Accelerating Tomato Breeding by Exploiting Genomic Selection Approaches


 , ,
, ,
Abstract
:1. The Tomato Genetic Background
2. Potential of GS in Plant
3. Lesson from Other Species
4. Tomato GS Schema Implementation
5. Applying GS in Tomato Crop Improvement
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Frusciante, L.; Barone, A.; Carputo, D.; Ercolano, M.R.; Della Rocca, F.; Esposito, S. Evaluation and use of plant biodiversity for food and pharmaceuticals. Fitoterapia 2000, 71, 66–72. [Google Scholar] [CrossRef]
- Sato, S.; Tabata, S.; Hirakawa, H.; Asamizu, E.; Shirasawa, K.; Isobe, S.; Kaneko, T.; Nakamura, Y.; Shibata, D.; Aoki, K. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar]
- Schouten, H.J.; Tikunov, Y.; Verkerke, W.; Finkers, R.; Bovy, A.; Bai, Y.; Visser, R.G.F. Breeding has Increased the Diversity of Cultivated Tomato in The Netherlands. Fron. Plant. Sci. 2019, 10, 1606. [Google Scholar] [CrossRef] [PubMed]
- Souza, L.M.; Paterniani, M.; Melo, P.C.T.; Melo, A.M.T. Diallel cross among fresh market tomato inbreeding lines. Hortic. Bras. 2012, 30, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Cappetta, E.; Andolfo, G.; Di Matteo, A.; Ercolano, M.R. Empowering crop resilience to environmental multiple stress through the modulation of key response components. J. Plant. Physiol. 2020, 246, 153134. [Google Scholar] [CrossRef] [PubMed]
- Ercolano, M.R.; Sanseverino, W.; Carli, P.; Ferriello, F.; Frusciante, L. Genetic and genomic approaches for R-gene mediated disease resistance in tomato: Retrospects and prospects. Plant. Cell Rep. 2012, 31, 973–985. [Google Scholar] [CrossRef] [Green Version]
- Sacco, A.; Di Matteo, A.; Lombardi, N.; Trotta, N.; Punzo, B.; Mari, A.; Barone, A. Quantitative trait loci pyramiding for fruit quality traits in tomato. Mol. Breed. 2013, 31, 217–222. [Google Scholar] [CrossRef] [Green Version]
- D’Esposito, D.; Cappetta, E.; Andolfo, G.; Ferriello, F.; Borgonuovo, C.; Caruso, G.; De Natale, A.; Frusciante, L.; Ercolano, M.R. Deciphering the biological processes underlying tomato biomass production and composition. Plant. Physiol. Bioch. 2019, 143, 50–60. [Google Scholar] [CrossRef]
- Available online: https://solgenomics.net/cview/index.pl (accessed on 15 April 2020).
- Hamilton, J.P.; Sim, S.; Stoffel, K.; Van Deynze, A.; Buell, C.R.; Francis, D.M. Single nucleotide polymorphism discovery in cultivated tomato via sequencing by synthesis. Plant. Genome 2012, 5, 17–29. [Google Scholar] [CrossRef]
- Sacco, A.; Ruggieri, V.; Parisi, M.; Festa, G.; Rigano, M.M.; Picarella, M.E.; Mazzucato, A.; Barone, A. Exploring a tomato landraces collection for fruit-related traits by the aid of a high-throughput genomic platform. PLoS ONE 2015, 10, e0137139. [Google Scholar] [CrossRef] [Green Version]
- Esposito, S.; Cardi, T.; Campanelli, G.; Sestili, S.; Díez, M.J.; Soler, S.; Prohens, J.; Tripodi, P. ddRAD sequencing-based genotyping for population structure analysis in cultivated tomato provides new insights into the genomic diversity of Mediterranean ‘da serbo’ type long shelf-life germplasm. Hortic. Res. 2020, 7, 134. [Google Scholar] [CrossRef] [PubMed]
