292 | 193 | 120 | 702 | 651 | 519 | 301 | 111 | 702 | 368 | |||
| a-S1-CN | 1319.30 | 18 | 70 | 97 | 302 | 224 | 209 | 145 | 51 | 721 | 586 | 544 | 256 | 51 | 721 | 314 | ||||||||
| a-S1-CN | 1397.14 | 17 | 52 | 51 | 247 | 259 | 254 | 259 | 51 | 259 | 214 | |||||||||||||
| a-S1-CN | 1480.59 | 16 | 36 | 46 | 472 | 464 | 459 | 251 | 46 | 472 | 338 | |||||||||||||
| a-S1-CN | mean | 414 | 728 | 891 | 125 | 387 | 375 | 366 | 356 | 104 | 363 | 288 | 251 | 169 | 82 | 639 | 590 | 488 | 274 | 324 | ||||
| BSA | all | n.a. | n.a. | 84 | 436 | 84 | 436 | 260 | ||||||||||||||||
| BSA | 953.93 | 72 | 72 | 0 | 161 | 58 | 124 | 58 | 0 | 232 | 238 | 113 | 85 | 0 | 238 | 107 | ||||||||
| BSA | 994.98 | 69 | 76 | 0 | 182 | 150 | 153 | 157 | 0 | 196 | 227 | 121 | 87 | 0 | 227 | 127 | ||||||||
| BSA | 1061.50 | 65 | 68 | 0 | 203 | 177 | 196 | 223 | 0 | 359 | 409 | 352 | 277 | 0 | 409 | 220 | ||||||||
| BSA | 1188.08 | 59 | 44 | 0 | 109 | 96 | 101 | 125 | 0 | 125 | 86 | |||||||||||||
| BSA | mean | 84 | 436 | 0 | 165 | 141 | 150 | 168 | 0 | 260 | 234 | 238 | 168 | 0 | 214 | 233 | 117 | 86 | 145 |
| MS/MS Mode | SID | SID | SID | CID | CID | CID | CID | CID | ETD | ETD | ETD | ETD | ETD | HCD | HCD | HCD | HCD | HCD | min | max | mean | No. AAs | % max | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NCE | 15 | 60 | 100 | 30 | 35 | 40 | 45 | 50 | 5 | 10 | 15 | 20 | 25 | 10 | 15 | 20 | 25 | 30 | ||||||||
| Protein | m/z | z | RI (%) 1 | |||||||||||||||||||||||
| Myoglobin | all | n.a. | n.a. | 1 | 19 | 20 | 1 | 20 | 13 | 153 | 13 | |||||||||||||||
| Myoglobin | 848.51 | 20 | 100 | 10 | 12 | 11 | 10 | 19 | 25 | 17 | 28 | 40 | 28 | 2 | 4 | 9 | 17 | 17 | 2 | 40 | 17 | 153 | 26 | |||
| Myoglobin | 893.22 | 19 | 98 | 4 | 8 | 8 | 9 | 12 | 6 | 24 | 17 | 45 | 48 | 3 | 2 | 11 | 11 | 11 | 2 | 48 | 15 | 153 | 31 | |||
| Myoglobin | 942.68 | 18 | 96 | 10 | 12 | 14 | 14 | 14 | 17 | 36 | 45 | 57 | 53 | 2 | 5 | 22 | 33 | 22 | 2 | 57 | 24 | 153 | 37 | |||
| Myoglobin | 1211.79 | 14 | 38 | 27 | 42 | 44 | 39 | 36 | 5 | 24 | 29 | 36 | 26 | 1 | 2 | 12 | 48 | 52 | 1 | 52 | 28 | 153 | 34 | |||
| Myoglobin | 1304.93 | 13 | 24 | 13 | 41 | 40 | 44 | 44 | 2 | 21 | 20 | 21 | 19 | 1 | 4 | 7 | 55 | 47 | 1 | 55 | 25 | 153 | 36 | |||
| Myoglobin | mean | 1 | 19 | 20 | 13 | 23 | 23 | 23 | 25 | 11 | 24 | 28 | 40 | 35 | 2 | 3 | 12 | 33 | 30 | 2 | 45 | 20 | 153 | 30 | ||
| b-LG A | all | n.a. | n.a. | 2 | 27 | 66 | 2 | 66 | 32 | 162 | 41 | |||||||||||||||
| b-LG A | 972.19 | 19 | 46 | 11 | 17 | 20 | 20 | 21 | 8 | 20 | 14 | 20 | 20 | 1 | 14 | 19 | 15 | 21 | 1 | 21 | 16 | 162 | 13 | |||
| b-LG A | 1026.15 | 18 | 74 | 11 | 18 | 19 | 20 | 17 | 4 | 9 | 9 | 14 | 11 | 6 | 28 | 24 | 22 | 20 | 4 | 28 | 15 | 162 | 17 | |||
