Clinical Proteomics in Colorectal Cancer, a Promising Tool for Improving Personalised Medicine



Abstract
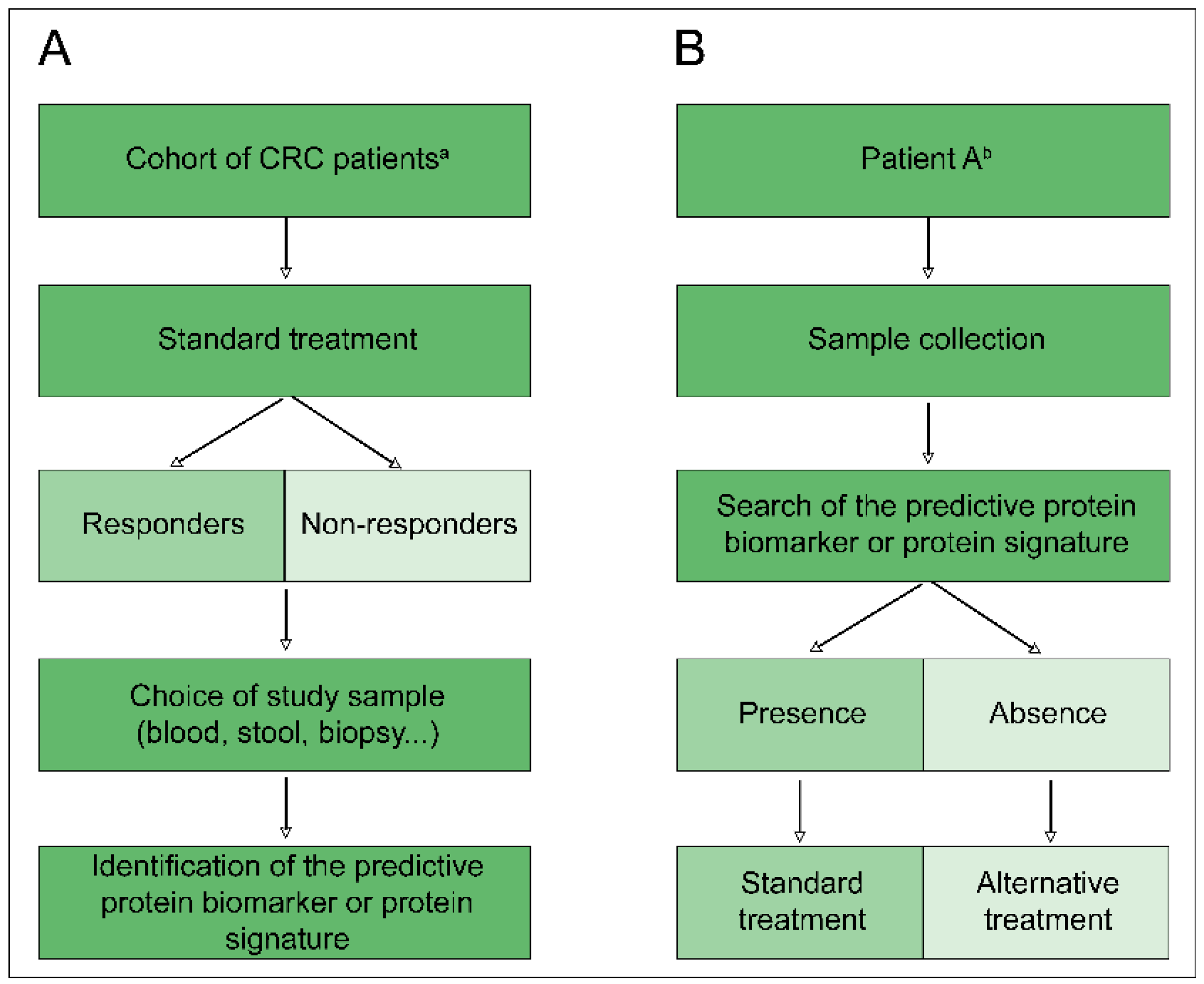
1. Introduction
1.1. Colorectal Cancer
1.2. Colorectal Cancer, a Plethora of Classifications
2. Proteomics for Biomarker Research
2.1. Biomarker Overview
2.2. Predictive Biomarkers and Proteomics
2.3. State-of-Art Tests Used in Clinical Practice
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- International Agency for Research on Cancer–World Health Organization. Colorectal Cancer Today. 2018. Available online: http://gco.iarc.fr/today/fact-sheets-cancers (accessed on 14 September 2018).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Edge, S.B.; Compton, C.C. The American Joint Committee on Cancer: The 7th Edition of the AJCC Cancer Staging Manual and the Future of TNM. Ann. Surg. Oncol. 2010, 17, 1471–1474. [Google Scholar] [CrossRef] [PubMed]
- Dukes, C.E. The classification of cancer of the rectum. J. Pathol. Bacteriol. 1932, 35, 323–332. [Google Scholar] [CrossRef]
- Astler, V.B.; Coller, F.A. The Prognostic Significance of Direct Extension of Carcinoma of the Colon and Rectum. Ann. Surg. 1954, 139, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, E.; Kastrinos, F. Familial CRC—Beyond the Lynch Syndrome. Clin. Gastroenterol. Hepatol. 2014, 12, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Samadder, N.J.; Jasperson, K.; Burt, R.W. Hereditary and Common Familial Colorectal Cancer: Evidence for Colorectal Screening. Dig. Dis. Sci. 2015, 60, 734–747. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Rozen, P.; Schuelke, G.S. Hereditary colon cancer: Polyposis and nonpolyposis variants. CA: Cancer J. Clin. 1985, 35, 95–114. [Google Scholar] [CrossRef]
- Poulogiannis, G.; Frayling, I.M.; Arends, M.J. DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology 2010, 56, 167–179. [Google Scholar] [CrossRef]
- Gardner, E.J. A genetic and clinical study of intestinal polyposis, a predisposing factor for carcinoma of the colon and rectum. Am. J. Hum. Genet. 1951, 3, 167–176. [Google Scholar]
- Polakis, P. Wnt signaling in cancer. Cold Spring Harb. Perspect Biol. 2012, 4, a008052. [Google Scholar] [CrossRef]
- Spier, I.; Horpaopan, S.; Vogt, S.; Uhlhaas, S.; Morak, M.; Stienen, D.; Draaken, M.; Ludwig, M.; Holinski-Feder, E.; Nöthen, M.M.; et al. Deep intronic APC mutations explain a substantial proportion of patients with familial or early-onset adenomatous polyposis. Hum. Mutat. 2012, 33, 1045–1050. [Google Scholar] [CrossRef]
- Weren, R.D.A.; Ligtenberg, M.J.L.; Geurts van Kessel, A.; De Voer, R.M.; Hoogerbrugge, N.; Kuiper, R.P. NTHL1 and MUTYH polyposis syndromes: Two sides of the same coin? J. Pathol. 2018, 244, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Win, A.K.; Dowty, J.G.; Cleary, S.P.; Kim, H.; Buchanan, D.D.; Young, J.P.; Clendenning, M.; Rosty, C.; Macinnis, R.J.; Giles, G.G.; et al. Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology 2014, 146, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Weren, R.D.A.; Ligtenberg, M.J.L.; Kets, C.M.; De Voer, R.M.; Verwiel, E.T.P.; Spruijt, L.; Van Zelst-Stams, W.A.G.; Jongmans, M.C.; Gilissen, C.; Hehir-Kwa, J.Y.; et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet. 2015, 47, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Brosens, L.A.A.; Langeveld, D.; van Hattem, W.A.; Giardiello, F.M.; Offerhaus, G.J.A. Juvenile polyposis syndrome. World J. Gastroenterol. 2011, 17, 4839–4844. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, T.T.; Abdelrahman, W.M.; Ristimki, A.; Lappalainen, M.; Lahermo, P.; Mecklin, J.; Jrvinen, H.J.; Peltomki, P. BMPR1A mutations in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology 2011, 141, 23–26. [Google Scholar] [CrossRef]
