Mapping the Proteome of the Synaptic Cleft through Proximity Labeling Reveals New Cleft Proteins

 , ,
, ,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1 Animals
2.2 Neuronal Cell Culture
2.3. HEK293T Cell Culture
2.4. Plasmids
2.5. Adeno-Associated Virus Production, Purification, and Titration
2.6. Transfection
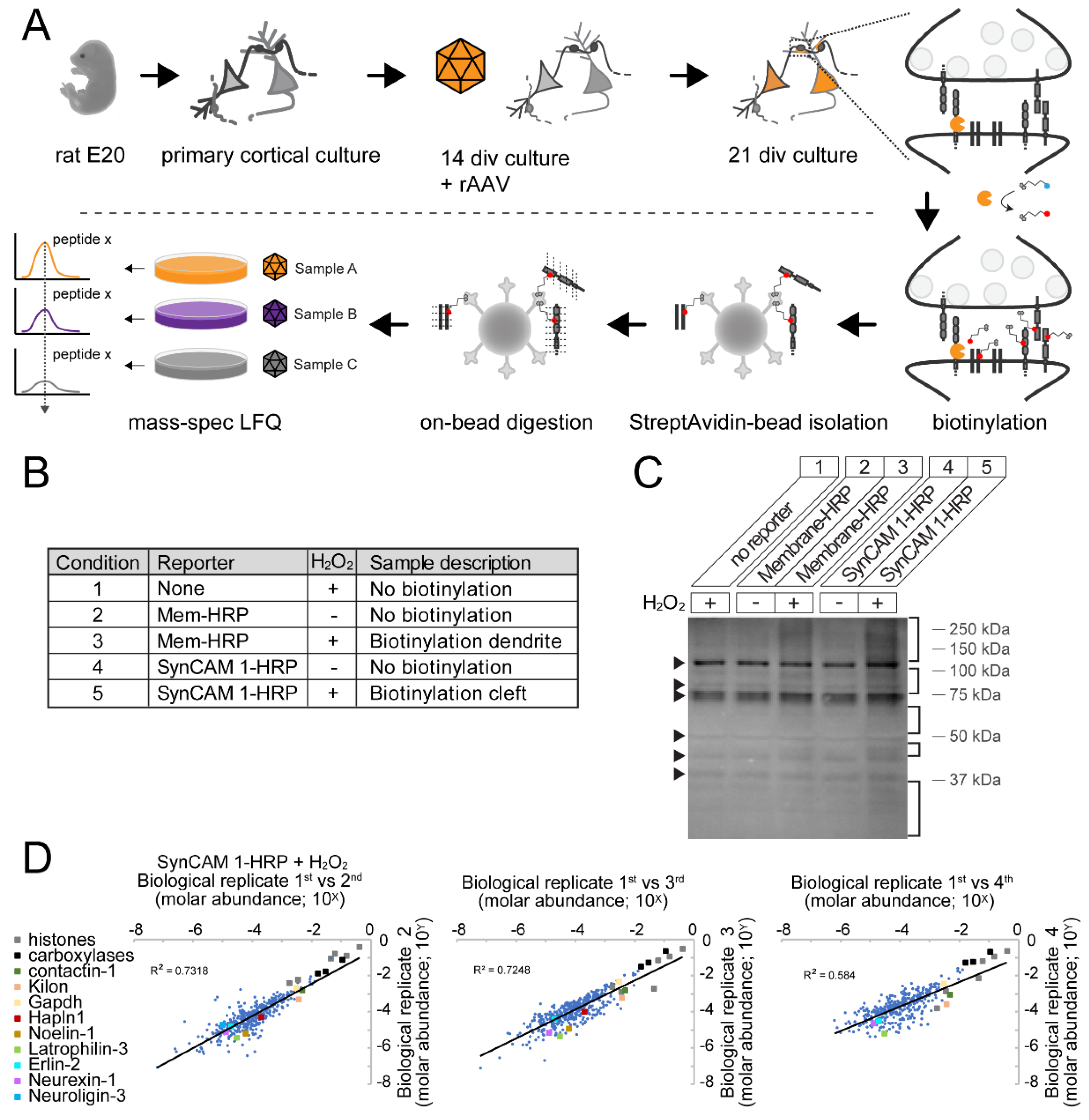
2.7. Peroxidase-Mediated Biotinylation
2.8 Sample Preparation for Mass Spectrometry
2.9 Mass Spectrometry and Label-Free Quantification
2.9.1. Mass Spectrometry Data Acquisition
2.9.2. Data Processing for Identification
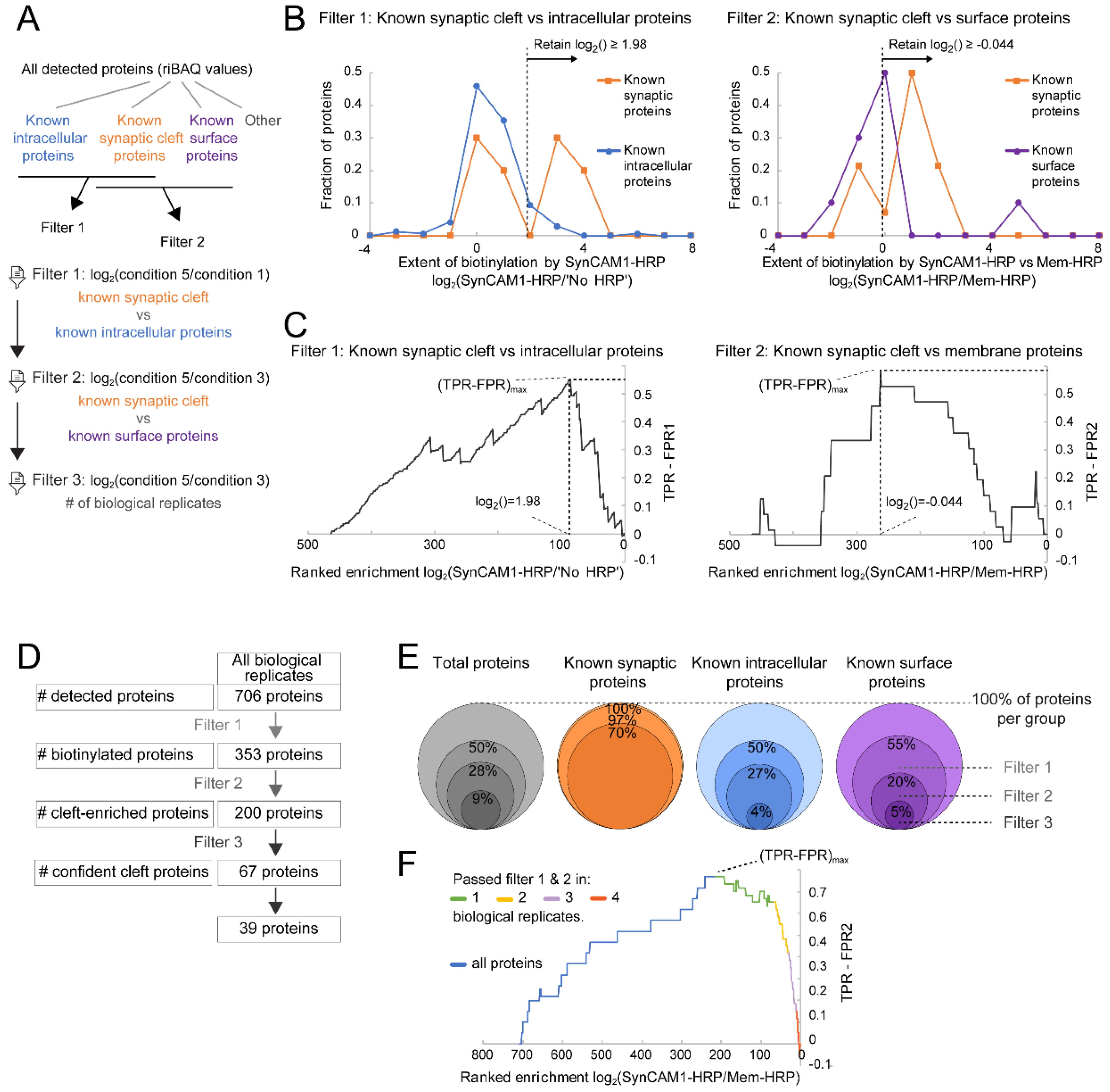
2.9.3 Post Hoc In-Silico Filtering/Data Analysis
Filter 1
Filter 2
Filter 3
2.9.4. Calculation of Depth of Coverage
2.10. Immunochemistry
2.11. Characterization of Biotinylation by SynCAM 1-HRP Using Western Blot
2.12. Staining for dSTORM, and dSTORM Imaging and Analysis of SynCAM 1-HRP
3. Results
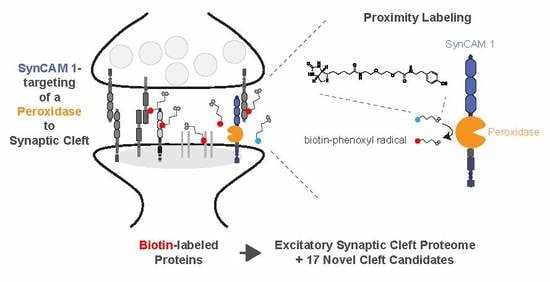
3.1. Peroxidase-Mediated Proximity Labeling Using a SynCAM 1-Horseradish Peroxidase (HRP)-Fusion Protein
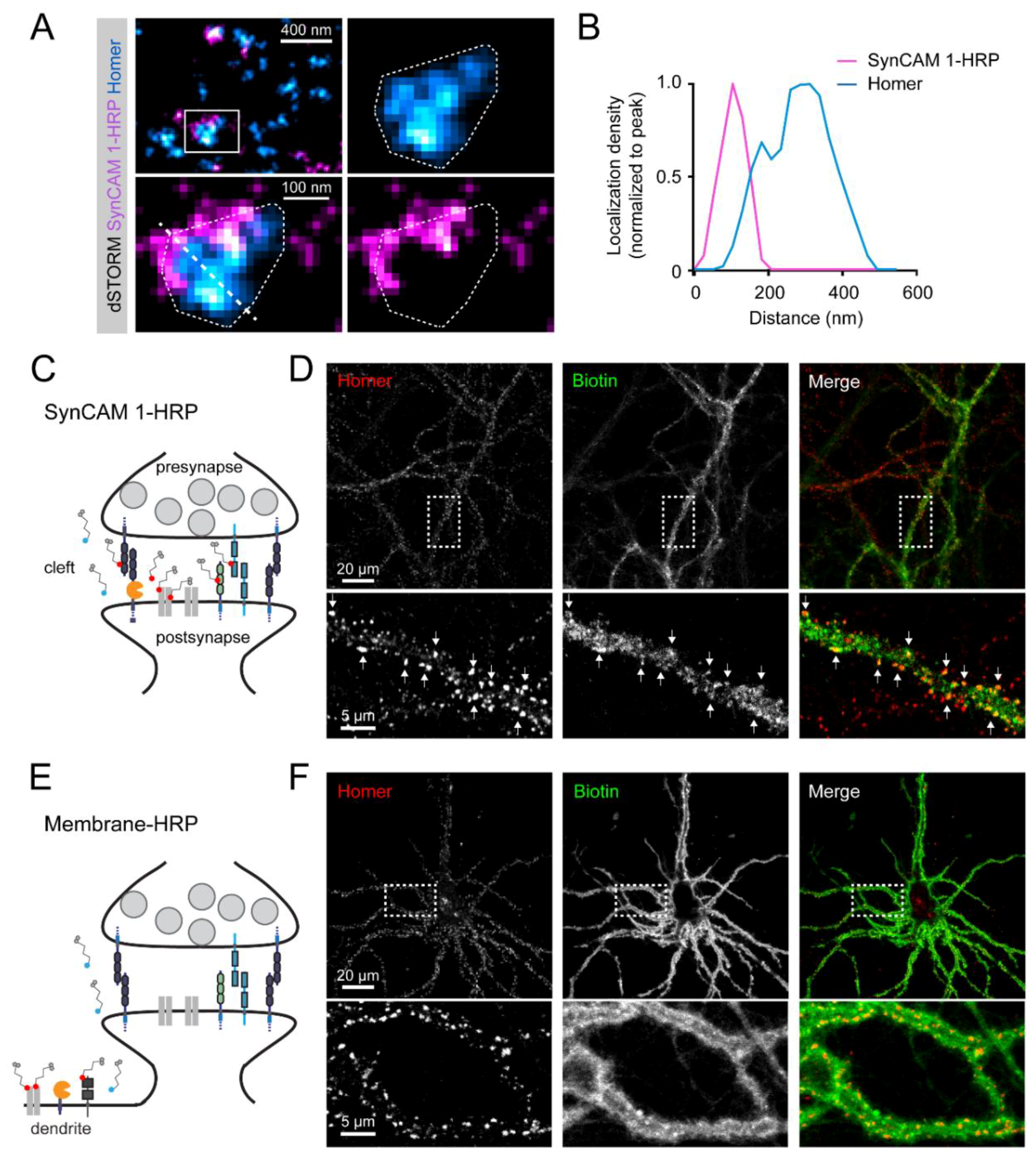
3.2. Subsynaptic Distribution of the SynCAM 1-HRP Reporter
3.3. SynCAM 1-HRP Validation for Synaptic Proximity Labeling
3.4. Robust Identification of Synaptic Cleft Candidate Proteins Using Proximity Labeling
3.5. Ratiometric Analysis of Proximity Labeled Protein Hits
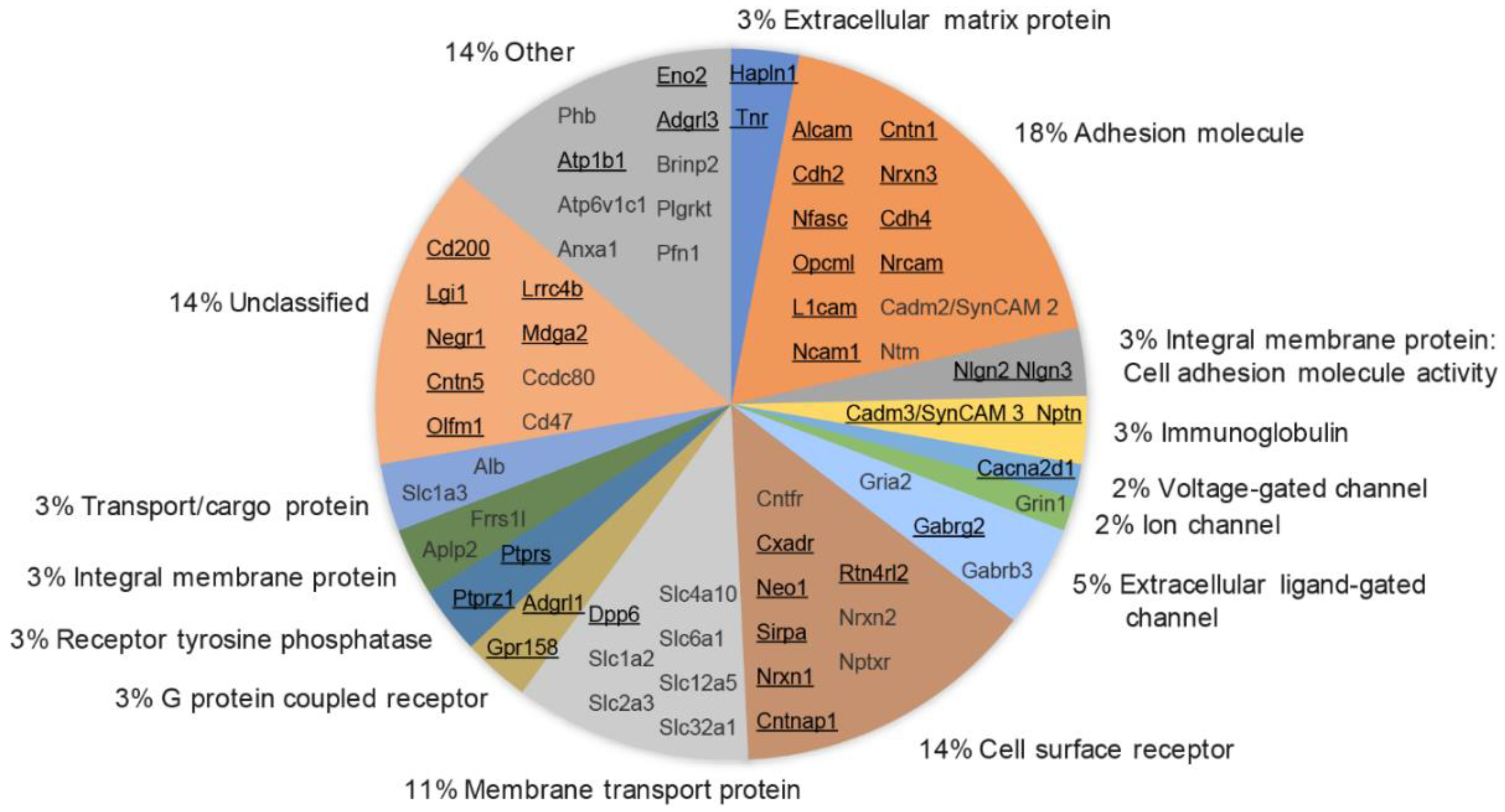
3.6. Molecular Class and Gene Ontology Analysis of Synaptic Cleft Hits
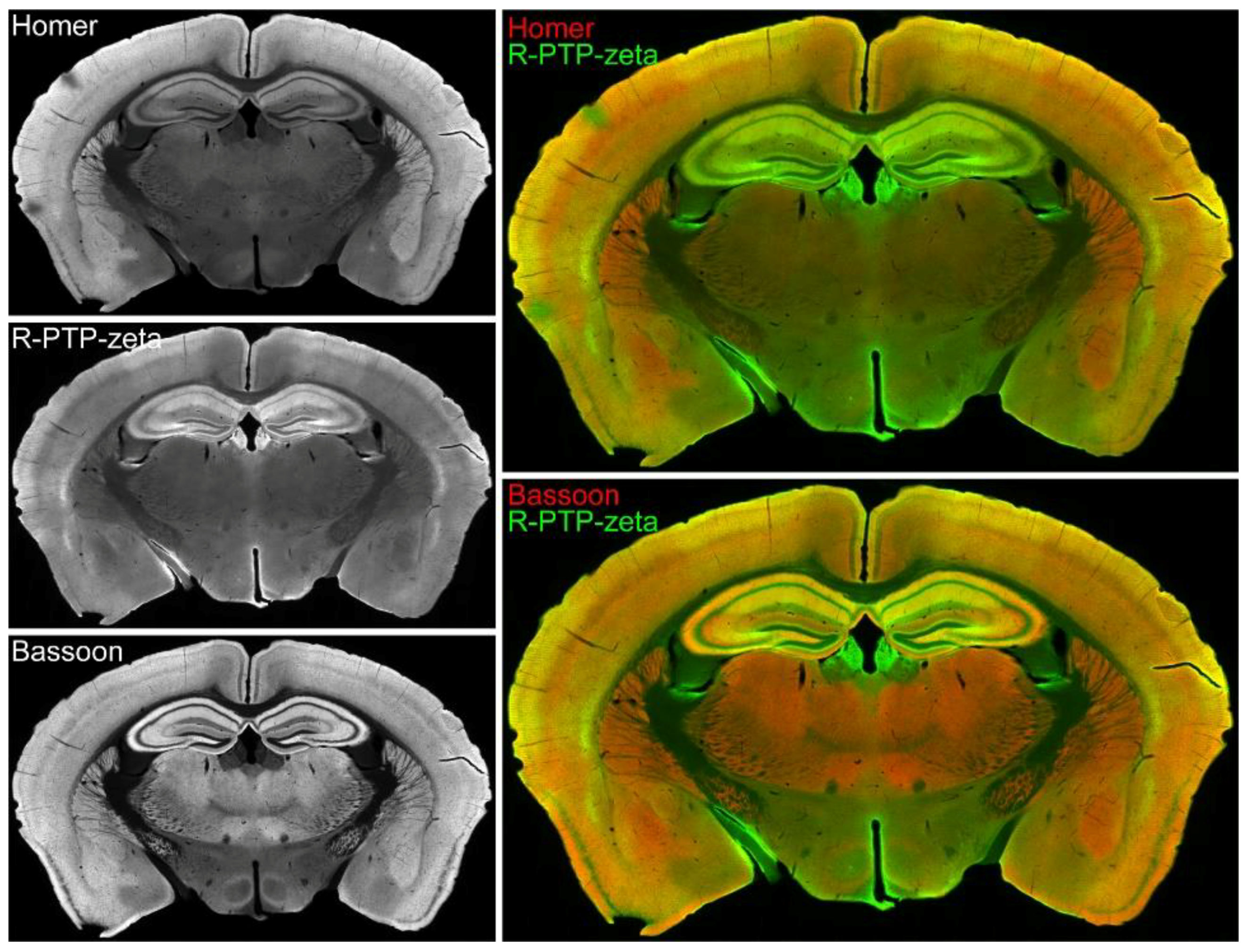
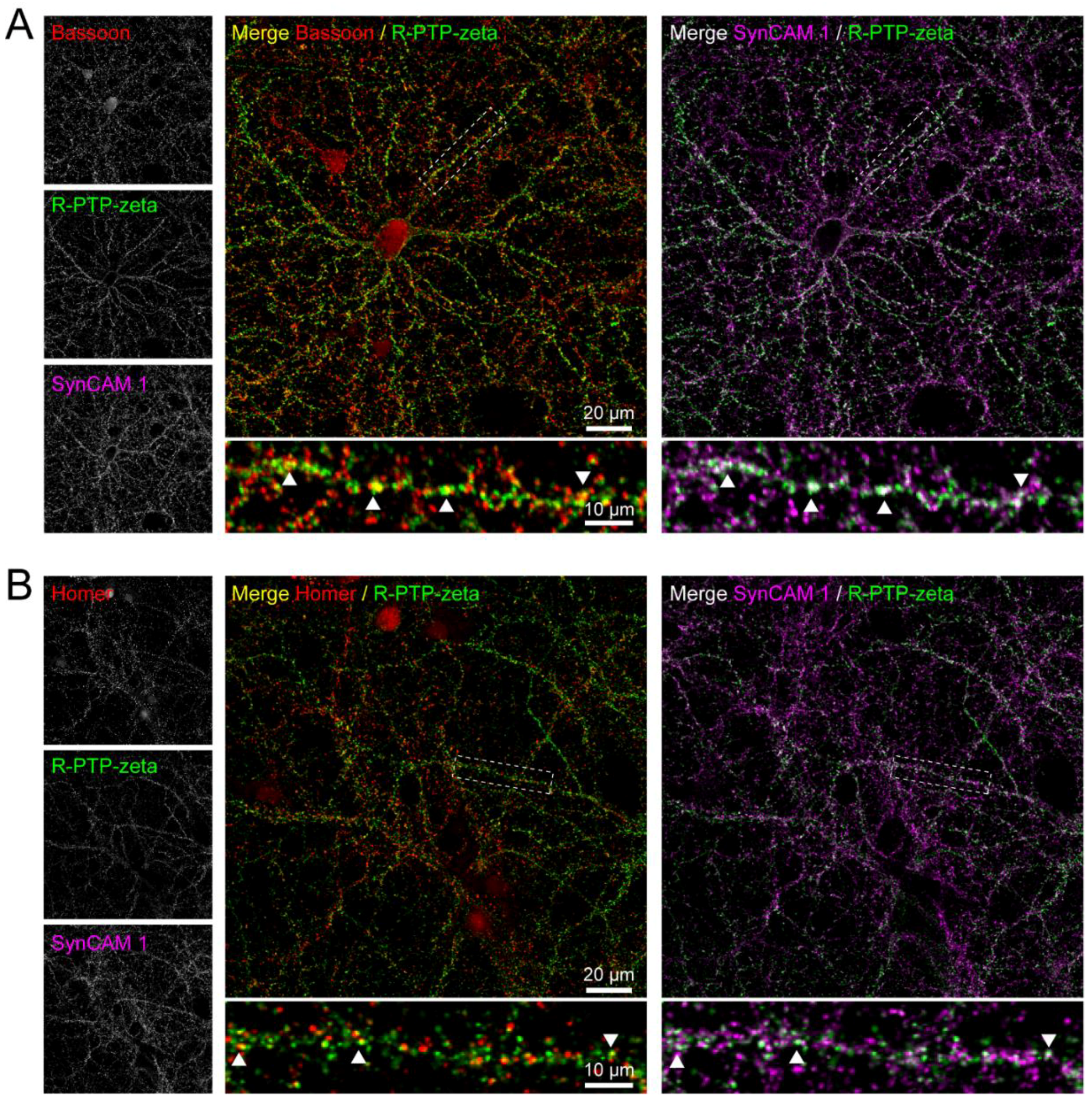
3.7. Validation of the Synaptic Cleft Candidate Receptor-Type Tyrosine-Protein Phosphatase Zeta
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sudhof, T.C. Synaptic Neurexin Complexes: A Molecular Code for the Logic of Neural Circuits. Cell 2017, 171, 745–769. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, N.A.; Weiler, N.C.; Micheva, K.D.; Smith, S.J. Deep molecular diversity of mammalian synapses: why it matters and how to measure it. Nat. Rev. Neurosci. 2012, 13, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Emes, R.D.; Grant, S.G. Evolution of synapse complexity and diversity. Annu. Rev. Neurosci. 2012, 35, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Missler, M.; Sudhof, T.C.; Biederer, T. Synaptic cell adhesion. Cold Spring Harb. Perspect. Biol. 2012, 4, a005694. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Scheiffele, P. Genetics and cell biology of building specific synaptic connectivity. Annu. Rev. Neurosci. 2010, 33, 473–507. [Google Scholar] [CrossRef] [PubMed]
- Maeder, C.I.; Shen, K. Genetic dissection of synaptic specificity. Curr. Opin. Neurobiol. 2011, 21, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Goda, Y.; Davis, G.W. Mechanisms of synapse assembly and disassembly. Neuron 2003, 40, 243–264. [Google Scholar] [CrossRef]