| b-LG A | 1091.40 | 17 | 80 | 11 | 24 | 23 | 26 | 18 | 5 | 34 | 29 | 28 | 26 | 5 | 29 | 22 | 162 | 18 | ||||||||
| b-LG A | 1232.84 | 15 | 100 | 23 | 23 | 21 | 23 | 22 | 4 | 8 | 12 | 13 | 19 | 3 | 17 | 27 | 27 | 21 | 3 | 23 | 18 | 162 | 14 | |||
| b-LG A | mean | 2 | 27 | 66 | 14 | 21 | 21 | 22 | 20 | 5 | 12 | 12 | 16 | 17 | 4 | 23 | 25 | 23 | 22 | 3 | 33 | 21 | 162 | 21 | ||
| a-S1-CN | all | n.a. | n.a. | 1 | 3 | 7 | 1 | 7 | 4 | 199 | 4 | |||||||||||||||
| a-S1-CN | 1139.60 | 21 | 94 | 4 | 7 | 8 | 7 | 17 | 1 | 24 | 37 | 43 | 37 | 1 | 43 | 19 | 199 | 22 | ||||||||
| a-S1-CN | 1193.38 | 20 | 100 | 2 | 10 | 9 | 10 | 6 | 3 | 23 | 25 | 24 | 25 | 2 | 32 | 41 | 37 | 36 | 2 | 41 | 19 | 199 | 21 | |||
| a-S1-CN | 1319.30 | 18 | 70 | 0 | 13 | 15 | 19 | 18 | 1 | 30 | 35 | 39 | 38 | 0 | 39 | 23 | 199 | 20 | ||||||||
| a-S1-CN | 1397.14 | 17 | 52 | 6 | 12 | 12 | 9 | 15 | 6 | 15 | 11 | 199 | 8 | |||||||||||||
| a-S1-CN | 1480.59 | 16 | 36 | 1 | 28 | 33 | 39 | 38 | 1 | 39 | 28 | 199 | 20 | |||||||||||||
| a-S1-CN | mean | 1 | 3 | 7 | 4 | 10 | 10 | 9 | 13 | 2 | 18 | 20 | 22 | 22 | 1 | 29 | 37 | 40 | 37 | 2 | 31 | 17 | 199 | 15 | ||
| BSA | all | n.a. | n.a. | 1 | 4 | 1 | 4 | 2 | 583 | 1 | ||||||||||||||||
| BSA | 953.93 | 72 | 72 | 0 | 6 | 4 | 8 | 7 | 0 | 9 | 13 | 11 | 9 | 0 | 13 | 7 | 583 | 2 | ||||||||
| BSA | 994.98 | 69 | 76 | 0 | 4 | 5 | 5 | 1 | 0 | 3 | 11 | 12 | 11 | 0 | 12 | 5 | 583 | 2 | ||||||||
| BSA | 1061.50 | 65 | 68 | 0 | 6 | 5 | 5 | 6 | 0 | 4 | 8 | 4 | 8 | 0 | 8 | 5 | 583 | 1 | ||||||||
| BSA | 1188.08 | 59 | 44 | 0 | 4 | 2 | 3 | 7 | 0 | 7 | 3 | 583 | 1 | |||||||||||||
| BSA | mean | 1 | 4 | 0 | 5 | 4 | 4 | 5 | 0 | 5 | 6 | 6 | 8 | 0 | 6 | 12 | 12 | 10 | 0 | 9 | 4 | 583 | 2 |
| (A) | (B) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tech. Rep. | bud 1 | bud 2 | bud 3 | Mean | SD | Method | bud 1 | bud 2 | bud 3 | Mean | SD |
| Replicate 1 | 442 | 483 | 483 | 469 | 19 | “Low” | 1157 | 1169 | 1208 | 1178 | 22 |
| Replicate 2 | 474 | 486 | 453 | 471 | 14 | “Mid” | 1173 | 1193 | 1226 | 1197 | 22 |
| mean | 458 | 485 | 468 | 470 | 17 | “High” | 1149 | 1192 | 1225 | 1189 | 31 |
| SD | 16 | 2 | 15 | mean | 1160 | 1185 | 1220 | ||||
| SD | 10 | 11 | 8 |
| Charge State | No. of Precursors | Min. m/z | Max. m/z | Min. Mass (Da) | Max. Mass (Da) | No. of MS/MS Events |
|---|---|---|---|---|---|---|
| 2 | 34 | 714.18 | 1500.37 | 1426.36 | 2998.73 | 63 |
| 3 | 8 | 848.75 | 1176.15 | 2543.23 | 3525.44 | 32 |
| 4 | 45 | 714.08 | 1380.06 | 2852.31 | 5516.21 | 143 |
| 5 | 39 | 803.49 | 1325.52 | 4012.42 | 6622.58 | 120 |
| 6 | 43 | 775.62 | 1458.49 | 4647.67 | 8744.89 | 109 |