- Collard, B.C.Y.; Mackill, D.J. Marker-assisted selection: An approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2008, 363, 557–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andolfo, G.; Sanseverino, W.; Aversano, R.; Frusciante, L.; Ercolano, M.R. Genome-wide identification and analysis of candidate genes for disease resistance in tomato. Mol. Breed. 2014, 33, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Andolfo, G.; Jupe, F.; Witek, K.; Etherington, G.J.; Ercolano, M.R.; Jones, J.D.G. Defining the full tomato NB-LRR resistance gene repertoire using genomic and cDNA RenSeq. BMC Plant. Biol. 2014, 14, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Donato, A.; Andolfo, G.; Ferrarini, A.; Delledonne, M.; Ercolano, M.R. Investigation of orthologous pathogen recognition gene-rich regions in solanaceous species. Genome 2017, 60, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Capuozzo, C.; Formisano, G.; Iovieno, P.; Andolfo, G.; Tomassoli, L.; Barbella, M.M.; Pico, B.; Paris, H.S.; Ercolano, M.R. Inheritance analysis and identification of SNP markers associated with ZYMV resistance in Cucurbita pepo. Mol. Breed. 2017, 37, 1–12. [Google Scholar] [CrossRef]
- Kissoudis, C.; Chowdhury, R.; van Heusden, S.; van de Wiel, C.; Finkers, R.; Visser, R.F.; Bai, Y.; van der Linden, G. Combined biotic and abiotic stress resistance in tomato. Euphytica 2015, 202, 317–332. [Google Scholar] [CrossRef] [Green Version]
- Osei, M.K.; Prempeh, R.; Adjebeng, J.; Opoku, J.; Danquah, A.; Danquah, E.; Blay, E.; Adu-Dapaah, H. Marker-Assisted Selection (MAS): A Fast-Track Tool in Tomato Breeding. In Recent Advances in Tomato Breeding and Production; IntechOpen: London, UK, 2018; pp. 93–113. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, J.C.M.; Hospital, F. The use of molecular genetics in the improvement of agricultural populations. Nat. Rev. Genet. 2002, 3, 22–32. [Google Scholar] [CrossRef]
- Heffner, E.L.; Sorrells, M.E.; Jannink, J. Genomic Selection for Crop Improvement. Crop. Sci. 2009, 49, 1–12. [Google Scholar] [CrossRef]
- Duangjit, J.; Causse, M.; Sauvage, C. Efficiency of genomic selection for tomato fruit quality. Mol. Breed. 2016, 36, 29. [Google Scholar] [CrossRef]
- Yamamoto, E.; Matsunaga, A.; Onogi, A.; Kajiya-Kanegae, H.; Minamikawa, M.; Suzuki, A.; Shirasawa, K.; Hirakawa, H.; Nunome, T.; Yamaguchi, H.; et al. A simulation-based breeding design that uses whole-genome prediction in tomato. Sci. Rep. 2016, 6, 19454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, E.; Matsunaga, H.; Onogi, A.; Ohyama, A.; Miyatake, K.; Yamaguchi, H.; Nunome, T.; Iwata, H.; Fukuoka, H. Efficiency of genomic selection for breeding population design and phenotype prediction in tomato. Heredity 2017, 118, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, D.; Sim, S.C.; Francis, D.M. Comparison of marker-based genomic estimated breeding values and phenotypic evaluation for selection of bacterial spot resistance in tomato. Phytopathology 2018, 108, 392–401. [Google Scholar] [CrossRef] [Green Version]
- Robertsen, C.D.; Hjotrtshøj, R.L.; Janss, L.L. Genomic Selection in Cereal Breeding. Agronomy 2019, 9, 95. [Google Scholar] [CrossRef] [Green Version]
- Crossa, J.; De Los Campos, G.; Pérez, P.; Gianola, D.; Burgueño, J.; Araus, J.L.; Makumbi, D.; Singh, R.P.; Dreisigacker, S.; Yan, J.; et al. Prediction of genetic values of quantitative traits in plant breeding using pedigree and molecular markers. Genetics 2010, 186, 713–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, A.J.; Chao, S.; Asoro, F.G.; Heffner, E.L.; Hayashi, T.; Iwata, H.; Hayashi, T.; Iwata, H.; Smith, K.P.; Sorrells, M.E.; et al. Genomic Selection in Plant Breeding. Knowledge and Prospects. Adv. Agron. 2011, 110, 77–123. [Google Scholar]