- Andreev, V.P.; Petyuk, V.A.; Brewer, H.M.; Karpievitch, Y.V.; Xie, F.; Clarke, J.; Camp, D.; Smith, R.D.; Lieberman, A.P.; Albin, R.L.; et al. Label-free quantitative LC-MS proteomics of Alzheimer’s disease and normally aged human brains. J. Proteome Res. 2012, 11, 3053–3067. [Google Scholar] [CrossRef] [PubMed]
- Moron, J.A.; Devi, L.A. Use of proteomics for the identification of novel drug targets in brain diseases. J. Neurochem. 2007, 102, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Abul-Husn, N.S.; Devi, L.A. Neuroproteomics of the synapse and drug addiction. J. Pharmacol. Exp. Ther. 2006, 318, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Abul-Husn, N.S.; Bushlin, I.; Moron, J.A.; Jenkins, S.L.; Dolios, G.; Wang, R.; Iyengar, R.; Ma’ayan, A.; Devi, L.A. Systems approach to explore components and interactions in the presynapse. Proteomics 2009, 9, 3303–3315. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.M.; Weinberg, R.J. Ultrastructure of synapses in the mammalian brain. Cold Spring Harb. Perspect. Biol. 2012, 4, a005587. [Google Scholar] [CrossRef] [PubMed]
- Biederer, T.; Kaeser, P.S.; Blanpied, T.A. Transcellular Nanoalignment of Synaptic Function. Neuron 2017, 96, 680–696. [Google Scholar] [CrossRef] [PubMed]
- Perez de Arce, K.; Schrod, N.; Metzbower, S.W.R.; Allgeyer, E.; Kong, G.K.; Tang, A.H.; Krupp, A.J.; Stein, V.; Liu, X.; Bewersdorf, J.; et al. Topographic Mapping of the Synaptic Cleft into Adhesive Nanodomains. Neuron 2015, 88, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Biesemann, C.; Gronborg, M.; Luquet, E.; Wichert, S.P.; Bernard, V.; Bungers, S.R.; Cooper, B.; Varoqueaux, F.; Li, L.; Byrne, J.A.; et al. Proteomic screening of glutamatergic mouse brain synaptosomes isolated by fluorescence activated sorting. EMBO J. 2014, 33, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Morciano, M.; Burre, J.; Corvey, C.; Karas, M.; Zimmermann, H.; Volknandt, W. Immunoisolation of two synaptic vesicle pools from synaptosomes: a proteomics analysis. J. Neurochem. 2005, 95, 1732–1745. [Google Scholar] [CrossRef] [PubMed]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Gronborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brugger, B.; Ringler, P.; et al. Molecular anatomy of a trafficking organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Boyken, J.; Gronborg, M.; Riedel, D.; Urlaub, H.; Jahn, R.; Chua, J.J. Molecular profiling of synaptic vesicle docking sites reveals novel proteins but few differences between glutamatergic and GABAergic synapses. Neuron 2013, 78, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Morciano, M.; Beckhaus, T.; Karas, M.; Zimmermann, H.; Volknandt, W. The proteome of the presynaptic active zone: from docked synaptic vesicles to adhesion molecules and maxi-channels. J. Neurochem. 2009, 108, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Bayes, A.; Collins, M.O.; Croning, M.D.; van de Lagemaat, L.N.; Choudhary, J.S.; Grant, S.G. Comparative study of human and mouse postsynaptic proteomes finds high compositional conservation and abundance differences for key synaptic proteins. PLoS One 2012, 7, e46683. [Google Scholar] [CrossRef] [PubMed]