- Woodford-Richens, K.L.; Rowan, A.J.; Gorman, P.; Halford, S.; Bicknell, D.C.; Wasan, H.S.; Roylance, R.R.; Bodmer, W.F.; Tomlinson, I.P. SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 9719–9723. [Google Scholar] [CrossRef]
- Pino, M.S.; Chung, D.C. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef]
- Boland, R.C.; Goel, A. Microsatellite Instability in Colorectal Cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef]
- Mojarad, E.; Kuppen, P.; Aghdaei, H.; Zali, M. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol. Hepatol. Bed Bench. 2013, 6, 120–128. [Google Scholar]
- Rodriguez-Salas, N.; Dominguez, G.; Barderas, R.; Mendiola, M.; García-Albéniz, X.; Maurel, J.; Batlle, J.F. Clinical relevance of colorectal cancer molecular subtypes. Crit. Rev. Oncol. Hematol. 2017, 109, 9–19. [Google Scholar] [CrossRef] [PubMed]
- De Sousa E Melo, F.; Wang, X.; Jansen, M.; Fessler, E.; Trinh, A.; De Rooij, L.P.M.H.; De Jong, J.H.; De Boer, O.J.; Van Leersum, R.; Bijlsma, M.F.; et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat. Med. 2013, 19, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Gonzales Ostos, L.C.; Lannon, W.A.; Grotzinger, C.; Del Rio, M.; et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Marisa, L.; de Reyniès, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene Expression Classification of Colon Cancer into Molecular Subtypes: Characterization, Validation, and Prognostic Value. PLoS Med. 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Roepman, P.; Schlicker, A.; Tabernero, J.; Majewski, I.; Tian, S.; Moreno, V.; Snel, M.H.; Chresta, C.M.; Rosenberg, R.; Nitsche, U.; et al. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int. J. Cancer. 2013, 134, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Salazar, R.; Roepman, P.; Capella, G.; Moreno, V.; Simon, I.; Dreezen, C.; Lopez-Doriga, A.; Santos, C.; Marijnen, C.; Westerga, J.; et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J. Clin. Oncol. 2011, 29, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef]
- Monti, S.; Tamayo, P.; Mesirov, J.; Golub, T. Consensus Clustering: A Resampling-Based Method for Class Discovery and Visualization of Gene Expression Microarray Data. Mach. Learn. 2003, 52, 91–118. [Google Scholar] [CrossRef]
- Brændengen, M.; Tveit, K.; Berglund, Å.; Birkemeyer, E.; Frykholm, G.; Påhlman, L.; Wiig, J.N.; Byström, P.; Bujko, K.; Glimelius, B. Randomized Phase III Study Comparing Preoperative Radiotherapy with Chemoradiotherapy in Nonresectable Rectal Cancer. J. Clin. Oncol. 2008, 26, 3687–3694. [Google Scholar] [CrossRef]
- Roh, M.S.; Colangelo, L.H.; O’Connell, M.J.; Yothers, G.; Deutsch, M.; Allegra, C.J.; Kahlenberg, M.S.; Baez-Diaz, L.; Ursiny, C.S.; Petrelli, N.J.; et al. Preoperative multimodality therapy improves disease-free survival in patients with carcinoma of the rectum: NSABP R.-03. J. Clin. Oncol. 2009, 27, 5124–5130. [Google Scholar] [CrossRef] [PubMed]
- García-Flórez, L.J.; Gómez-Álvarez, G.; Frunza, A.M.; Barneo-Serra, L.; Martínez-Alonso, C.; Fresno-Forcelledo, M.F. Predictive markers of response to neoadjuvant therapy in rectal cancer. J. Surg. Res. 2015, 194, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, A.; Wang, C.S.; Geha, S.; Garde-Granger, P.; Mathieu, A.A.; Lacasse, V.; Boisvert, F.M. The response to neoadjuvant chemoradiotherapy with 5-fluorouracil in locally advanced rectal cancer patients: A predictive proteomic signature. Clin. Proteomics. BioMed. Central 2018, 15, 16. [Google Scholar] [CrossRef] [PubMed]