- Voss-Fels, K.P.; Cooper, M.; Hayes, B.J. Accelerating crop genetic gains with genomic selection. Theor. Appl. Genet. 2018, 132, 669–686. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, X.; Fu, J.; Wang, H.; Wang, J.; Huang, C.; Prasanna, B.M.; Olsen, M.S.; Wang, G.; Zhang, A. Enhancing genetic gain through genomic selection: From livestock to plants. Plant. Commun. 2020, 1, 2641–2666. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar]
- Gaynor, R.C.; Gorjanc, G.; Bentley, A.R.; Ober, E.S.; Howell, P.; Jackson, R.; Mackay, I.J.; Hickey, J.M. A two-part strategy for using genomic selection to develop inbred lines. Crop. Sci. 2017, 57, 2372. [Google Scholar] [CrossRef] [Green Version]
- Gorjanc, G.; Gaynor, R.C.; Hickey, J.M. Optimal cross selection for long-term genetic gain in two-part programs with rapid recurrent genomic selection. Theor. Appl. Genet. 2018, 131, 1953–1966. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Cogan, N.O.I.; Pembleton, L.W.; Spangenberg, G.C.; Forster, J.W.; Hayes, B.J.; Daetwyler, H.D. Genetic gain and inbreeding from genomic selection in a simulated commercial breeding program for perennial ryegrass. Plant. Genome 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Voss-Fels, K.P.; Herzog, E.; Dreisigacker, S.; Sukurmaran, S.; Watson, A.; Frisch, M.; Hayes, B.J.; Hickey, L.T. Speed GS to accelerate genetic gain in spring wheat. In Applications of Genetic and Genomic Research in Cereals, 1st ed.; Miedaner, T., Korzun, V., Eds.; Woodhead Publishing: Cambridge, MA, USA, 2018. [Google Scholar]
- Heslot, N.; Jannink, J.L.; Sorrells, M.E. Perspectives for genomic selection applications and research in plants. Crop. Sci. 2015, 55, 1–12. [Google Scholar] [CrossRef]
- Heslot, N.; Yang, H.-P.; Sorrells, M.E.; Jannink, J.-L. Genomic selection in plant breeding. A comparison of models. Crop. Sci. 2012, 52, 146. [Google Scholar] [CrossRef]
- Jannink, J.-L.; Lorenz, A.J.; Iwata, H. Genomic selection in plant breeding. From theory to practice. Brief. Funct. Genom. 2010, 9, 166–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, A.J.; Smith, K.P. Adding genetically distant individuals to training populations reduces genomic prediction accuracy in barley. Crop. Sci. 2015, 55, 2657. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.; Messina, C.D.; Podlich, D.; Totir, L.R.; Baumgarten, A.; Hausmann, N.J.; Wright, D.; Graham, G. Predicting the future of plant breeding. Complementing empirical evaluation with genetic prediction. Crop. Pasture Sci. 2014, 65, 311. [Google Scholar] [CrossRef] [Green Version]
- Riedelsheimer, C.; Endelman, J.B.; Stange, M.; Sorrells, M.E.; Jannink, J.L.; Melchinger, A.E. Genomic predictability of interconnected biparental maize populations. Genetics 2013, 194, 493–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, A.; Lian, L.; Zhong, S.; Bernardo, R. General combining ability model for genomewide selection in a biparental cross. Crop. Sci. 2014, 54, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Brandariz, S.P.; Bernardo, R. Small ad hoc versus large general training populations for genomewide selection in maize biparental crosses. Theor. Appl. Genet. 2019, 132, 347–353. [Google Scholar] [CrossRef]