- Distler, U.; Schmeisser, M.J.; Pelosi, A.; Reim, D.; Kuharev, J.; Weiczner, R.; Baumgart, J.; Boeckers, T.M.; Nitsch, R.; Vogt, J.; et al. In-depth protein profiling of the postsynaptic density from mouse hippocampus using data-independent acquisition proteomics. Proteomics 2014, 14, 2607–2613. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.O.; Husi, H.; Yu, L.; Brandon, J.M.; Anderson, C.N.; Blackstock, W.P.; Choudhary, J.S.; Grant, S.G. Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J. Neurochem. 2006, 97, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Selimi, F.; Cristea, I.M.; Heller, E.; Chait, B.T.; Heintz, N. Proteomic studies of a single CNS synapse type: the parallel fiber/purkinje cell synapse. PLoS Biol. 2009, 7, e83. [Google Scholar] [CrossRef] [PubMed]
- Heller, E.A.; Zhang, W.; Selimi, F.; Earnheart, J.C.; Slimak, M.A.; Santos-Torres, J.; Ibanez-Tallon, I.; Aoki, C.; Chait, B.T.; Heintz, N. The biochemical anatomy of cortical inhibitory synapses. PLoS One 2012, 7, e39572. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Li, J.; Ting, A.Y. Proximity labeling: spatially resolved proteomic mapping for neurobiology. Curr. Opin. Neurobiol. 2018, 50, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Perrimon, N. Proximity-dependent labeling methods for proteomic profiling in living cells. Wiley Interdiscip. Rev. Dev. Biol. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Varnaite, R.; MacNeill, S.A. Meet the neighbors: Mapping local protein interactomes by proximity-dependent labeling with BioID. Proteomics 2016, 16, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.H.; Stawski, P.S.; Draycott, A.S.; Udeshi, N.D.; Lehrman, E.K.; Wilton, D.K.; Svinkina, T.; Deerinck, T.J.; Ellisman, M.H.; Stevens, B.; et al. Proteomic Analysis of Unbounded Cellular Compartments: Synaptic Clefts. Cell 2016, 166, 1295–1307.e21. [Google Scholar] [CrossRef] [PubMed]
- Reinke, A.W.; Balla, K.M.; Bennett, E.J.; Troemel, E.R. Identification of microsporidia host-exposed proteins reveals a repertoire of rapidly evolving proteins. Nat. Commun. 2017, 8, 14023. [Google Scholar] [CrossRef] [PubMed]
- Reinke, A.W.; Mak, R.; Troemel, E.R.; Bennett, E.J. In vivo mapping of tissue- and subcellular-specific proteomes in Caenorhabditis elegans. Sci. Adv. 2017, 3, e1602426. [Google Scholar] [CrossRef] [PubMed]