- Corbo, C.; Cevenini, A.; Salvatore, F. Biomarker discovery by proteomics-based approaches for early detection and personalized medicine in colorectal cancer. Proteomics Clin. Appl. 2017, 11, 15–17. [Google Scholar]
- Jimenez, C.; Knol, J.; Meijer, G.; Fijneman, R. Proteomics of colorectal cancer: Overview of discovery studies and identification of commonly identified cancer-associated proteins and candidate CRC serum markers. J. Proteomics. 2010, 73, 1873–1895. [Google Scholar] [CrossRef] [PubMed]
- de Wit, M.; Fijneman, R.J.A.; Verheul, H.M.W.; Meijer, G.A.; Jimenez, C.R. Proteomics in colorectal cancer translational research: Biomarker discovery for clinical applications. Clin. Biochem. 2013, 46, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Henry, N.L.; Hayes, D.F. Cancer biomarkers. Mol. Oncol. 2012, 6, 140–146. [Google Scholar] [CrossRef]
- Masucci, G.V.; Cesano, A.; Hawtin, R.; Janetzki, S.; Zhang, J.; Kirsch, I.; Dobbin, K.K.; Alvarez, J.; Robbins, P.B.; Selvan, S.R.; et al. Validation of biomarkers to predict response to immunotherapy in cancer: Volume I—Pre-analytical and analytical validation. J. Immunother. Cancer. 2016, 4, 76. [Google Scholar] [CrossRef] [PubMed]
- Dobbin, K.K.; Cesano, A.; Alvarez, J.; Hawtin, R.; Janetzki, S.; Kirsch, I.; Masucci, G.V.; Robbins, P.B.; Selvan, S.R.; Streicher, H.Z.; et al. Validation of biomarkers to predict response to immunotherapy in cancer: Volume II—Clinical validation and regulatory considerations. J. Immunother. Cancer 2016, 4, 77. [Google Scholar] [CrossRef] [PubMed]
- Mischak, H.; Ioannidis, J.P.A.; Argiles, A.; Attwood, T.K.; Bongcam-Rudloff, E.; Broenstrup, M.; Charonis, A.; Chrousos, G.P.; Delles, C.; Dominiczak, A.; et al. Implementation of proteomic biomarkers: Making it work. Eur. J. Clin. Invest. 2012, 42, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.; Van Deerlin, V.M.; Gulley, M.L. Recommended Principles and Practices for Validating. Arch. Pathol. Lab Med. 2009, 133, 743–755. [Google Scholar] [PubMed]
- Duffy, M.J.; O’Donovan, N.; Crown, J. Use of molecular markers for predicting therapy response in cancer patients. Cancer Treat. Rev. Elsevier Ltd. 2011, 37, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Lièvre, A.; Bachet, J.B.; Boige, V.; Cayre, A.; Le Corre, D.; Buc, E.; Ychou, M.; Bouché, O.; Landi, B.; Louvet, C.; et al. KRAS Mutations as an Independent Prognostic Factor in Patients with Advanced Colorectal Cancer Treated with Cetuximab. J. Clin. Oncol. 2008, 26, 374–379. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, F.; Blanchard, F.; Charbonnier, F.; Le Pessot, F.; Lamy, A.; Galais, M.P.; Bastit, L.; Killian, A.; Sesboüé, R.; Tuech, J.J.; et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br. J. Cancer 2007, 96, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Benvenuti, S.; Sartore-Bianchi, A.; Di Nicolantonio, F.; Zanon, C.; Moroni, M.; Veronese, S.; Siena, S.; Bardelli, A. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007, 67, 2643–2648. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Tortora, G. EGFR Antagonists in Cancer Treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef]
- Takano, M.; Sugiyama, T. UGTIAI polymorphisms in cancer: Impact on irinotecan treatment. Pharmgenomics. Pers. Med. 2017, 10, 61–68. [Google Scholar]