| 7 | 61 | 747.77 | 1534.29 | 5227.35 | 10,732.96 | 222 |
| 8 | 86 | 787.70 | 1429.84 | 6293.52 | 11,430.63 | 341 |
| 9 | 69 | 700.41 | 1564.79 | 6294.62 | 14,074.01 | 262 |
| 10 | 48 | 756.92 | 1729.69 | 7559.16 | 17,286.78 | 195 |
| 11 | 32 | 726.96 | 1338.87 | 7985.51 | 14,716.50 | 113 |
| 12 | 30 | 710.98 | 1338.68 | 8519.65 | 16,052.07 | 99 |
| 13 | 32 | 762.47 | 1256.51 | 9898.99 | 16,321.52 | 114 |
| 14 | 36 | 732.89 | 1318.67 | 10,246.31 | 18,447.31 | 125 |
| 15 | 32 | 738.60 | 1099.47 | 11,063.95 | 16,433.03 | 109 |
| 16 | 29 | 708.10 | 1153.96 | 11,269.49 | 18,447.30 | 105 |
| 17 | 29 | 737.28 | 1129.03 | 12,516.63 | 19,176.39 | 86 |
| 18 | 27 | 754.89 | 1163.66 | 13,569.88 | 20,927.81 | 96 |
| 19 | 37 | 715.21 | 1135.96 | 13,569.85 | 21,564.03 | 124 |
| 20 | 38 | 710.24 | 1240.59 | 14,184.59 | 24,791.58 | 126 |
| 21 | 34 | 723.89 | 1185.04 | 15,180.59 | 24,864.66 | 106 |
| 22 | 28 | 701.95 | 1155.10 | 15,420.70 | 25,390.00 | 92 |
| 23 | 14 | 711.74 | 1104.83 | 16,346.79 | 25,387.98 | 31 |
| 24 | 8 | 746.08 | 1036.99 | 17,881.77 | 24,863.64 | 18 |
| 25 | 3 | 745.98 | 992.59 | 18,624.23 | 24,789.59 | 3 |
| No. | Accession | Score | Mass (Da) | RT (min) | No. of Matches | Description | Species | Database | Proteoform 1 | BUP 2 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A0A0U2H3S7 | 111 | 11721 | 7.5 | 4 | 30S ribosomal protein S14, chloroplastic | Humulus lupulus | Uniprot | NME, O | no |
| 2 | AKP55264.1 | 111 | 10442 | 40.5 | 1 | 30S ribosomal protein S16, chloroplastic | Cannabis sativa | NCBI | no | |
| 3 | csa_locus_3973_iso_2_len_767_ver_2 | 41 | 15430 | 18.8 | 4 | 40S ribosomal protein S24 | Actinidia chinensis | MPGR | NME, NA | no |
| 4 | csa_locus_3786_iso_3_len_389_ver_2 | 345 | 6796 | 2.9 | 3 | 40S ribosomal protein S30 | Dichanthelium oligosanthes | MPGR | no | |
| 5 | A0A0H3W8B6 | 21 | 15440 | 47.5 | 1 | 50S ribosomal protein L16 | Cannabis sativa | Uniprot | M | yes |
| 6 | A0A0C5ARQ5 | 180 | 8020 | 100.3 | 9 | ATP synthase CF0 C subunit | Cannabis sativa | Uniprot | NA, O | no |
| 7 | A0A0C5AUH9 | 62 | 14615 | 75.0 | 3 | ATP synthase CF1 epsilon subunit | Cannabis sativa | Uniprot | NME, NA, O | yes |
| 8 | A0A0U2H159 | 54 | 14697 | 75.0 | 3 | ATP synthase epsilon chain, chloroplastic | Humulus lupulus | Uniprot | NA, O, D | no |
| 9 | I6XT51 | 80 | 17490 | 52.6 | 6 | Betv1-like protein | Cannabis sativa | Uniprot | NME, NA, O, M, P | yes |
| 10 | A0A0C5ARS8 | 1641 | 9257 | 96.4 | 29 | Cytochrome b559 subunit alpha | Cannabis sativa | Uniprot | NME, NA | yes |
| 11 | A0A0U2GZT5 | 902 | 9237 | 96.4 | 1 | Cytochrome b559 subunit alpha | Humulus lupulus | Uniprot | NME | yes |