- Uezu, A.; Kanak, D.J.; Bradshaw, T.W.; Soderblom, E.J.; Catavero, C.M.; Burette, A.C.; Weinberg, R.J.; Soderling, S.H. Identification of an elaborate complex mediating postsynaptic inhibition. Science 2016, 353, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Fogel, A.I.; Akins, M.R.; Krupp, A.J.; Stagi, M.; Stein, V.; Biederer, T. SynCAMs organize synapses through heterophilic adhesion. J. Neurosci. 2007, 27, 12516–12530. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.A.; Akins, M.R.; Biederer, T. Expression and adhesion profiles of SynCAM molecules indicate distinct neuronal functions. J. Comp. Neurol. 2008, 510, 47–67. [Google Scholar] [CrossRef] [PubMed]
- Biederer, T.; Sara, Y.; Mozhayeva, M.; Atasoy, D.; Liu, X.; Kavalali, E.T.; Sudhof, T.C. SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science 2002, 297, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- Robbins, E.M.; Krupp, A.J.; Perez de Arce, K.; Ghosh, A.K.; Fogel, A.I.; Boucard, A.; Sudhof, T.C.; Stein, V.; Biederer, T. SynCAM 1 adhesion dynamically regulates synapse number and impacts plasticity and learning. Neuron 2010, 68, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Biederer, T.; Scheiffele, P. Mixed-culture assays for analyzing neuronal synapse formation. Nat. Protoc. 2007, 2, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Zolotukhin, S.; Byrne, B.J.; Mason, E.; Zolotukhin, I.; Potter, M.; Chesnut, K.; Summerford, C.; Samulski, R.J.; Muzyczka, N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999, 6, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Hommel, J.D.; Sears, R.M.; Georgescu, D.; Simmons, D.L.; DiLeone, R.J. Local gene knockdown in the brain using viral-mediated RNA interference. Nat. Med. 2003, 9, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, Y.; Obata, Y.; Ishibashi, K.; Tamura, N.; Kikuchi, I.; Aoyama, K.; Hattori, Y.; Tsuda, K.; Nakayama, Y.; Yamaguchi, N. Cost-effective gene transfection by DNA compaction at pH 4.0 using acidified, long shelf-life polyethylenimine. Cytotechnology 2010, 62, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Krey, J.F.; Wilmarth, P.A.; Shin, J.B.; Klimek, J.; Sherman, N.E.; Jeffery, E.D.; Choi, D.; David, L.L.; Barr-Gillespie, P.G. Accurate label-free protein quantitation with high- and low-resolution mass spectrometers. J. Proteome Res. 2014, 13, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Hung, V.; Udeshi, N.D.; Lam, S.S.; Loh, K.H.; Cox, K.J.; Pedram, K.; Carr, S.A.; Ting, A.Y. Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat. Protoc. 2016, 11, 456–475. [Google Scholar] [CrossRef] [PubMed]
- Stanly, T.A.; Fritzsche, M.; Banerji, S.; Garcia, E.; Bernardino de la Serna, J.; Jackson, D.G.; Eggeling, C. Critical importance of appropriate fixation conditions for faithful imaging of receptor microclusters. Biol. Open 2016, 5, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Ovesny, M.; Krizek, P.; Borkovec, J.; Svindrych, Z.; Hagen, G.M. ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 2014, 30, 2389–2390. [Google Scholar] [CrossRef] [PubMed]