- Schulz, C.; Heinemann, V.; Schalhorn, A.; Moosmann, N.; Zwingers, T.; Boeck, S.; Giessen, C.; Stemmler, H.J. UGT1A1 gene polymorphism: Impact on toxicity and efficacy of irinotecan-based regimens in metastatic colorectal cancer. World J. Gastroenterol. 2009, 15, 5058–5066. [Google Scholar] [CrossRef]
- Rastelli, F.; Crispino, S. Factors predictive of response to hormone therapy in breast cancer. Tumori 2008, 94, 370–383. [Google Scholar] [CrossRef]
- Lipton, A.; Köstler, W.J.; Leitzel, K.; Ali, S.M.; Sperinde, J.; Weidler, J.; Paquet, A.; Sherwood, T.; Huang, W.; Bates, M. Quantitative HER2 protein levels predict outcome in fluorescence in situ hybridization-positive patients with metastatic breast cancer treated with trastuzumab. Cancer 2010, 116, 5168–5178. [Google Scholar] [CrossRef] [PubMed]
- Köstler, W.J.; Schwab, B.; Singer, C.F.; Neumann, R.; Rücklinger, E.; Brodowicz, T.; Tomek, S.; Niedermayr, M.; Hejna, M.; Steger, G.G.; et al. Monitoring of Serum Her-2/neu Predicts Response and Progression-Free Survival to Trastuzumab-Based Treatment in Patients with Metastatic Breast Cancer. Clin. Cancer Res. 2004, 10, 1618–1624. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Aguilar, J.; Chik, J.; Nicholson, J.; Semaan, C.; Mckay, M.J.; Molloy, M.P. Quantitative mass spectrometry for colorectal cancer proteomics. Proteomics Clin. Appl. 2013, 7, 42–54. [Google Scholar]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteomics 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.A.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed Protein Quantitation in Saccharomyces cerevisiae Using Amine-reactive Isobaric Tagging Reagents. Mol. Cell. Proteomics 2004, 3, 1154–1169. [Google Scholar] [CrossRef]
- Xiang, F.; Ye, H.; Chen, R.; Fu, Q.; Li, L.N. N-dimethyl leucines as novel isobaric tandem mass tags for quantitative proteomics and peptidomics. Anal. Chem. 2010, 82, 2817–2825. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Li, S. Deuterium isobaric amine-reactive tags for quantitative proteomics. Anal. Chem. 2010, 82, 7588–7595. [Google Scholar] [CrossRef]
- Katsila, T.; Juliachs, M.; Gregori, J.; Macarulla, T.; Villarreal, L.; Bardelli, A.; Torrance, C.; Elez, E.; Tabernero, J.; Villanueva, J. Circulating pegfr is a candidate response biomarker of cetuximab therapy in colorectal cancer. Clin. Cancer Res. 2014, 20, 6346–6356. [Google Scholar] [CrossRef]
- Martin, P.; Noonan, S.; Mullen, M.P.; Scaife, C.; Tosetto, M.; Nolan, B.; Wynne, K.; Hyland, J.; Sheahan, K.; Elia, G.; et al. Predicting response to vascular endothelial growth factor inhibitor and chemotherapy in metastatic colorectal cancer. BMC Cancer 2014, 14, 1–14. [Google Scholar] [CrossRef]
- Croner, R.S.; Sevim, M.; Metodiev, M.V.; Jo, P.; Ghadimi, M.; Schellerer, V.; Brunner, M.; Geppert, C.; Rau, T.; Stürzl, M.; et al. Identification of predictive markers for response to neoadjuvant chemoradiation in rectal carcinomas by proteomic isotope coded protein label (ICPL) analysis. Int. J. Mol. Sci. 2016, 17, 209. [Google Scholar] [CrossRef]
- Repetto, O.; De Re, V.; De Paoli, A.; Belluco, C.; Alessandrini, L.; Canzonieri, V.; Cannizzaro, R. Identification of protein clusters predictive of tumor response in rectal cancer patients receiving neoadjuvant chemoradiotherapy. Oncotarget. 2017, 8, 28328–28341. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.M.; Peng, X.C.; Tan, B.X.; Ge, J.; Chen, X.; Chen, Y.; Xu, F.; Bi, F.; Hou, J.M.; Liu, J.Y. Comparative proteomic analysis of irinotecan-sensitive colorectal carcinoma cell line and its chemoresistant counterpart. Anticancer drugs 2011, 22, 500–506. [Google Scholar] [CrossRef]