- Hickey, L.T.; Hafeez, A.N.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.M.; Tester, M.; Gao, C.; Godwin, I.D.; Hayes, B.J.; Wulff, B.B.H. Breeding crops to feed 10 billion. Nat. Biotechnol. 2019, 37, 744–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeinizade, S.; Hu, G.; Wang, L.; Schnable, P.S. Optimizing selection and mating in genomic selection with a look-ahead approach: An operations research framework. G3 Genes Genom. Genet. 2019, 9, 2123–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Kadarmideen, H.N.; Dekkers, J.C. Selection on multiple QTL with control of gene diversity and inbreeding for long-term benefit. J. Anim. Breed. Genet. 2008, 125, 320–329. [Google Scholar] [CrossRef]
- Goddard, M.E.; Hayes, B.J. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 2009, 10, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Guijarro-Real, C.; Navarro, A.; Esposito, S.; Festa, G.; Macellaro, R.; Di Cesare, C.; Fita, A.; Rodríguez-Burruezo, A.; Cardi, T.; Prohens, J.; et al. Large scale phenotyping and molecular analysis in a germplasm collection of rocket salad (Eruca vesicaria) reveal a differentiation of the gene pool by geographical origin. Euphytica 2020, 216, 53. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Hu, Z.; Xu, C. Genomic selection methods for crop improvement: Current status and prospects. Crop. J. 2018, 6, 330–340. [Google Scholar] [CrossRef]
- Wendel, A.; Underwood, J.; Walsh, K. Maturity estimation of mangoes using hyperspectral imaging from a ground based mobile platform. Comput. Electron. Agric. 2018, 155, 298–313. [Google Scholar] [CrossRef]
- Westling, F.; Underwood, J.; Orn, S. Light interception modelling using unstructured LiDAR data in avocado orchards. Comput. Electron. Agric. 2018, 153, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Gil-Docampo, M.L.; Arza-Garcia, M.; Ortiz-Sanz, J.; Martinez-Rodriguez, S.; Marcos-Robles, J.L.; Sanchez-Sastre, L.F. Above-ground biomass estimation of arable crops using UAV-based SfM photogrammetry. Geocarto Int. 2018, 35, 687–699. [Google Scholar] [CrossRef]
- Heffner, E.L.; Jannink, J.L.; Sorrells, M.E. Genomic selection accuracy using multifamily prediction models in a wheat breeding program. Plant. Genome 2011, 4, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, V.; Lehermeier, C.; Albrecht, T.; Auinger, H.J.; Wang, Y.; Schon, C.C. Genome-wide prediction of traits with different genetic architecture through efficient variable selection. Genetics 2013, 195, 573–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Rodriguez, P.; Gianola, D.; González-Camacho, J.M.; Crossa, J.; Manes, Y.; Dreisigacker, S. Comparison between linear and non-parametric regression models for genome-enabled prediction in wheat. G3 Genes Genom. Genet. 2012, 2, 1595–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crossa, J.; Perez, P.; Campos, G.D.L.; Mahuku, G.; Dreisigacker, S.; Magorokosho, C. Genomic selection and prediction in plant breeding. J. Crop. Improv. 2011, 25, 239–261. [Google Scholar] [CrossRef]