- Udeshi, N.D.; Pedram, K.; Svinkina, T.; Fereshetian, S.; Myers, S.A.; Aygun, O.; Krug, K.; Clauser, K.; Ryan, D.; Ast, T.; et al. Antibodies to biotin enable large-scale detection of biotinylation sites on proteins. Nat. Methods 2017, 14, 1167–1170. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.S.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Stagi, M.; Fogel, A.I.; Biederer, T. SynCAM 1 participates in axo-dendritic contact assembly and shapes neuronal growth cones. Proc. Natl. Acad. Sci. USA 2010, 107, 7568–7573. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Berglund, G.I.; Carlsson, G.H.; Smith, A.T.; Szoke, H.; Henriksen, A.; Hajdu, J. The catalytic pathway of horseradish peroxidase at high resolution. Nature 2002, 417, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Hung, V.; Zou, P.; Rhee, H.W.; Udeshi, N.D.; Cracan, V.; Svinkina, T.; Carr, S.A.; Mootha, V.K.; Ting, A.Y. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 2014, 55, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Lemeer, S.; Savitski, M.M.; Kuster, B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Analyt. Bioanal. Chem. 2012, 404, 939–965. [Google Scholar] [CrossRef] [PubMed]
- Schulze, W.X.; Usadel, B. Quantitation in mass-spectrometry-based proteomics. Annu. Rev. Plant Biol. 2010, 61, 491–516. [Google Scholar] [CrossRef] [PubMed]
- Ankney, J.A.; Muneer, A.; Chen, X. Relative and Absolute Quantitation in Mass Spectrometry-Based Proteomics. Annu. Rev. Analyt. Chem. 2018, 11, 49–77. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Y.I.; Zempleni, J. A novel, enigmatic histone modification: biotinylation of histones by holocarboxylase synthetase. Nutr. Rev. 2008, 66, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Kuroishi, T.; Rios-Avila, L.; Pestinger, V.; Wijeratne, S.S.; Zempleni, J. Biotinylation is a natural, albeit rare, modification of human histones. Mol. Genet. Metab. 2011, 104, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Jitrapakdee, S.; Wallace, J.C. The biotin enzyme family: conserved structural motifs and domain rearrangements. Curr. Protein Pept. Sci. 2003, 4, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Tytgat, H.L.; Schoofs, G.; Driesen, M.; Proost, P.; Van Damme, E.J.; Vanderleyden, J.; Lebeer, S. Endogenous biotin-binding proteins: an overlooked factor causing false positives in streptavidin-based protein detection. Microb. Biotechnol. 2015, 8, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Peri, S.; Navarro, J.D.; Amanchy, R.; Kristiansen, T.Z.; Jonnalagadda, C.K.; Surendranath, V.; Niranjan, V.; Muthusamy, B.; Gandhi, T.K.; Gronborg, M.; et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003, 13, 2363–2371. [Google Scholar] [CrossRef] [PubMed]
- Mishra, G.R.; Suresh, M.; Kumaran, K.; Kannabiran, N.; Suresh, S.; Bala, P.; Shivakumar, K.; Anuradha, N.; Reddy, R.; Raghavan, T.M.; et al. Human protein reference database—2006 update. Nucleic Acids Res. 2006, 34, D411–D414. [Google Scholar] [CrossRef] [PubMed]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database--2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed]