- Sakai, A.; Otani, M.; Miyamoto, A.; Yoshida, H.; Furuya, E.; Tanigawa, N. Identification of phosphorylated serine-15 and -82 residues of HSPB1 in 5-fluorouracil-resistant colorectal cancer cells by proteomics. J. Proteomics Elsevier B.V. 2012, 75, 806–818. [Google Scholar] [CrossRef]
- Monteleone, F.; Rosa, R.; Vitale, M.; D’Ambrosio, C.; Succoio, M.; Formisano, L.; Nappi, L.; Romano, M.F.; Scaloni, A.; Tortora, G.; et al. Increased anaerobic metabolism is a distinctive signature in a colorectal cancer cellular model of resistance to antiepidermal growth factor receptor antibody. Proteomics 2013, 13, 866–877. [Google Scholar] [CrossRef] [PubMed]
- McKinley, E.T.; Liu, H.; McDonald, W.H.; Luo, W.; Zhao, P.; Coffey, R.J.; Hanks, S.K.; Manning, H.C. Global phosphotyrosine proteomics identifies PKCδ as a marker of responsiveness to Src inhibition in colorectal cancer. PLoS ONE 2013, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Siolas, D.; Hannon, G.J. Patient-derived tumor xenografts: Transforming clinical samples into mouse models. Cancer Res. 2013, 73, 5315–5319. [Google Scholar] [CrossRef]
- Clark, C.R.; Starr, T.K. Mouse models for the discovery of colorectal cancer driver genes. World J. Gastroenterol. 2016, 22, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Rosfjord, E.; Lucas, J.; Li, G.; Gerber, H.P. Advances in patient-derived tumor xenografts: From target identification to predicting clinical response rates in oncology. Biochem. Pharmacol. 2014, 91, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.T.; Tan, S.; Lin, Q.; Lim, T.K.; Hew, C.L.; Chung, M.C.M. Quantitative and temporal proteome analysis of butyrate-treated colorectal cancer cells. Mol. Cell. Proteomics 2008, 7, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Van Houdt, W.; Emmink, B.; Pham, T.; Piersma, S.; Verheem, A.; Vries, R.; Fratantoni, S.A.; Pronk, A.; Clevers, H.; Borel Rinkes, I.H.M.; et al. Comparative Proteomics of Colon Cancer Stem Cells and Differentiated Tumor Cells Identifies BIRC6 as a Potential Therapeutic Target. Mol. Cell. Proteomics 2011, 10, M111011353. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt–villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Dedhia, P.H.; Bertaux-Skeirik, N.; Zavros, Y.; Spence, J.R. Organoid Models of Human Gastrointestinal Development and Disease. Gastroenterology 2016, 150, 1098–1112. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van Den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Koo, B.; Stange, D.; Sato, T.; Karthaus, W.; Farin, H.; Huch, M.; Van Es, J.H.; Clevers, H. Controlled gene expression in primary Lgr5 organoid cultures. Nat. Methods 2012, 9, 81–83. [Google Scholar] [CrossRef]
- Hynds, R.E.; Giangreco, A. Concise Review: The Relevance of Human Stem Cell-Derived Organoid Models for Epithelial Translational Medicine. Stem Cells 2013, 31, 417–422. [Google Scholar] [CrossRef]
- Allison, J.E.; Lawson, M. Screening tests for colorectal cancer: A menu for options remains relevant. Curr. Oncol. Rep. 2006, 8, 492–498. [Google Scholar] [CrossRef]
- Allison, J.E.; Sakoda, L.C.; Levin, T.R.; Tucker, J.P.; Tekawa, I.S.; Cuff, T.; Pauly, M.P.; Shlager, L.; Palitz, A.M.; Zhao, W.K.; et al. Screening for colorectal neoplasms with new fecal occult blood tests: Update on performance characteristics. J. Natl. Cancer Inst. 2007, 99, 1462–1470. [Google Scholar] [CrossRef]
- Whitlock, E.P.; Lin, J.S.; Liles, E.; Beil, T.L.; Fu, R. Screening for Colorectal Cancer: A. Targeted, Updated Systematic. Ann. Intern. Med. 2008, 149, 638–658. [Google Scholar] [CrossRef]
- Lieberman, D. Screening for Colorectal Cancer. N. Engl. J. Med. 2009, 361, 1179–1187. [Google Scholar] [CrossRef]