| 12 | A0A0C5AUI2 | 163 | 4392 | 71.9 | 15 | Cytochrome b559 subunit beta | Cannabis sativa | Uniprot | no | |
| 13 | A0A0H3W844 | 24 | 17381 | 99.2 | 1 | Cytochrome b6-f complex subunit 4 | Cannabis sativa | Uniprot | NME | no |
| 14 | A0A0C5APY4 | 27 | 4195 | 101.1 | 1 | Cytochrome b6-f complex subunit 5 | Cannabis sativa | Uniprot | no | |
| 15 | csa_locus_2489_iso_3_len_603_ver_2 | 20 | 14971 | 35.6 | 2 | Furry | Trema orientale | MPGR | NME, NA | no |
| 16 | csa_locus_15285_iso_1_len_577_ver_2 | 43 | 8673 | 27.6 | 3 | GAG1At protein | Trema orientale | MPGR | NME, NA, O | no |
| 17 | csa_locus_3395_iso_3_len_637_ver_2 | 14 | 10650 | 31.8 | 2 | GroES-like protein | Corchorus capsularis | MPGR | NME, NA | no |
| 18 | csa_locus_4170_iso_1_len_735_ver_2 | 203 | 13905 | 59.9 | 20 | Histone H2A | Morus notabilis | MPGR | NME, A, P | no |
| 19 | csa_locus_3458_iso_4_len_603_ver_2 | 87 | 14959 | 47.1 | 4 | Histone H2B | Cicer arietinum | MPGR | NME, O | no |
| 20 | csa_locus_1853_iso_2_len_1208_ver_2 | 17 | 17895 | 49.2 | 1 | Histone H3.2 | Triticum aestivum | MPGR | no | |
| 21 | csa_locus_1853_iso_1_len_474_ver_2 | 44 | 15234 | 50.7 | 5 | Histone H3.2 | Triticum aestivum | MPGR | NME, M, A, P | no |
| 22 | csa_locus_11346_iso_1_len_965_ver_2 | 143 | 15352 | 15.5 | 10 | Histone H3.3 | Meleagris gallopavo | MPGR | NME, NA, O, M, A | no |
| 23 | csa_locus_61264_iso_2_len_414_ver_2 | 303 | 11322 | 41.5 | 27 | Histone H4 | Phanerochaete chrysosporium | MPGR | NME, M, A, P, S | no |
| 24 | csa_locus_4104_iso_2_len_741_ver_2 | 151 | 17736 | 53.3 | 7 | Major latex/Bet v I type allergen | Parasponia andersonii | MPGR | NME, NA, A | no |
| 25 | csa_locus_2495_iso_2_len_611_ver_2 | 37 | 9659 | 75.0 | 4 | Mitochondrial outer membrane translocase complex, subunit Tom | Trema orientale | MPGR | NME, NA, O | no |
| 26 | W0U0V5 | 28 | 9563 | 26.2 | 2 | Non-specific lipid-transfer protein (Fragment) | Cannabis sativa | Uniprot | M | yes |
| 27 | csa_locus_1463_iso_3_len_564_ver_2 | 45 | 7624 | 18.7 | 1 | Non-specific lipid-transfer protein AKCS9 | Parasponia andersonii | MPGR | yes | |
| 28 | I6WU39 | 174 | 11901 | 65.9 | 11 | Olivetolic acid cyclase | Cannabis sativa | Uniprot | NME, NA, O, M, A | yes |
| 29 | A0A0C5AS17 | 1538 | 9415 | 31.0 | 39 | Photosystem I iron-sulfur centre | Cannabis sativa | Uniprot | NME, O | yes |
| 30 | A0A0C5AS04 | 15 | 4814 | 98.9 | 2 | Photosystem I reaction centre subunit IX | Cannabis sativa | Uniprot | O, A | no |
| 31 | A0A0C5AS00 | 30 | 4094 | 99.1 | 2 | Photosystem I reaction centre subunit VIII | Cannabis sativa | Uniprot | O, P | no |
| 32 | A0A0C5B2J7 | 1167 | 7515 | 99.6 | 11 | Photosystem II reaction centre protein H | Cannabis sativa | Uniprot | NME, O | no |