- Poland, J.; Endelman, J.; Dawson, J.; Rutkoski, J.; Wu, S.; Manes, Y.; Dreisigackere, S.; Crossae, J.; Sanchez-Villedae, H.; Sorrells, M.; et al. Genomic selection in wheat breeding using genotyping-by-sequencing. Plant. Genome 2012, 5, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.Z. Genetic mapping and genomic selection using recombination breakpoint data. Genetics 2013, 195, 1103–1115. [Google Scholar] [CrossRef] [Green Version]
- Lorenzana, R.E.; Bernardo, R. Accuracy of genotypic value predictions for marker-based selection in biparental plant populations. Theor. Appl. Genet. 2009, 120, 151–161. [Google Scholar] [CrossRef]
- Stewart-Brown, B.B.; Song, Q.; Vaughn, J.N.; Li, Z. Genomic selection for yield and seed composition traits within an applied soybean breeding program. G3 Genes Genom. Genet. 2019, 9, 2253–2265. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; Chang, H.X.; Brown, P.J.; Domier, L.L.; Hartman, G.L. Genome-wide association and genomic prediction identifies soybean cyst nematode resistance in common bean including a syntenic region to soybean Rhg1 locus. Hortic. Res. 2019, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Windhausen, V.S.; Atlin, G.N.; Hickey, J.M.; Crossa, J.; Jannink, J.L.; Sorrells, M.E.; Raman, B.; Cairns, J.E.; Tarekegne, A.; Semagn, K.; et al. Effectiveness of genomic prediction of maize hybrid performance in different breeding populations and environments. G3 Genes Genom. Genet. 2012, 2, 1427–1436. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, A.J.; Smith, K.P.; Jannink, J.L. Potential and optimization of genomic selection for fusarium head blight resistance in six-row barley. Crop. Sci. 2012, 52, 1609–1621. [Google Scholar] [CrossRef]
- Gaffney, J.; Schussler, J.; Löffler, C.; Cai, W.; Paszkiewicz, S.; Messina, C.; Groeteke, J.; Keaschall, J.; Cooper, M. Industry-scale evaluation of maize hybrids selected for increased yield in drought-stress conditions of the US corn belt. Crop. Sci. 2015, 55, 1608–1618. [Google Scholar] [CrossRef]
- Cao, S.; Loladze, A.; Yuan, Y.; Wu, Y.; Zhang, A.; Chen, J.; Huestis, G.; Cao, J.; Chaikam, V.; Olsen, M.; et al. Genome-wide analysis of tar spot complex resistance in maize using genotyping-by-sequencing SNPs and whole-genome prediction. Plant. Genome 2017, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Clark, S.; van der Werf, J.H.J. Estimation of genomic prediction accuracy from reference populations with varying degrees of relationship. PLoS ONE 2017, 12, e0189775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Wang, X.; Ding, X.; Zheng, X.; Yang, Z.; Xu, C.; Hu, Z. Genomic selection of agronomic traits in hybrid rice using an NCII population. Rice 2018, 11, 32. [Google Scholar] [CrossRef] [Green Version]
- Juliana, P.; Poland, J.; Huerta-Espino, J.; Shrestha, S.; Crossa, J.; Crespo-Herrera, L.; Toledo, F.H.; Govindan, V.; Mondal, S.; Kumar, U.; et al. Improving grain yield, stress resilience and quality of bread wheat using large-scale genomics. Nat. Genet. 2019, 51, 1530–1539. [Google Scholar] [CrossRef]
- Liu, X.; Wang, H.; Wang, H.; Guo, Z.; Xu, X.; Liu, J.; Wang, S.; Li, W.-X.; Zou, C.; Prasanna, B.M.; et al. Factors affecting genomic selection revealed by empirical evidence in maize. Crop. J. 2018, 6, 341–352. [Google Scholar] [CrossRef]