- Allison, J.E.; Fraser, C.G.; Halloran, S.P.; Young, G.P. Population screening for colorectal cancer means getting FIT: The past, present, and future of colorectal cancer screening using the fecal immunochemical test for hemoglobin (FIT). Gut Liver 2014, 8, 117–130. [Google Scholar] [CrossRef]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Levin, T.R.; Lavin, P.; Lidgard, G.P.; Ahlquist, D.A.; Berger, B.M. Multitarget Stool DNA Testing for Colorectal-Cancer Screening. N. Engl. J. Med. 2014, 370, 1287–1297. [Google Scholar] [CrossRef]
- Ahlquist, D.A. Multi-Target Stool DNA Test: A New High Bar for Noninvasive Screening. Dig. Dis. Sci. 2015, 60, 623–633. [Google Scholar] [CrossRef]
- Ned, R.M.; Melillo, S.; Marrone, M. Fecal DNA testing for colorectal cancer screening: The ColoSureTM test. PLoS Curr. 2011, 3, RNN220. [Google Scholar] [CrossRef]
- Kanthan, R.; Senger, J.L.; Kanthan, S.C. Fecal Molecular Markers for Colorectal Cancer Screening. Gastroenterol. Res. Pract. 2012, 2012, 1–15. [Google Scholar] [CrossRef]
- Bailey, J.R.; Aggarwal, A.; Imperiale, T.F. Colorectal cancer screening: Stool DNA and other non-invasive modalities. Gut Liver. 2016, 10, 204–211. [Google Scholar] [CrossRef]
- Castro, G.; Azrak, M.F.; Seeff, L.C.; Royalty, J. Outpatient colonoscopy complications in the CDC’s Colorectal Cancer Screening Demonstration Program: A prospective analysis. Cancer 2013, 119, 2849–2854. [Google Scholar] [CrossRef]
- Su, B.-B.; Shi, H.; Wan, J. Role of serum carcinoembryonic antigen in the detection of colorectal cancer before and after surgical resection. World J. Gastroenterol. 2012, 18, 2121. [Google Scholar] [CrossRef]
- Tanaka, T.; Tanaka, M.; Tanaka, T.; Ishigamori, R. Biomarkers for colorectal cancer. Int. J. Mol. Sci. 2010, 11, 3209–3225. [Google Scholar] [CrossRef]
- Fakih, M.; Padmanabhan, A. CEA Monitoring in Colorectal Cancer. What you should know. Oncology 2006, 20, 579–587. [Google Scholar]
- Park, I.J.; Choi, G.S.; Lim, K.H.; Kang, B.M.; Jun, S.H. Serum carcinoembryonic antigen monitoring after curative resection for colorectal cancer: Clinical significance of the preoperative level. Ann. Surg. Oncol. 2009, 16, 3087–3093. [Google Scholar] [CrossRef]
- Tan, E.; Gouvas, N.; Nicholls, R.J.; Ziprin, P.; Xynos, E.; Tekkis, P.P. Diagnostic precision of carcinoembryonic antigen in the detection of recurrence of colorectal cancer. Surg. Oncol. 2009, 18, 15–24. [Google Scholar] [CrossRef]
- Araujo, R.L.C.; Gönen, M.; Allen, P.; DeMatteo, R.; Kingham, P.; Jarnagin, W.; D’Angelica, M.; Fong, Y. Positive Postoperative CEA is a Strong Predictor of Recurrence for Patients After Resection for Colorectal Liver Metastases. Ann. Surg. Oncol. 2015, 22, 3087–3093. [Google Scholar] [CrossRef][Green Version]
- Aichler, M.; Walch, A. MALDI Imaging mass spectrometry: Current frontiers and perspectives in pathology research and practice. Lab. Investig. 2015, 95, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Veselkov, K.A.; Mirnezami, R.; Strittmatter, N.; Goldin, R.D.; Kinross, J.; Speller, A.V.M.; Abramov, T.; Jones, E.A.; Darzi, A.; Holmes, E.; et al. Chemo-informatic strategy for imaging mass spectrometry-based hyperspectral profiling of lipid signatures in colorectal cancer. Proc. Natl. Acad Sci. USA 2014, 111, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Chaurand, P.; Caprioli, R.M. Direct profiling and imaging of peptides and proteins from mammalian cells and tissue sections by mass spectrometry. Electrophoresis 2002, 23, 3125–3135. [Google Scholar] [CrossRef]