| 33 | A0A0C5APX7 | 249 | 4193 | 100.1 | 5 | Photosystem II reaction centre protein I | Cannabis sativa | Uniprot | NA, O, M | no |
| 34 | A0A0C5APY3 | 66 | 4193 | 100.3 | 1 | Photosystem II reaction centre protein J | Cannabis sativa | Uniprot | NA | no |
| 35 | A0A0H3W8G1 | 25 | 4363 | 99.2 | 2 | Photosystem II reaction centre protein L | Cannabis sativa | Uniprot | NME | no |
| 36 | A0A0U2DTK8 | 1438 | 3843 | 99.7 | 25 | Photosystem II reaction centre protein T | Cannabis sativa | Uniprot | no | |
| 37 | A9XV92 | 1243 | 3843 | 99.7 | 18 | Photosystem II reaction centre protein T | Cannabis sativa | Uniprot | no | |
| 38 | A0A0C5AUI5 | 72 | 7780 | 4.5 | 1 | Ribosomal protein L33 | Cannabis sativa | Uniprot | NME | no |
| 39 | A0A0C5B2H7 | 53 | 11720 | 7.5 | 2 | Ribosomal protein S14 | Cannabis sativa | Uniprot | NME, A | no |
| 40 | A0A0H3W6G0 | 82 | 10443 | 40.5 | 3 | Ribosomal protein S16 | Cannabis sativa | Uniprot | M, A | no |
| 41 | csa_locus_3039_iso_2_len_611_ver_2 | 125 | 9779 | 42.5 | 5 | Small zinc finger/mitochondrial import inner membrane translocase subunit TIM10-like | Juglans regia | MPGR | NME, NA, O, A | no |
| 42 | csa_locus_13354_iso_1_len_537_ver_2 | 31 | 9073 | 55.7 | 2 | Small zinc finger/Tim10/DDP family zinc finger | Parasponia andersonii | MPGR | O | no |
| 43 | csa_locus_2526_iso_1_len_396_ver_2 | 227 | 6442 | 54.2 | 10 | Ubiquinol-cytochrome c reductase complex 6.7 kDa protein | Trema orientale | MPGR | NME | no |
| 44 | csa_locus_5849_iso_5_len_634_ver_2 | 178 | 13600 | 31.1 | 4 | Uncharacterized protein | Fagus sylvatica | MPGR | NME, NA, A | no |
| 45 | csa_locus_6096_iso_1_len_715_ver_2 | 21 | 7326 | 45.4 | 1 | Uncharacterized protein | Trema orientale | MPGR | NME | no |
| 46 | csa_locus_3129_iso_3_len_709_ver_2 | 17 | 11165 | 34.2 | 2 | Uncharacterized protein | Aquilegia coerulea | MPGR | NME, NA | no |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vincent, D.; Binos, S.; Rochfort, S.; Spangenberg, G. Top-Down Proteomics of Medicinal Cannabis. Proteomes 2019, 7, 33. https://doi.org/10.3390/proteomes7040033
Vincent D, Binos S, Rochfort S, Spangenberg G. Top-Down Proteomics of Medicinal Cannabis. Proteomes. 2019; 7(4):33. https://doi.org/10.3390/proteomes7040033
Chicago/Turabian StyleVincent, Delphine, Steve Binos, Simone Rochfort, and German Spangenberg. 2019. "Top-Down Proteomics of Medicinal Cannabis" Proteomes 7, no. 4: 33. https://doi.org/10.3390/proteomes7040033
APA StyleVincent, D., Binos, S., Rochfort, S., & Spangenberg, G. (2019). Top-Down Proteomics of Medicinal Cannabis. Proteomes, 7(4), 33. https://doi.org/10.3390/proteomes7040033