- Weingarten, J.; Lassek, M.; Mueller, B.F.; Rohmer, M.; Lunger, I.; Baeumlisberger, D.; Dudek, S.; Gogesch, P.; Karas, M.; Volknandt, W. The proteome of the presynaptic active zone from mouse brain. Mol. Cell. Neurosci. 2014, 59, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Pocklington, A.J.; Cumiskey, M.; Armstrong, J.D.; Grant, S.G. The proteomes of neurotransmitter receptor complexes form modular networks with distributed functionality underlying plasticity and behaviour. Mol. Syst. Biol. 2006, 2, 0023. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, J.; Baehrens, D.; Haupt, A.; Bildl, W.; Boudkkazi, S.; Roeper, J.; Fakler, B.; Schulte, U. Regional diversity and developmental dynamics of the AMPA-receptor proteome in the mammalian brain. Neuron 2014, 84, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, N.; Oohira, A.; Miyata, S. Synaptic localization of receptor-type protein tyrosine phosphatase ζ/β in the cerebral and hippocampal neurons of adult rats. Brain Res. 2005, 1050, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Sorokina, O.; McLean, C.; Tapia-Gonzalez, S.; DeFelipe, J.; Armstrong, J.D.; Grant, S.G.N. Regional Diversity in the Postsynaptic Proteome of the Mouse Brain. Proteomes 2018, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Cizeron, M.; Qiu, Z.; Benavides-Piccione, R.; Kopanitsa, M.V.; Skene, N.G.; Koniaris, B.; DeFelipe, J.; Fransen, E.; Komiyama, N.H.; et al. Architecture of the Mouse Brain Synaptome. Neuron 2018, 99, 781–799.e10. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Sorokina, O.; Skene, N.; Simonnet, C.; Mazzo, F.; Zwart, R.; Sher, E.; Smith, C.; Armstrong, J.D.; Grant, S.G.N. Proteomic analysis of postsynaptic proteins in regions of the human neocortex. Nat. Neurosci. 2018, 21, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Carlyle, B.C.; Kitchen, R.R.; Kanyo, J.E.; Voss, E.Z.; Pletikos, M.; Sousa, A.M.M.; Lam, T.T.; Gerstein, M.B.; Sestan, N.; Nairn, A.C. A multiregional proteomic survey of the postnatal human brain. Nat. Neurosci. 2017, 20, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.A.; Muralidhar, S.; Wang, Z.; Cervantes, D.C.; Basu, R.; Taylor, M.R.; Hunter, J.; Cutforth, T.; Wilke, S.A.; Ghosh, A.; et al. The intellectual disability gene Kirrel3 regulates target-specific mossy fiber synapse development in the hippocampus. Elife 2015, 4, e09395. [Google Scholar] [CrossRef] [PubMed]
- Sylwestrak, E.L.; Ghosh, A. Elfn1 regulates target-specific release probability at CA1-interneuron synapses. Science 2012, 338, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.E.; de Wit, J.; Ghosh, A. Molecular mechanisms of synaptic specificity in developing neural circuits. Neuron 2010, 68, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.E.; Wilke, S.A.; Daggett, A.; Davis, E.; Otto, S.; Ravi, D.; Ripley, B.; Bushong, E.A.; Ellisman, M.H.; Klein, G.; et al. Cadherin-9 regulates synapse-specific differentiation in the developing hippocampus. Neuron 2011, 71, 640–655. [Google Scholar] [CrossRef] [PubMed]