- Hao, Y.; Wang, H.; Yang, X.; Zhang, H.; He, C.; Li, D.; Li, H.; Wang, G.; Wang, J.; Fu, J. Genomic prediction using existing historical data contributing to selection in biparental populations: A study of kernel oil in maize. Plant. Genome 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Contaldi, F.; Cappetta, E.; Esposito, S. Practical workflow from High Throughput Genotyping to Genomic Estimated Breeding Values (GEBVs). In Crop Breeding Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2021; in press. [Google Scholar]
- Esposito, S.; Carputo, D.; Cardi, T.; Tripodi, P. Applications and Trends of Machine Learning in Genomics and Phenomics for Next-Generation Breeding. Plants 2020, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Araus, J.L.; Cairns, J.E. Field high-throughput phenotyping: The new crop breeding frontier. Trends Plant. Sci. 2014, 19, 52–61. [Google Scholar] [CrossRef]
- Panthee, D.R.; Labate, J.A.; McGrath, M.T.; Breksa, A.P.; Robertson, L.D. Genotype and environmental interaction for fruit quality traits in vintage tomato varieties. Euphytica 2013, 193, 169–182. [Google Scholar] [CrossRef]
- Daniel, I.O.; Atinsola, K.O.; Ajala, M.O.; Popoola, A.R. Phenotyping a Tomato Breeding Population by Manual Field Evaluation and Digital Imaging Analysis. Int. J. Plant. Breed. Genet. 2017, 11, 19–24. [Google Scholar] [CrossRef]
- Meuwissen, T.; Hayes, B.; Goddard, M. Accelerating improvement of livestock with genomic selection. Annu. Rev. Anim. Biosci. 2013, 1, 21–237. [Google Scholar] [CrossRef] [PubMed]
- Sarinelli, J.M.; Murphy, J.P.; Tyagi, P.; Holland, J.B.; Johnson, J.W.; Mergoum, M.; Mason, R.E.; Babar, A.; Harrison, S.; Sutton, R.; et al. Training population selection and use of fixed effects to optimize genomic predictions in a historical USA winter wheat panel. Theor. Appl. Genet. 2019, 132, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schopp, P.; Müller, D.; Technow, F.; Melchinger, A.E. Accuracy of genomic prediction in synthetic populations depending on the number of parents, relatedness, and ancestral linkage disequilibrium. Genetics 2017, 205, 441–454. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.M.; Buntjer, J.B.; Jackson, R.; Bentley, A.R.; Lage, J.; Byrne, E.; Burt, C.; Jack, P.; Berry, S.; Flatman, E.; et al. The effects of training population design on genomic prediction accuracy in wheat. Theor. Appl. Genet. 2019, 132, 1943–1952. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, N.H.; Jahoor, A.; Jensen, D.; Orabi, J.; Cericola, F.; Edriss, V.; Jensen, J. Genomic prediction of seed quality traits using advanced barley breeding lines. PLoS ONE 2016, 1, e0164494. [Google Scholar] [CrossRef] [Green Version]
- Cericola, F.; Jahoor, A.; Orabi, J.; Andersen, J.R.; Janss, L.L.; Jensen, J. Optimizing training population size and genotyping strategy for genomic prediction using association study results and pedigree information. A case of study in advanced wheat breeding lines. PLoS ONE 2017, 12, e0169606. [Google Scholar] [CrossRef]
- Zhang, H.; Yin, L.; Wang, M.; Yuan, X.; Liu, X. Factors affecting the accuracy of genomic selection for agricultural economic traits in maize, cattle, and pig populations. Front. Genet. 2019, 10, 189. [Google Scholar] [CrossRef] [Green Version]
- Alabady, M.S.; Rogers, W.L.; Malmberg, R.L. Development of transcriptomic markers for population analysis using restriction site associated RNA sequencing (RARseq). PLoS ONE 2015, 10, e0134855. [Google Scholar] [CrossRef]