- Taktas, Z.; Wiseman, J.M.; Gologan, B.; Cooks, R.G. Mass Spectrometry Sampling Under Ambient Conditions with Desorption Electrospray Ionization. Science 2004, 306, 471–473. [Google Scholar] [CrossRef]
- Fletcher, J.S. Cellular imaging with secondary ion mass spectrometry. Analyst 2009, 134, 2204–2215. [Google Scholar] [CrossRef]
- Aichler, M.; Elsner, M.; Ludyga, N.; Feuchtinger, A.; Zangen, V.; Maier, S.K.; Balluff, B.; Schöne, C.; Hierber, L.; Braselmann, H.; et al. Clinical response to chemotherapy in oesophageal adenocarcinoma patients is linked to defects in mitochondria. J. Pathol. 2013, 230, 410–419. [Google Scholar] [CrossRef]
- Balluff, B.; Rauser, S.; Meding, S.; Elsner, M.; Schöne, C.; Feuchtinger, A.; Schuhmacher, C.; Novotny, A.; Jütting, U.; Maccarrone, G.; et al. MALDI Imaging Identifies Prognostic Seven-Protein Signature of Novel Tissue Markers in Intestinal-Type Gastric Cancer. Am. J. Pathol. 2011, 179, 2720–2729. [Google Scholar] [CrossRef]
- Diamandis, E.P. The failure of protein cancer biomarkers to reach the clinic: Why, and what can be done to address the problem? BMC Med. 2012, 10, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Chen, G.; Guo, M. Mass spectrometry based translational proteomics for biomarker discovery and application in colorectal cancer. Proteomics Clin. Appl. 2016, 10, 503–515. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Anatomic Stage | |||||
|---|---|---|---|---|---|
| Stage | T | N | M | Duke’s | MAC |
| 0 | Tis | N0 | M0 | - | - |
| I | T1, T2 | N0 | M0 | A | A B1 |
| IIA | T3 | N0 | M0 | B | B2 |
| IIB | T4a | N0 | M0 | B | B2 |
| IIC | T4b | N0 | M0 | B | B3 |
| IIIA | T1–T2, T1 | N1/Nc N2a | M0 | C | C1 |
| IIIB | T3–T4a, T2–T3, T1–T2 | N1/N1c, N2a, N2b | M0 | C | C2, C1/C2, C1 |
| IIIC | T4a, T3–T4a, T4b | N2a, N2b, N1–N2 | M0 | C | C2, C2, C3 |
| IVA | Any T | Any N | M1a | - | - |
| IVB | Any T | Any N | M1b | - | - |
| Terms | Definitions |
|---|---|
| Analytical Validation | |
| Accuracy | Agreement between a test result of a quantity and its reference value |
| Repeatability | Describes test results performed under the same conditions |
| Reproducibility | Describes test results performed under different conditions |
| Analytical Sensitivity | The ability of the assay to obtain a concordance in positive results between assay and reference method |
| Analytical Specificity | The ability of the assay to obtain a concordance in negative results between assay and reference method |
| Linearity | The ability of the assay to yield a proportional effect between test values and concentrations of the analyte in the sample |
| Limit of Detection | The lowest concentration of analyte significantly different from zero or negative control |
| Robustness | Test precision following deliberate changes in assay conditions (temperature, storage, etc.) |
| Clinical Validation | |
| Clinical Sensitivity | Ability of a biomarker to predict a change in a clinical endpoint (relationship between the magnitude of change in the biomarker and the magnitude of change in the clinical endpoint) |
| Clinical Specificity | Ability of a biomarker to distinguish responders and NR patients in terms of changes in clinical endpoints |
| Relative Risk | Ratio of the probability of an event (e.g., disease recurrence, death) occurring in the treated group to the probability of the event occurring in the control group |
| Biological Sample Type | Proteomic Approach | Treatment | Identified Candidate Biomarkers | Reference |
|---|---|---|---|---|
| Secretome | LC-MS/MS | Cetuximab + FOLFIRI | Phospho-epidermal growth factor receptor (pEGFR) [P00533] | [60] |