- de Wit, J.; Ghosh, A. Specification of synaptic connectivity by cell surface interactions. Nat. Rev. Neurosci. 2016, 17, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, M.; Sanes, J.R. Dscam and Sidekick proteins direct lamina-specific synaptic connections in vertebrate retina. Nature 2008, 451, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Krishnaswamy, A.; De la Huerta, I.; Sanes, J.R. Type II cadherins guide assembly of a direction-selective retinal circuit. Cell 2014, 158, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Linhoff, M.W.; Lauren, J.; Cassidy, R.M.; Dobie, F.A.; Takahashi, H.; Nygaard, H.B.; Airaksinen, M.S.; Strittmatter, S.M.; Craig, A.M. An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron 2009, 61, 734–749. [Google Scholar] [CrossRef] [PubMed]
- De Wit, J.; Sylwestrak, E.; O’Sullivan, M.L.; Otto, S.; Tiglio, K.; Savas, J.N.; Yates, J.R.; Comoletti, D.; Taylor, P.; Ghosh, A. LRRTM2 interacts with Neurexin1 and regulates excitatory synapse formation. Neuron 2009, 64, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Chamma, I.; Letellier, M.; Butler, C.; Tessier, B.; Lim, K.H.; Gauthereau, I.; Choquet, D.; Sibarita, J.B.; Park, S.; Sainlos, M.; et al. Mapping the dynamics and nanoscale organization of synaptic adhesion proteins using monomeric streptavidin. Nat. Commun. 2016, 7, 10773. [Google Scholar] [CrossRef] [PubMed]
- Santuy, A.; Rodriguez, J.R.; DeFelipe, J.; Merchan-Perez, A. Study of the Size and Shape of Synapses in the Juvenile Rat Somatosensory Cortex with 3D Electron Microscopy. eNeuro 2018, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Schikorski, T.; Stevens, C.F. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J. Neurosci. 1997, 17, 5858–5867. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Fukada, M.; Fujikawa, A.; Noda, M. Protein tyrosine phosphatase receptor type Z is involved in hippocampus-dependent memory formation through dephosphorylation at Y1105 on p190 RhoGAP. Neurosci. Lett. 2006, 399, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Niisato, K.; Fujikawa, A.; Komai, S.; Shintani, T.; Watanabe, E.; Sakaguchi, G.; Katsuura, G.; Manabe, T.; Noda, M. Age-dependent enhancement of hippocampal long-term potentiation and impairment of spatial learning through the Rho-associated kinase pathway in protein tyrosine phosphatase receptor type Z-deficient mice. J. Neurosci. 