- Barchi, L.; Acquadro, A.; Alonso, D.; Aprea, G.; Bassolino, L.; Demurtas, O.; Ferrante, P.; Gramazio, P.; Mini, P.; Portis, E.; et al. Single Primer Enrichment Technology (SPET) for High-Throughput Genotyping in Tomato and Eggplant Germplasm. Front. Plant. Sci. 2019, 10, 1005. [Google Scholar] [CrossRef] [Green Version]
- Sim, S.C.; Van Deynze, A.; Stoffel, K.; Douches, D.S.; Zarka, D.; Ganal, M.W.; Chetelat, R.T.; Hutton, S.F.; Scott, J.W.; Gardner, R.G.; et al. High-Density SNP Genotyping of Tomato (Solanum lycopersicum L.) Reveals Patterns of Genetic Variation Due to Breeding. PLoS ONE 2012, 7, e45520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smykal, P.; Nelson, M.N.; Berger, J.D.; von Wettberg, E.J.B. The impact of genetic changes during crop domestication. Agron 2018, 8, 119. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Gonda, I.; Sun, H.; Ma, Q.; Bao, K.; Tieman, D.M.; Burzynski-Chang, E.A.; Fish, T.L.; Stromberg, K.A.; Sacks, G.L.; et al. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 2019, 51, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Gonda, I.; Ashrafi, H.; Lyon, D.A.; Strickler, S.R.; Hulse-Kemp, A.M.; Ma, Q.; Sun, H.; Stoffel, K.; Powell, A.F.; Futrell, S.; et al. Sequencing-based bin map construction of a tomato mapping population, facilitating high-resolution quantitative trait loci detection. Plant. Genome 2019, 12, 180010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltecca, C.; Parker, K.L.; Cassady, J.P. Application of multiple shrinkage methods to genomic predictions. J. Anim. Sci. 2012, 90, 1777–1787. [Google Scholar] [CrossRef]
- Heslot, N.; Rutkoski, J.; Poland, J.; Jannink, J.L.; Sorrells, M.E. Impact of marker ascertainment bias on genomic selection accuracy and estimates of genetic diversity. PLoS ONE 2013, 8, e74612. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, J.C.; Thompson, R.; Denham, M.C. Marker-assisted selection using ridge regression. Genet. Res. 2000, 75, 249–252. [Google Scholar] [CrossRef]
- De los Campos, G.; Hickey, J.M.; Pong-Wong, R.; Daetwyler, H.D.; Calus, M.P.L. Whole-genome regression and prediction methods applied to plant and animal breeding. Genetics 2013, 193, 327–345. [Google Scholar] [CrossRef] [Green Version]
- Bassi, F.M.; Bentley, A.R.; Charmet, G.; Ortiz, R.; Crossa, J. Breeding Schemes for the Implementation of Genomic Selection in Wheat (Triticum Spp.). Plant. Sci. 2016, 242, 23–36. [Google Scholar] [CrossRef]
- Falk, D.E. Generating and maintaining diversity at the elite level in crop breeding. Genome 2010, 53, 982–991. [Google Scholar] [CrossRef]
- Rahim, M.S.; Bhandawat, A.; Rana, N.; Sharma, H.; Parveen, A.; Kumar, P.; Madhawan, A.; Bisht, A.; Sonah, H.; Sharma, T.R.; et al. Genomic Selection in Cereal Crops: Methods and Applications. In Accelerated Plant Breeding: Cereal Crops; Gosal, S.S., Wani, S.H., Eds.; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Ercolano, M.R.; Sacco, A.; Ferriello, F.; D’Alessandro, R.; Tononi, P.; Traini, A.; Barone, A.; Zago, E.; Chiusano, M.L.; Buson, G.; et al. Patchwork sequencing of tomato San Marzano and Vesuviano varieties highlights genome-wide variations. BMC Genom. 2014, 15, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanbar, A.; Kondo, K.; Shashidhar, H.E. Comparative efficiency of pedigree, modified bulk and single seed descent breeding methods of selection for developing high-yielding lines in rice (Oryza sativa L.) under aerobic condition. Electron. J. Plant. Breed. 2011, 2, 184–193. [Google Scholar]