| Serum | 2D-DIGE + LC-MS/MS | Bevacizumab + XELOX or FOLFOX | Apolipoprotein E (APOE) [P02649] *, angiotensinogen (AGT) [P01019] *, D site-binding protein (DBP) [Q10586] | [61] |
| Tumour biopsy | ICPL + LC-MS/MS | NRCT 5-FU/capecitabine ± oxaliplatin | Plectin (PLEC1) [Q15149], transketolase (TKT) [P29401], trifunctional enzyme subunit alpha, mitochondrial (HADHA) [P40939], transgelin-2 (TAGLN) [P37802] * | [62] |
| Tumour biopsy | 2-DIGE + LC-MS | NRCT 5-FU/capecitabine ± oxaliplatin | Fibrinogen ß chain (FGB) [P02675], actin (three isoforms), serpin B5 (SERPINB5) [P36952], serpin B9 (SERPINB9) [P50453], peroxiredoxin-4 (PRDX4) [Q13162] *, cathepsin D (CTSD) [P07339] * | [63] |
| Tumour biopsy | LC-MS/MS | NRCT 5-FU/capecitabine | Caldesmon (CALD1) [Q05682] *, mast cell carboxypeptidase 4 (CPA3) [P15088] *, beta-1,3-galactosyltransferase 5 (B3GALT5) [Q9Y2C3], CD177 antigen (CD177) [Q8N6Q3], receptor-interacting serine/threonine-protein kinase 1 (RIPK1) [Q13546] *, dihydropyrimidine dehydrogenase (DPYD) [Q12882], NDUF proteins (complex 1 of the mitochondrial respiratory chain) *, ribosomal proteins (small/large subunits) * | [34] |
| CRC Cell Line | Proteomic Approach | Study Focus | Identified Candidate Biomarkers | Reference |
|---|---|---|---|---|
| HCT-116 | iTRAQ, ICAT; LC MALDI-TOF/TOF MS | Butyrate response | Heat shock protein HSP 90-β (HSP90AB1) [P08238] *, galectin-1 (LGALS1) [P09382] *, A-kinase anchor protein 12 (AKAP12) [Q02952] *, vesicle-trafficking protein SEC22b (SEC22B) [O75396] *, cytochrome c oxidase 6b1 (COX6B1) [P14854] * | [71] |
| SW620 | LC MALDI-Q-TOF MS/MS | Irinotecan resistance | α-enolase (ENO1) [P06733], cofilin (CFL1) [P23528], peroxiredoxin-2 (PRDX2) [P32119] * | [64] |
| Colonospheres derived from liver metastases | LC-MS/MS | Cisplatin and oxaliplatin resistance | Baculoviral IAP repeat-containing protein 6 (BIRC6) [Q9NR09] * | [72] |
| DLD-1 | 2-DIGE; LC MALDI-TOF/TOF MS | 5-FU resistance | Heat shock protein beta-1 (HSPB1) [P04792] *, proteasome subunit α type-5 (PSMA5) [P28066], transitional endoplasmic reticulum, ATPase (VCP) [P55072] *, 14-3-3 protein β (YWHAB) [P31946], 14-3-3 protein γ (YWHAG) [P61981], 14-3-3 protein σ (SFN) [P31947], phosphoglycerate kinase 1 (PGK1) [P00558] | [65] |
| GEO | 2-DIGE; LC-MS | Cetuximab resistance | Glucose-6-phosphate 1-dehydrogenase (G6PD) [P11413] *, L-lactate dehydrogenase B chain (LDHB) [P07195], pyruvate dehydrogenase E1 component subunit alpha, somatic form, mitochondrial (PDHA1) [P08559], transketolase (TKT) [P29401] | [66] |
| HCT-116 | LC-MS/MS | Dasatinib (Src-selective inhibitor) resistance | pY313-protein kinase C delta type (PRKCD) [Q05655] * | [67] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chauvin, A.; Boisvert, F.-M. Clinical Proteomics in Colorectal Cancer, a Promising Tool for Improving Personalised Medicine. Proteomes 2018, 6, 49. https://doi.org/10.3390/proteomes6040049
Chauvin A, Boisvert F-M. Clinical Proteomics in Colorectal Cancer, a Promising Tool for Improving Personalised Medicine. Proteomes. 2018; 6(4):49. https://doi.org/10.3390/proteomes6040049
Chicago/Turabian StyleChauvin, Anaïs, and François-Michel Boisvert. 2018. "Clinical Proteomics in Colorectal Cancer, a Promising Tool for Improving Personalised Medicine" Proteomes 6, no. 4: 49. https://doi.org/10.3390/proteomes6040049
APA StyleChauvin, A., & Boisvert, F.-M. (2018). Clinical Proteomics in Colorectal Cancer, a Promising Tool for Improving Personalised Medicine. Proteomes, 6(4), 49. https://doi.org/10.3390/proteomes6040049