2005, 25, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.D.; Georgieva, L.; Young, J.J.; Plescia, C.; Kajiwara, Y.; Jiang, Y.; Moskvina, V.; Norton, N.; Peirce, T.; Williams, H.; et al. Molecular dissection of NRG1-ERBB4 signaling implicates PTPRZ1 as a potential schizophrenia susceptibility gene. Mol. Psychiatry 2008, 13, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Sakurai, T.; Bozdagi-Gunal, O.; Dorr, N.P.; Moy, J.; Krug, L.; Gama-Sosa, M.; Elder, G.A.; Koch, R.J.; Walker, R.H.; et al. Increased expression of receptor phosphotyrosine phosphatase-β/ζ is associated with molecular, cellular, behavioral and cognitive schizophrenia phenotypes. Transl. Psychiatry 2011, 1, e8. [Google Scholar] [CrossRef] [PubMed]
- Cressant, A.; Dubreuil, V.; Kong, J.; Kranz, T.M.; Lazarini, F.; Launay, J.M.; Callebert, J.; Sap, J.; Malaspina, D.; Granon, S.; et al. Loss-of-function of PTPR γ and ζ, observed in sporadic schizophrenia, causes brain region-specific deregulation of monoamine levels and altered behavior in mice. Psychopharmacology 2017, 234, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Nishiwaki, T.; Maeda, N.; Noda, M. Characterization and Developmental Regulation of Proteoglycan-Type Protein Tyrosine Phosphatase ζ/RPTP β Isoforms. J. Biochem. 1998, 123, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.P.; Fujikawa, A.; Shimizu, H.; Suzuki, R.; Noda, M. Metalloproteinase- and γ-secretase-mediated cleavage of protein-tyrosine phosphatase receptor type Z. J. Biol. Chem. 2008, 283, 30879–30889. [Google Scholar] [CrossRef] [PubMed]
- Canoll, P.D.; Petanceska, S.; Schlessinger, J.; Musacchio, J.M. Three forms of RPTP-β are differentially expressed during gliogenesis in the developing rat brain and during glial cell differentiation in culture. J. Neurosci. Res. 1996, 44, 199–215. [Google Scholar] [CrossRef]
- Giza, J.I.; Jung, Y.; Jeffrey, R.A.; Neugebauer, N.M.; Picciotto, M.R.; Biederer, T. The synaptic adhesion molecule SynCAM 1 contributes to cocaine effects on synapse structure and psychostimulant behavior. Neuropsychopharmacology 2013, 38, 628–638. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cijsouw, T.; Ramsey, A.M.; Lam, T.T.; Carbone, B.E.; Blanpied, T.A.; Biederer, T. Mapping the Proteome of the Synaptic Cleft through Proximity Labeling Reveals New Cleft Proteins. Proteomes 2018, 6, 48. https://doi.org/10.3390/proteomes6040048
Cijsouw T, Ramsey AM, Lam TT, Carbone BE, Blanpied TA, Biederer T. Mapping the Proteome of the Synaptic Cleft through Proximity Labeling Reveals New Cleft Proteins. Proteomes. 2018; 6(4):48. https://doi.org/10.3390/proteomes6040048
Chicago/Turabian StyleCijsouw, Tony, Austin M. Ramsey, TuKiet T. Lam, Beatrice E. Carbone, Thomas A. Blanpied, and Thomas Biederer. 2018. "Mapping the Proteome of the Synaptic Cleft through Proximity Labeling Reveals New Cleft Proteins" Proteomes 6, no. 4: 48. https://doi.org/10.3390/proteomes6040048
APA StyleCijsouw, T., Ramsey, A. M., Lam, T. T., Carbone, B. E., Blanpied, T. A., & Biederer, T. (2018). Mapping the Proteome of the Synaptic Cleft through Proximity Labeling Reveals New Cleft Proteins. Proteomes, 6(4), 48. https://doi.org/10.3390/proteomes6040048