- Breseghello, F.; Morais, O.P.; Castro, E.M.; Prabhu, S.A.; Bassinello, P.Z.; Pereira, J.P.; Utumi, M.M.; Ferreira, M.E.; Soares, A.A. Recurrent selection resulted in rapid genetic gain for upland rice in Brazil. Int. Rice Res. Notes 2009, 34, 1–4. [Google Scholar] [CrossRef]
- Shelton, A.; Tracy, W.; Shelton, A.C.; Tracy, W.F. Recurrent selection and participatory plant breeding for improvement of two organic open-pollinated sweet corn (Zea mays L.) populations. Sustainability 2015, 7, 5139–5152. [Google Scholar] [CrossRef] [Green Version]
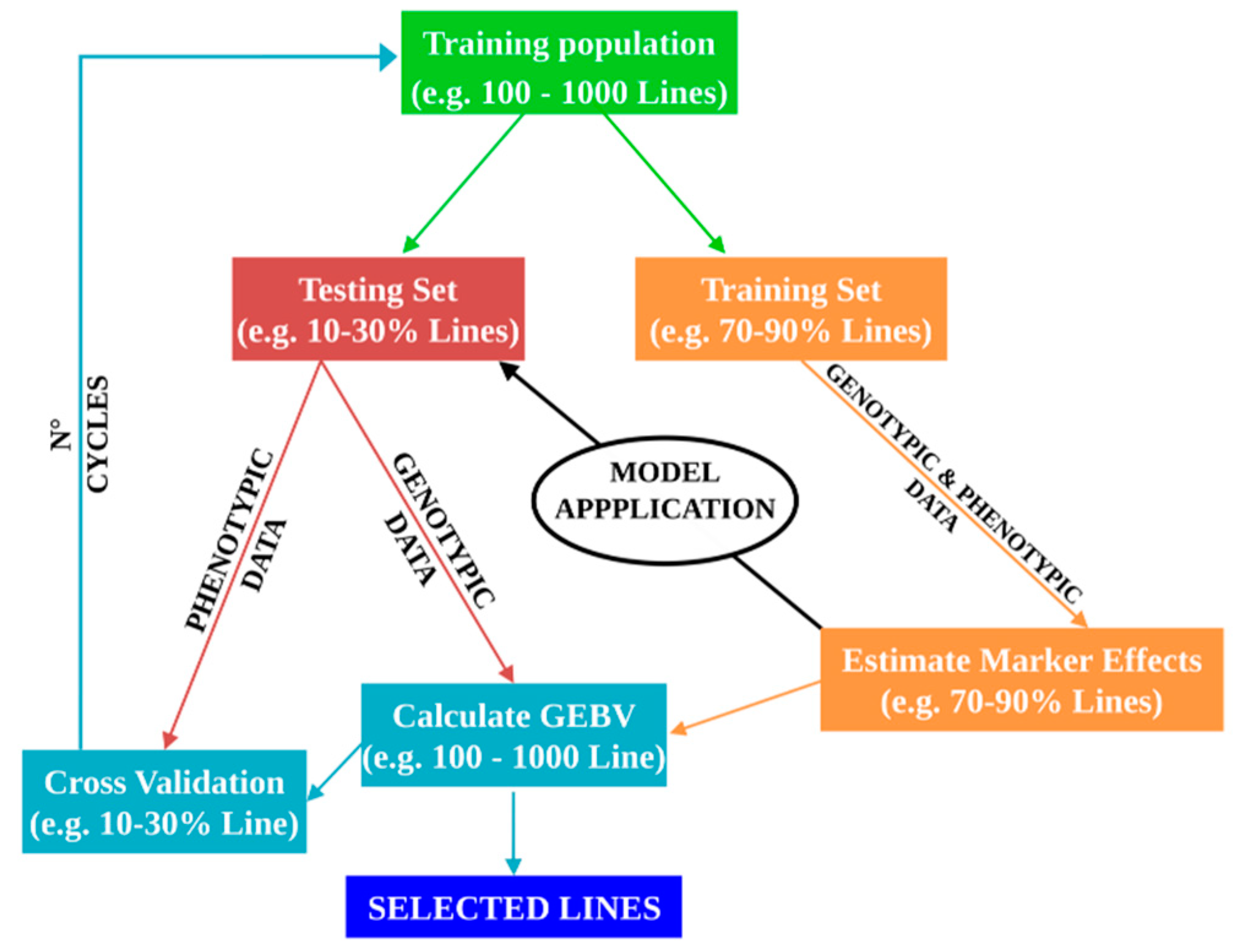

{kind=link}
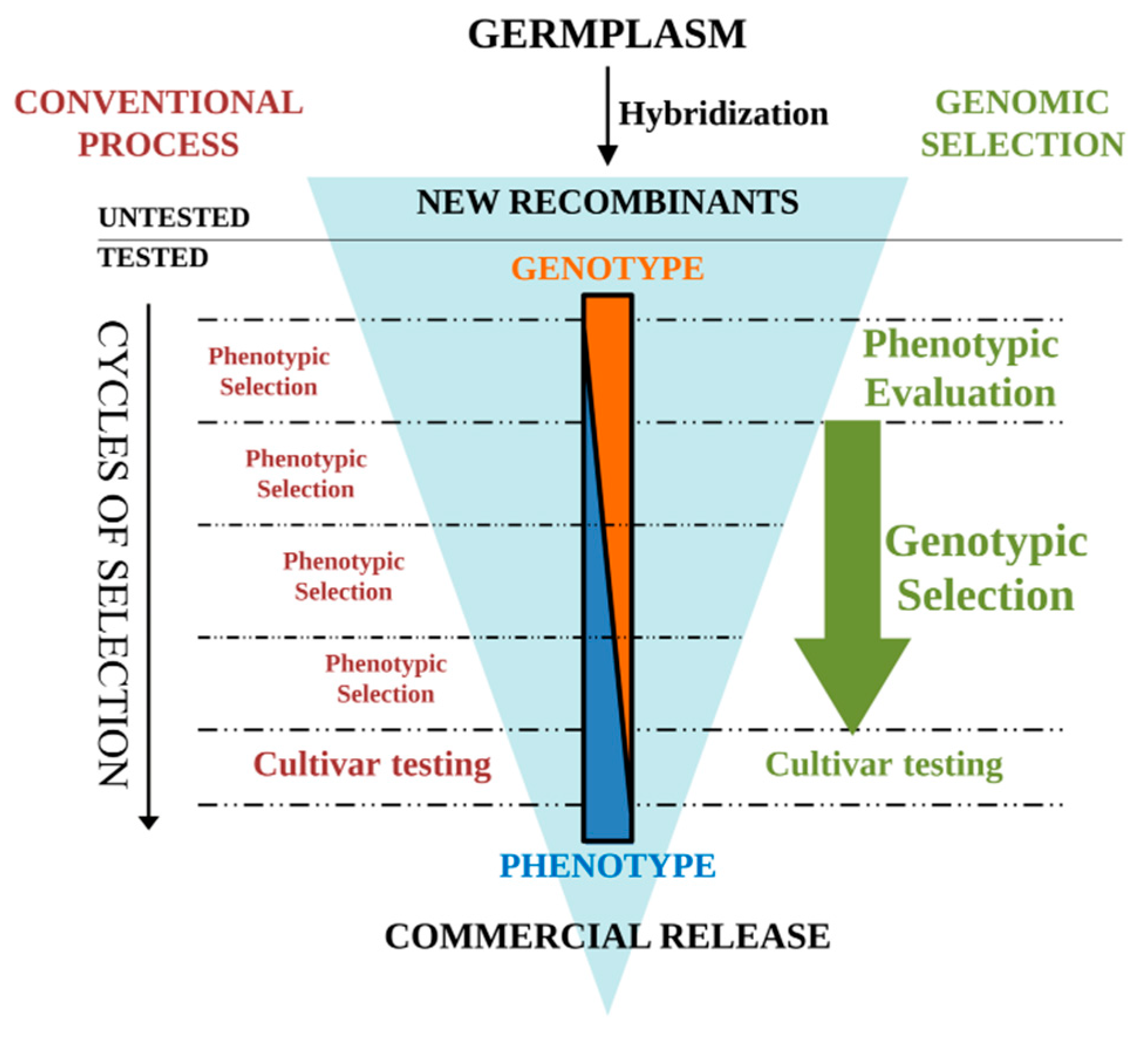
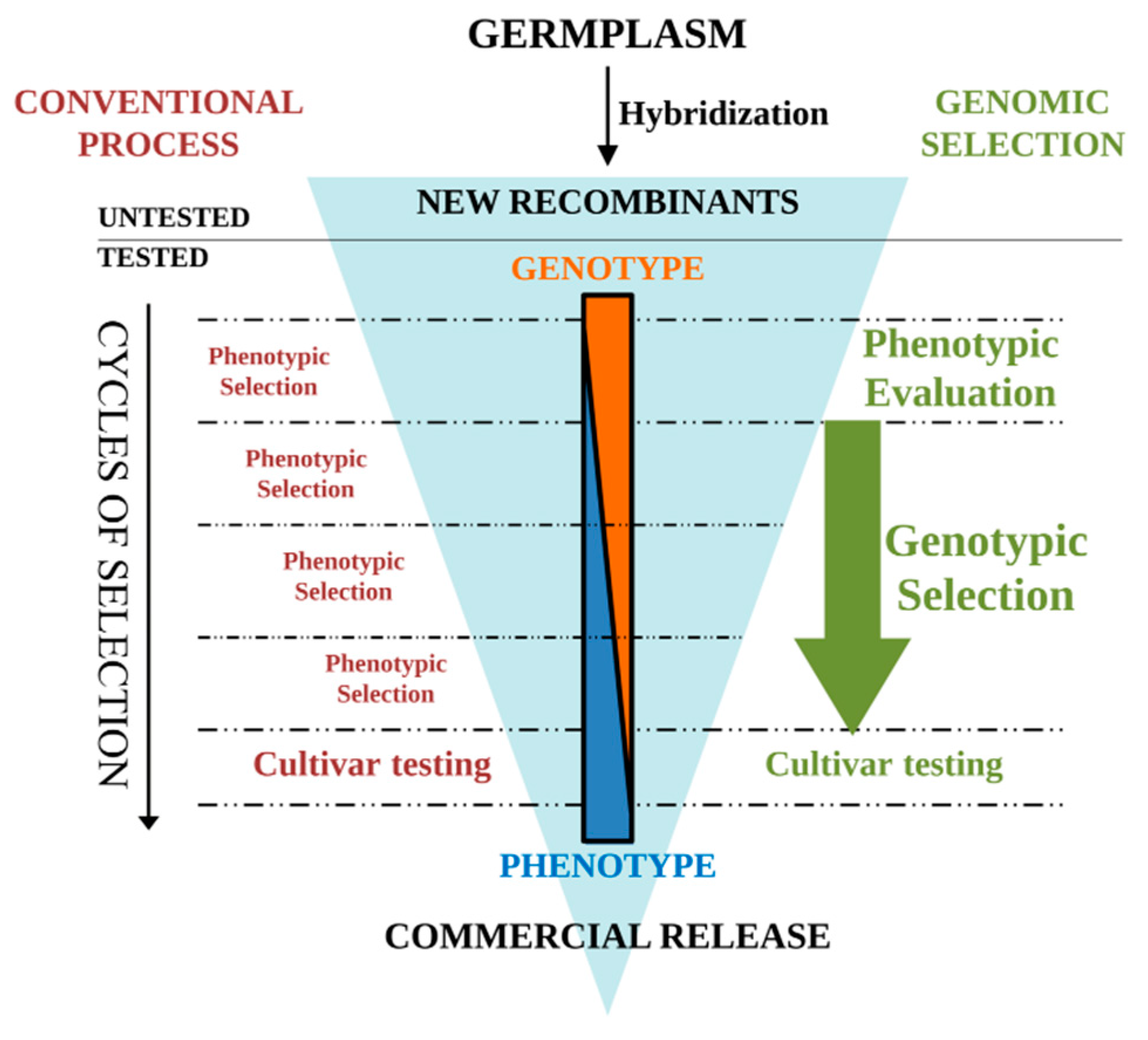{kind=link}
| Species | Traits | TRN Size and Type | No Markers | Statistical Model | Accuracy | References |
|---|---|---|---|---|---|---|
| Wheat | GY | 374 inbred lines | 1158 DArTs | RR-BLIP, Bayes-A, B, C | 0.21 | [53] |
| Wheat | GY, HD | 306 lines CIMMYT | 1717 DArTs | RR-BLUP, Bayes-A, B, LASSO, RKHS, RBFNN, BRNN | 0.7 0.5–0.6 | [55] |
| Wheat | GY | 254 lines CIMMYT | 2056 SNPs | LASSO, Bayes-b, RR-BLUP | 0.43–0.51 | [54] |
| Wheat | GY | 94 lines CIMMYT | 234 DArTs | Bayes-LASSO-RKHS | 0.43–0.79 | [56] |
| Wheat | GY | 254 lines CIMMYT | 34,749 SNPs | GBLUP | 0.2–0.4 | [57] |
| Rice | FT | 413 varietes | 36,901 SNPs | LASSO, Bayes-b, RR-BLUP | ~0.5 | [54] |
| Rice | YP, FT, WSY | 210 Inbred lines | 270,820 SNPs | LASSO | 0.16–0.26–0.98 | [58] |
| Arabidopsis | FT | 199 inbred lines | 215,908 SNPs | RR-BLUP | 0.65–0.75 | [54] |
| Arabidopsis | FT, DM | 415 RILs | 69 SSRs | BLUP | 0.90–0.93 | [59] |
| Soybean | YP, PO | 540 (RILs) | 2647 SNPs | RR-BLUP | 0.81, 0.71, 0.26 | [60] |
| Soybean | nematode resistance | 363 Genotypes | 84,416 SNPs | RR-BLUP | 0.41–0.52 | [61] |
| Maize | GY, ASI | 255 inbred lines | 37,403 SNPs | RR-BLUP | ~0.5 | [62] |
| Maize | GY, FF, MF, ASI | 300 lines CIMMYT | 1148 SNPs | M-BL | 0.42–0.79 | [27] |
| Barley | GY, AA | 150 DHs | 223 RFLPs | BLUP | 0.64–0.83 | [59] |
| Barley | PH, CC | 140 DHs | 107 RFLPs, AFLPs | BLUP | 0.66–0.85 | [59] |
| Tomato | SSC, FW | 96 F1 varietes | 337 SNPs | GBLUP, Bayesian Lasso, Wbsr, BayesC, RKHS, RF | 0.56–0.68 0.22–0.27 | [24] |
| Tomato | Metabolic and quality traits | 163 Genotypes | 5995 SNPs | RR-BLUP | 0.05–0.81 | [22] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappetta, E.; Andolfo, G.; Di Matteo, A.; Barone, A.; Frusciante, L.; Ercolano, M.R. Accelerating Tomato Breeding by Exploiting Genomic Selection Approaches. Plants 2020, 9, 1236. https://doi.org/10.3390/plants9091236
Cappetta E, Andolfo G, Di Matteo A, Barone A, Frusciante L, Ercolano MR. Accelerating Tomato Breeding by Exploiting Genomic Selection Approaches. Plants. 2020; 9(9):1236. https://doi.org/10.3390/plants9091236
Chicago/Turabian StyleCappetta, Elisa, Giuseppe Andolfo, Antonio Di Matteo, Amalia Barone, Luigi Frusciante, and Maria Raffaella Ercolano. 2020. "Accelerating Tomato Breeding by Exploiting Genomic Selection Approaches" Plants 9, no. 9: 1236. https://doi.org/10.3390/plants9091236
APA StyleCappetta, E., Andolfo, G., Di Matteo, A., Barone, A., Frusciante, L., & Ercolano, M. R. (2020). Accelerating Tomato Breeding by Exploiting Genomic Selection Approaches. Plants, 9(9), 1236. https://doi.org/10.3390/plants9091236